
Stimulating Mesoporous Characteristics of Activated Carbon through Pyrolysis of Compacted Hydroxyethyl Cellulose—A Showcase for H2S Removal


Abstract

1. Introduction
2. Materials and Methