1. Introduction
Graphite is the anode material of commercial Li-ion batteries because it offers low working potential, prolonged cycle life, and relatively low cost. However, during the cell formation a portion of the lithium made available from the positive electrode is consumed (irreversibly lost) to form a stable solid electrolyte interphase (or SEI) on the graphite surface [
1]. Therefore, the coulombic efficiency only reaches about 90% in the first cycle, thus resulting in a permanent capacity loss of the cell. To address the irreversible losses during the first cycle, graphite with low surface area (i.e., below 10 m
2g
−1) is generally preferred due to the lower extent of electrolyte decomposition reactions. Another limitation of graphite is its poor rate performance, especially at reduced (sub-ambient) temperatures where lithium metal deposition may occur. For this, reduced graphite particle size would positively affect the solid-state lithium diffusion, leading to higher delivered capacity at high rates [
2]. However, the corresponding higher surface area would result in higher SEI formation and, thus, higher first cycle irreversible capacity loss. Other factors such as particle morphology, electrode thickness, porosity, choice of binder, and electrolyte composition affect the rate capability of graphite as well [
3,
4,
5]. Furthermore, surface properties (e.g., presence of defects and resistivity of the passivation layer) are also important as a fast charge transfer at the interface of graphite with the electrolyte, and the current collector is a key factor for high rate performance.
These considerations drive the interest in graphite surface modification via polymers [
6], metals [
7], and carbon coatings [
8,
9,
10,
11]. Focusing on this latter case, it has been reported that amorphous carbon, being isotropic, allows random Li
+ intercalation, which may lead to a better rate capability of carbon-coated graphite [
11]. Improved cycling performance of carbon-coated graphite in the electrolytes containing propylene carbonate (PC) has also been reported to show the protective effect of the coating towards solvent co-intercalation [
12].
Several research groups have been working on the use of different polymers as carbon precursors. For example, Wang et al. [
11] coated artificial graphite with a thin carbon layer derived from glucose. They concluded that the coating could increase the specific capacity and initial coulombic efficiency. The best results were obtained using an aqueous solution (5 wt %) of glucose. Nozaki et al. [
10] rationalized the use of different thermoplastic polymers as carbon sources, dividing them into three categories, based on the amount of carbon residue, which directly influences the irreversible capacity loss during the first charge (lithiation) and cycling performance.
Based on the literature survey, the expected performance improvement upon carbon coating is not always observed as it strongly depends on the amount and nature of the carbon layer, the graphite characteristics, and the electrolyte composition [
5,
12,
13]. This makes data comparison very difficult, and thus each case has to be evaluated individually via a systematic investigation. In addition, the coating process should be cost competitive and not detrimental to the environmental. Thus, the carbon precursor should be readily available and the process should employ a relatively low temperature and reduced organic solvent amounts.
Along these lines, we systematically investigated the effect of different carbon precursors on the electrochemical performance of graphite anodes, focusing on non-toxic, cheap, and abundant compounds. The influence of the material preparation and the amount of carbon residue was evaluated as well. The electrochemical tests were conducted using graphite electrodes with loadings, appropriate for application in lithium-ion batteries (2.4–2.7 mAh cm−2).
2. Results
The choice of carbon precursors was based on the goal of limiting the environmental impact associated with the coating process. Therefore, precursors requiring processing in organic solvents or releasing toxic compounds during the thermal treatment (e.g., poly(vinyl chloride)) [
10] were not considered. Glucose, sucrose, and citric acid (CA) have been extensively studied as carbon sources for positive and negative electrode coatings [
14,
15,
16,
17]. Polymer-type precursors, such as poly(acrylic acid) (PAA) and poly(vinyl alcohol) (PVA), which possibly form a more homogeneous network around the graphite particles, thus leading to a better dispersion of carbon on the surface, were also investigated [
10,
18,
19]. For the selection of the most suitable coating method, sucrose was used as a carbon source and the graphite:sucrose weight ratio was set to 1:1.
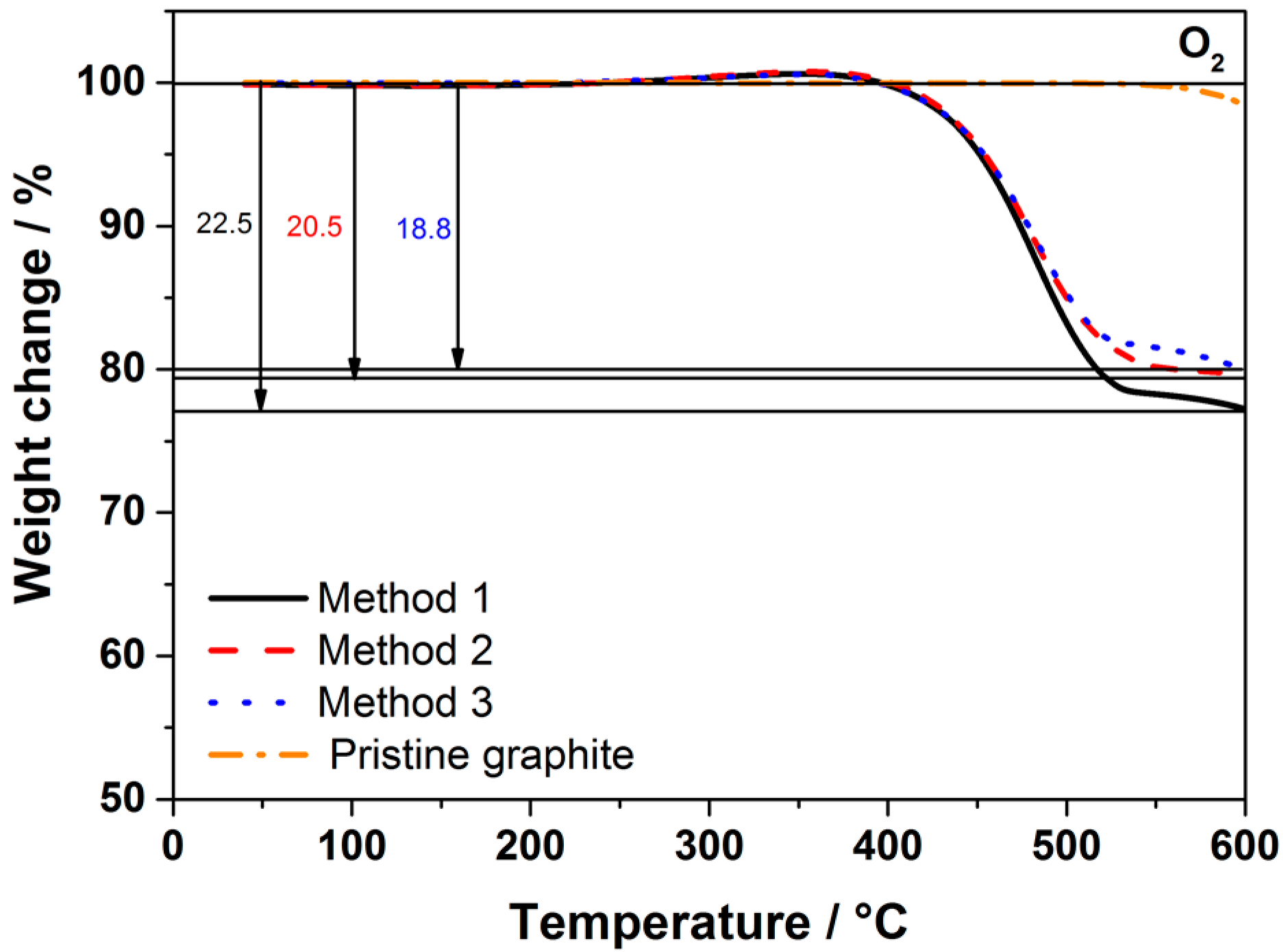
Figure 1 shows the Thermogravimetric analysis (TGA) traces of the carbon-coated graphite obtained using the three methods, which are described in the experimental section and summarized in
Table 1. Pristine graphite is stable up to 550 °C; therefore, the weight loss at lower temperatures is mainly attributed to the carbon coating. TGA results of the coated graphite using sucrose via three synthesis routes revealed that the coating accounts for about 20 wt % of the total mass, independent of the mixing method.
Figure 2 shows the scanning electron microscopy (SEM) images of the pristine and coated graphite materials. In all coated samples, but especially in those obtained via methods 2 and 3, the graphite particles are agglomerated. Method 1 was selected for further investigations since “wet” coating methods yield more homogeneous coating layers with respect to “dry” methods [
14], and the introduction of the ball milling step (method 3) did not lead to any clear advantage. SEM images of the carbon-coated samples show that all synthesis procedures resulted in the modification of the graphite surface, which appears rougher than that of pristine graphite. This indicates the presence of a coating layer, which is, however, difficult to visualize by SEM.
Method 1 was then extended to glucose and PAA as carbon sources, while maintaining the 1:1 graphite:precursor weight ratio. The carbon yield obtained with glucose was similar to that of sucrose (~20 wt %), whereas the use of PAA as a precursor yielded a lower amount of carbon (about 13 wt %, result not shown here). Since a high content of amorphous carbon can exacerbate the irreversible reactions taking place at the anode/electrolyte interface [
9,
10], 5 wt % of residual carbon was targeted for the coatings using all precursors. The characteristics of the coated samples are reported in
Table 2. It is interesting to note that, although the amount of carbon coating is almost the same when employing water solutions of glucose and citric acid, the Brunauer-Emmett-Teller (BET) surface area of the coated samples increases with respect to that of pristine graphite. In the case of sucrose, the surface area of the final product is three times higher. An inverse effect is obtained with the polymeric precursors, leading to comparable or smaller surface areas of the coated samples with respect to pristine graphite.
As it can be seen from the SEM images in
Figure 3, the 5 wt % carbon-coated samples appear to be less agglomerated than those in
Figure 2. The coated graphite particles have a similar morphology except for the sample coated using PAA, which displays the additional formation of needle-like carbon particles.
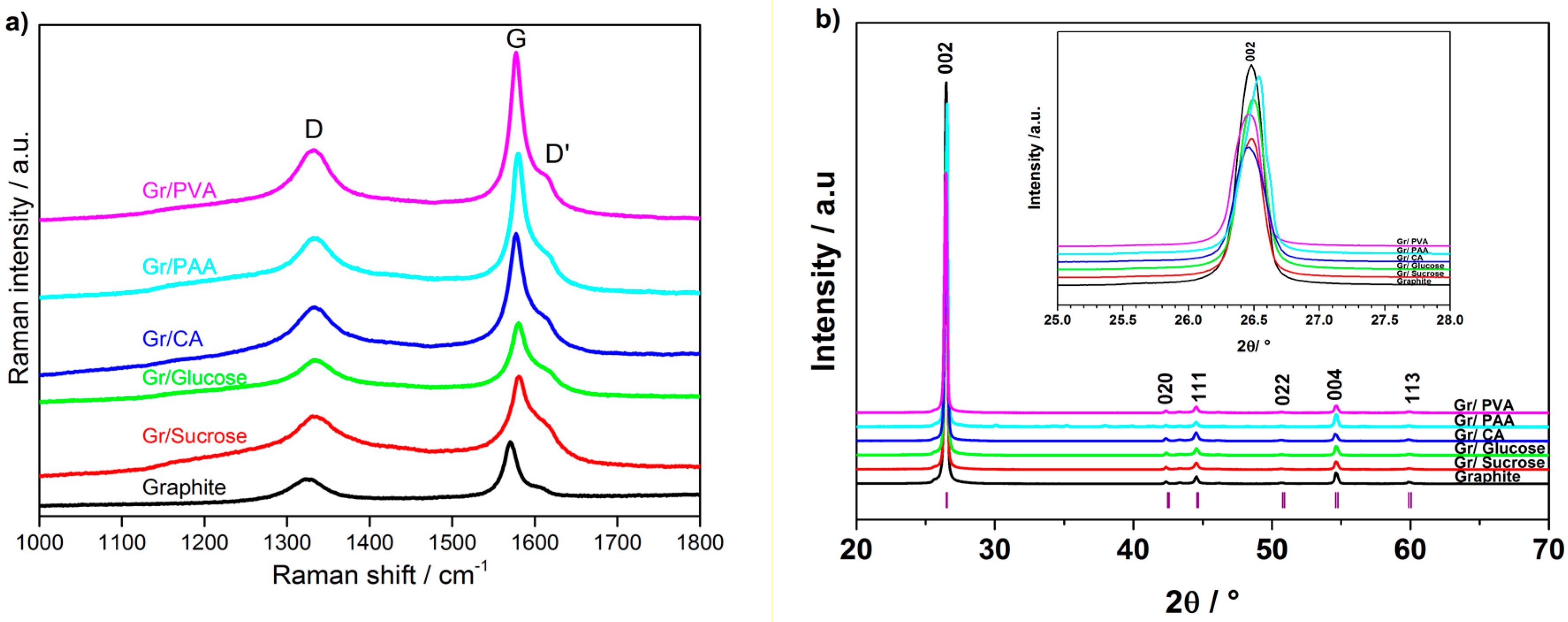
The change of the graphite crystallinity due to the formation of the amorphous carbon layer on its surface was evaluated using Raman spectroscopy. The Raman spectra of the pristine and carbon-coated graphite materials are shown in
Figure 4a. The spectrum of pristine graphite exhibits an intense peak at 1572 cm
−1, marked with G, which is associated with sp
2 carbon bonds stretching in the basal plane of graphite. The smaller peak at 1325 cm
−1, marked with D, is attributed to the breathing mode of sp
3 atoms at the edge sites of graphite [
20,
21]. The small shoulder at 1603 cm
−1, marked as D′, is also indicative of defects in the crystalline structure of graphite [
22]. The D/G area ratio obtained from Raman spectroscopy is commonly used as an indicator of the extent of surface disorder in carbonaceous materials [
8]. The coated samples, except that derived from PVA, have higher D/G ratios than graphite, thus confirming the surface modification due to the carbon coating.
Figure 4b reports the X-ray diffraction (XRD) patterns of pristine and carbon-coated graphite samples. The carbon coating does not change the graphite structure. However, a shift towards higher angles is observed for the materials prepared using citric acid and PAA, as shown in the inset of
Figure 4b.
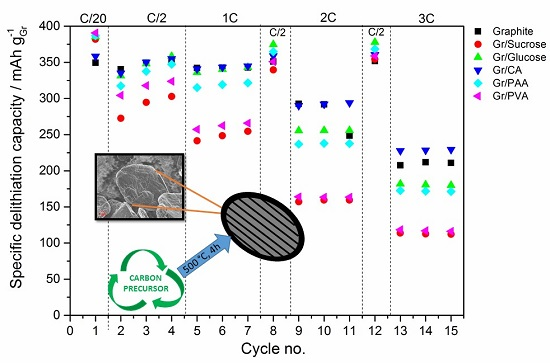
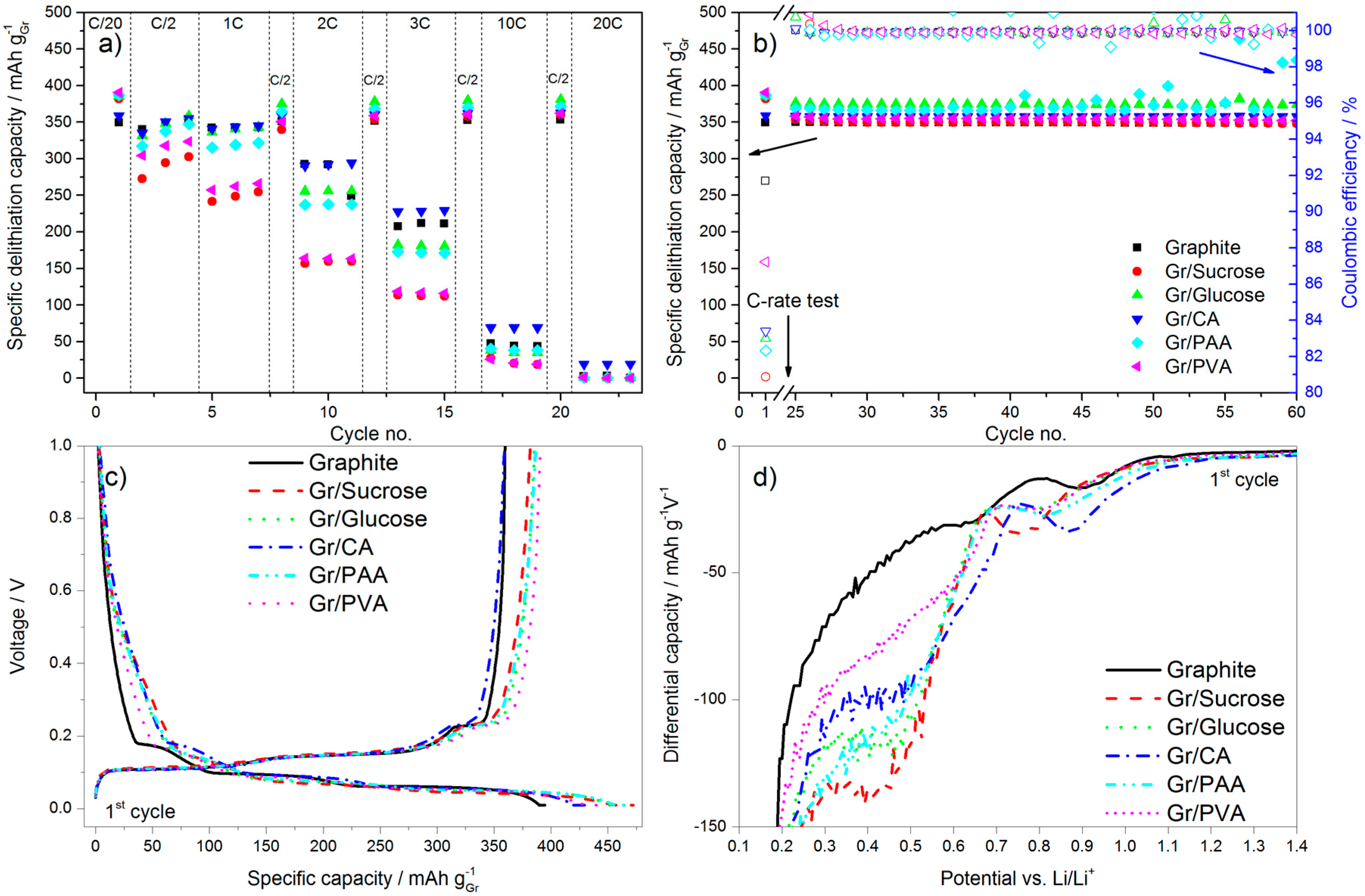
In terms of electrochemical performance, the effect of the carbon coating (~5 wt %) from the various precursors on the discharge rate of graphite is reported in
Figure 5. The galvanostatic charge rate was kept constant (C/2) with the addition of the constant voltage step at the end of the galvanostatic charge to accomplish the full lithiation of the anode. In
Figure 5a the results of the C-rate test are reported, while
Figure 5b shows the subsequent cycling at C/2.
Figure 5c,d reports, respectively, the first cycle voltage profile and a portion of the relative differential capacity plot.
During the first delithiation (discharge) the carbon-coated graphite electrodes deliver a higher capacity than the pristine material. However, the values of the first cycle coulombic efficiency (calculated as the ratio of the capacity delivered during delithiation divided by the capacity taken up during lithiation in the same cycle) obtained for the former electrodes are lower than that of pristine graphite, especially for the samples with the higher surface area (i.e., Gr/Sucrose, Gr/Glucose and Gr/CA). This is due to extended SEI formation occurring in the voltage region between 1.0 and 0.2 V. As shown in
Figure 5c,d, below 1.0 V the decomposition of fluoroethylene carbonate (FEC) takes place [
23], the extent of which is larger for the carbon-coated materials as indicated by the relative peaks in
Figure 5d being more intense. The reduction peak just below 0.7 V in graphite is related to the decomposition of the electrolyte solvents (mainly EC), which is not completely prevented by the addition of FEC [
24]. For the carbon-coated graphite electrodes, the electrolyte reduction peak is shifted to a lower voltage (~0.5 V) and its intensity is higher for the samples that have higher surface area than pristine graphite such as Gr/Glucose, Gr/Sucrose, and Gr/PAA.
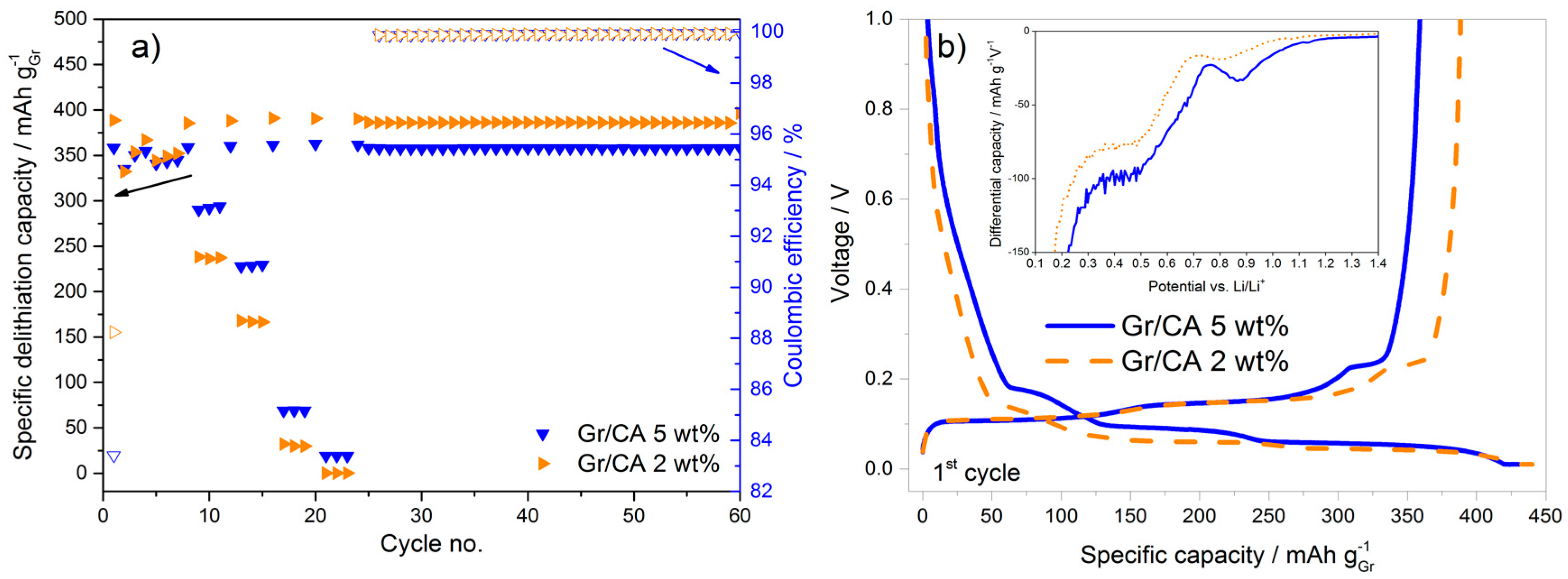
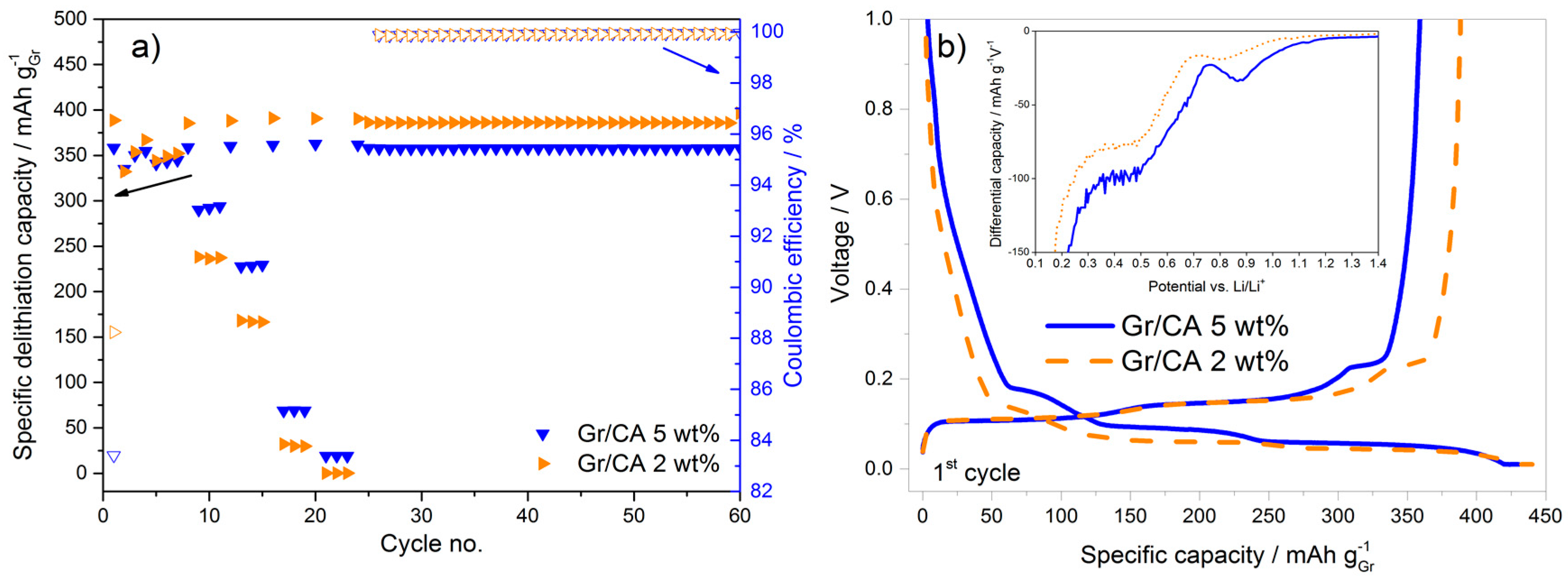
In terms of rate capability improvement, citric acid seems to be the most appropriate carbon precursor. In an attempt to further improve the performance, the amount of carbon coating was reduced from 5 to 2 wt %. This corresponds to the surface area reduction of 50% and, in fact, the first cycle coulombic efficiency increases (from 83.4% to 88.6%), as shown in
Figure 6a. This can be univocally attributed to the lower surface area available for the SEI formation as confirmed by the voltage profile and differential capacity plots in
Figure 6b. However, this approach leads to a decrease of the rate capability, as the discharge capacity at 3C for the sample with 2 wt % carbon-coating is 30% lower than that obtained using the 5 wt % carbon-coating. The higher capacity upon constant cycling at C/2 with 2 wt % carbon-coating is additionally contributed by the moderate variation of active material loading in the electrodes.
Carbon coating was reported to allow Li
+ intercalation/deintercalation into graphite even in non-SEI forming electrolytes (i.e., without ethylene carbonate (EC)) [
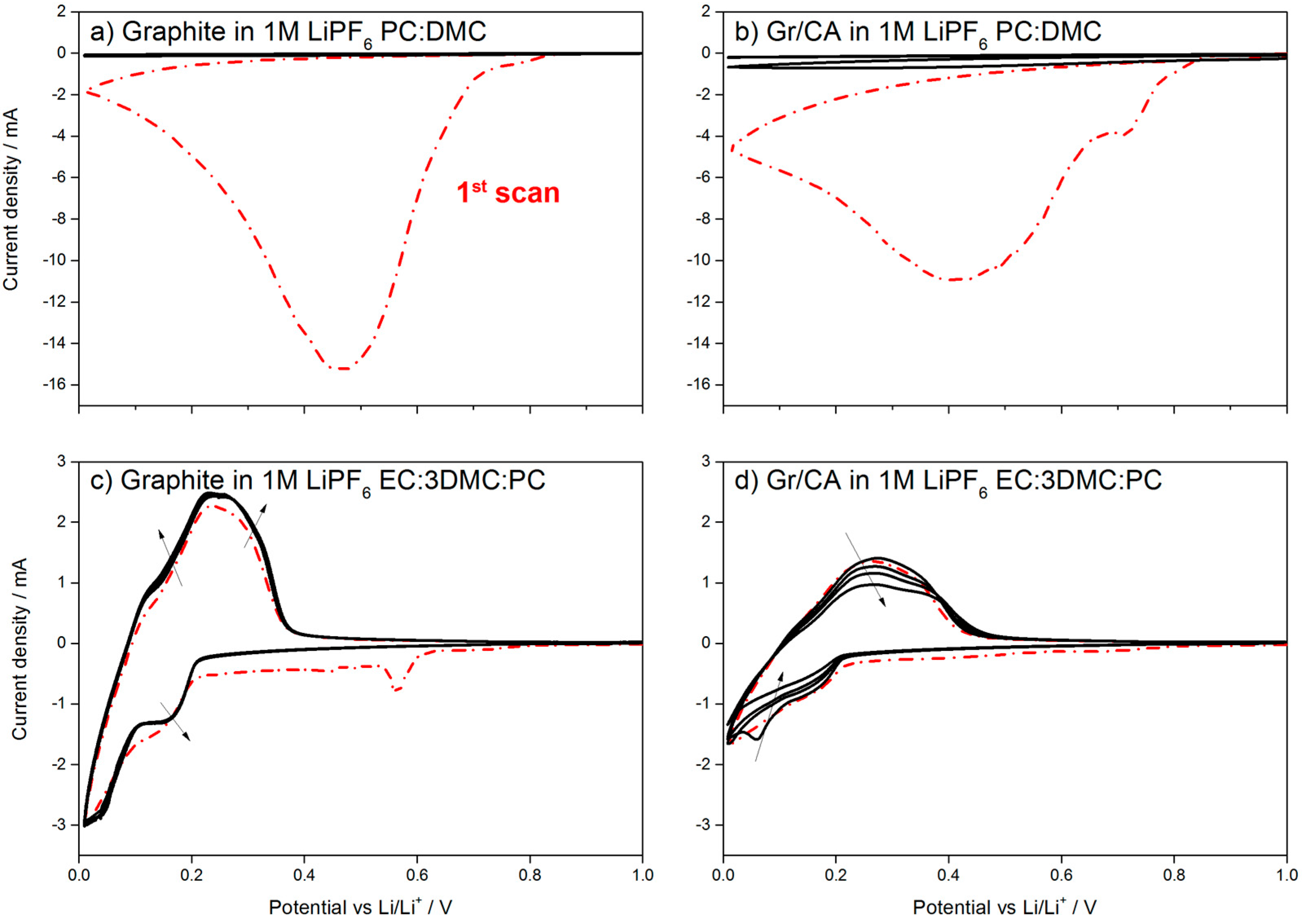
23]. Indeed, the possibility of using electrolytes with a high amount of PC in combination with dimethyl carbonate (DMC) would be beneficial for the low temperature performance as PC has a lower melting point than EC. However, the use of PC is commonly prevented by the occurrence of graphite exfoliation upon solvent co-intercalation, as shown in the cyclic voltammetry (CV) in
Figure 7a, where 1M LiPF
6 in PC:DMC was used as the electrolyte.
Figure 7b shows that the presence of 5 wt % of carbon coating is not sufficient to protect graphite. When the test is repeated using a ternary solvents mixture containing 16.67 wt % of PC (EC:DMC:PC (1/3/1,
w/
w/
w)), one peak in the cathodic sweep is observed at 0.6 V for the pristine graphite, as shown in
Figure 7c. This peak indicates the formation of the protective SEI [
24], which enables reversible cycling of the uncoated material. However, in the case of 5 wt % carbon-coating, no peak is seen until 0.2 V
vs. Li/Li
+. This results in the continuous Li
+ consumption, as shown by the constant decrease of the peak intensity in
Figure 7d.
3. Discussion
The change in the morphology and non-sp2 carbon content observed using different synthesis routes and carbon precursors confirms the successful formation of the carbon layer on the graphite surface. An increase in the surface disorder is supported by the broadening of D and D′ bands upon carbon coating, as indicated by Raman spectroscopy. The coated samples also have additional shoulders on both sides of the D band, which can be associated with coating-induced defects and/or the presence of heteroatoms on the graphite surface. The degree of surface “amorphization”, expressed by the D/G ratio, reaches a maximum with sucrose as the carbon precursor. Gr/Sucrose and Gr/PVA samples have rather different surface areas, highlighting the different impact of the carbon precursors on the final coated samples. The XRD patterns show that the carbon coating does not significantly change the graphite structure. However, the intensity decrease of the (002) diffraction peak and its broadening may indicate the reduced crystallinity of the coated materials with respect to the pristine graphite. Furthermore, the pattern of the sample using PAA contains additional peaks, which confirms the presence of crystalline impurities, i.e., needle-like particles, also observed in the SEM images, most likely arising from incomplete precursor decomposition.
The results of the cycling test in half-cells show that the presence of a high surface area amorphous carbon layer intensifies the electrolyte decomposition reactions during SEI formation, leading to lower values of first cycle coulombic efficiency. Indeed, for the sample coated with PVA, which has only half the surface area of pristine graphite, the coulombic efficiency also approaches that of the uncoated material. It should also be noted that the calculations of the current densities were only based on the graphite loading, although the carbon coating is also electrochemically active and contributes to the total delivered capacity values. This results in the slight capacity increase of 30 mAh g
−1 upon first lithiation (charge) and 10 mAh g
−1 upon the following cycling steps as compared to the pristine graphite. Upon increasing the cycling rates, only the sample coated with citric acid displays a slightly higher discharge capacity than pristine graphite. Surprisingly, the lowest capacity values are obtained from the graphite coated using sucrose and PVA, which have the highest and the lowest surface area, respectively. However, when the cycle rate is brought back to C/2, the Gr/PAA and Gr/Glucose electrodes deliver slightly higher capacities than pristine graphite, indicating that the carbon coating participates in the Li
+ storage process [
25]. This suggests that the amount of Li
+ stored in the coating varies with the precursor and the final carbon nature, thickness, and homogeneity of the layer [
9,
15].
Upon decreasing the carbon coating amount from 5 to 2 wt % the increase of the first cycle coulombic efficiency is obtained, but at the expense of the decreased rate capability. Therefore, the amount of carbon in the coating needs to be optimized in order to achieve the best tradeoff between these two parameters.
As shown in the CV test, 5 wt % of carbon coating on graphite is not sufficient to protect it from exfoliation in PC-rich electrolyte, which is in line with other reports, highlighting that more than 10 wt % of carbon coating is necessary [
12,
23,
25]. When the ternary electrolyte with EC is used, however, the carbon coating prevents the formation of the SEI on the graphite electrode, which leads to the continuous capacity fading upon cycling.
4. Materials and Methods
Commercial synthetic graphite, glucose (Sigma-Aldrich, St. Louis, MO, USA; purity ≥99.0%), sucrose (VWR Chemicals, Radnor, PA, USA; D(+) anhydrous), citric acid (Sigma-Aldrich, St. Louis, MO, USA; purity ≥99.5%), poly(acrylic acid) (PAA, Sigma-Aldrich, St. Louis, MO, USA; Mw~450,000), and poly(vinyl alcohol) (PVA, Sigma-Aldrich, St. Louis, MO, USA; Mw~130,000, purity ≥99.0% hydrolyzed) were used as received. Ethylene carbonate (EC), dimethyl carbonate (DMC), propylene carbonate (PC), fluoroethylene carbonate (FEC), were all obtained from UBE (Tokio, Japan), and lithium hexafluorophosphate (LiPF6, Sigma Aldrich, St. Louis, MO, USA) was also used as received. Electrolyte solutions were prepared in a glove-box (water and oxygen content below 0.1 ppm, MBraun, Stratham, NH, USA) by mixing the components and stirring them for 12 h.
Three different mixing methods of graphite and carbon precursor were investigated. In method 1, the precursor and graphite were mixed in ultra-pure water for 8 h. The resulting mixture was dried in an oven at 80 °C before thermal treatment. In method 2, dry mixing of the carbon precursor and graphite was performed using a mortar. In method 3, the two components were mixed in ultra-pure water, ball-milled (two periods of 45 min at 400 rpm with a 10-min rest step in between), followed by drying in the oven at 80 °C. In all three cases, the dry powders were ground and finally thermally treated at 500 °C for 4 h in an argon atmosphere [
15].
Thermogravimetric analysis, (TGA, TA Instruments, New Castle, DE, USA), was performed to determine the amount of carbon residue. The samples were thermally equilibrated at 40 °C for 30 min and then heated to 800 °C at a rate of 5 °C/min under oxygen flow. The morphology of the samples was examined using a scanning electron microscope (Zeiss LEO 1550 VP Field Emission Scanning Electron Microscope, Oberkochen, Germany). The total surface area was measured by gas adsorption (N2) using the Brunauer–Emmett–Teller method (Autosorb-iQ, Quantachrome, Boynton Beach, FL, USA). Raman spectra were collected using a confocal InVia Raman microspectrometer (Renishaw, Wotton-under-Edge, UK) with a laser wavelength of 633 nm. X-ray diffraction (XRD) patterns were recorded using a Brucker D8 Advance (Cu Kα radiation source, Brucker, Billerica, MA, USA) diffractometer.
Pristine and carbon-coated electrodes were prepared by first mixing the conductive carbon (Super C45, Imerys, Paris, France) with sodium carboxymethyl cellulose (Walocel Na-CMC, Mw~30,000, degree of substitution 0.9, DOW, Midland, MI, USA) water solution for 2 h. Next, the active material (pristine or carbon-coated graphite) was stepwise added to the mixture. The slurry was stirred for two additional hours before adding styrene butadiene rubber (SBR). The dispersion was mixed for 10 min using a blade mixer (Dremel, Racine, WI, USA), and then casted on 20 µm thick Cu foil (Carl SCHLENK AG, Roth, Germany). The electrode tapes were dried for 12 h at 80 °C. Electrode disks (1.13 cm2) were cut and finally dried under vacuum at 120 °C for at least 4 h. The weight percentage of the components in the dry electrode was: active material 95%, Super C45 1%, CMC 2% and styrene-butadiene rubber (SBR) 2%. The average active material mass loading was 2.4–2.7 mAh cm−2.
Two-electrode coin cells (type 2032) were assembled in a glove-box using Li metal foil (Rockwood Lithium-ALBEMARLE, Charlotte, NC, USA) as the counter electrode. The separator (Whatman® GF/D, GE Healthcare, Chicago, IL, USA) was soaked with 100 µL of electrolyte (1M LiPF6 in EC:DMC (1:1 w/w) + 2 wt % FEC). The combined galvanostatic-potentiostatic cycling tests, including the rate capability, were conducted at 20 ± 1 °C in a climatic chamber (Binder, Tuttlingen, Germany) using a Maccor 4300 Series Battery Tester (Maccor, Inc., Tulsa, OK, USA). The cycling rate of 1C corresponded to a specific current of 372 mA g−1. The delivered capacity was calculated considering only the weight of graphite by subtracting the amount of carbon in the coating, determined by TGA. Here, the convention of graphite electrode for Li-ion batteries is used, i.e., lithiation is referred as charge and delithiation as discharge. The cells were initially equilibrated at 20 °C for 16 h, before the graphite electrodes were lithiated (charged) to 0.01 V at C/20 followed by a constant voltage step (or CV step) until the current reached a value corresponding to C/40. The subsequent graphite delithiation was conducted at C/20 to 1.00 V. The rate test was conducted by cycling the electrodes between 0.01 and 1.00 V. The charge rate was C/2 (followed by the CV step), while the discharge rate was varied from C/2 to 20 C. Before changing the discharge rate, a fingerprint cycle at C/2 was performed to check the material integrity. At the end of the rate test, the cells were left to cycle at C/2, including the CV step at the end of each lithiation (charge) step. The cyclic voltammetry tests were conducted, using three-electrode Swagelock cells with Li as a reference and counter electrode, by means of VMP-3 potentiostat (BioLogic, Inc., Cary, NC, USA). The same kind of separator was soaked with 120 µL of either 1M LiPF6 in PC:DMC (1/1, v/v) or 1M LiPF6 in EC:DMC:PC (1/3/1, w/w/w). The measurements were performed with a scan rate of 0.1 mV s−1 between 0.01 and 1.00 V vs. Li/Li+ for 10 cycles.

 and
and 



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}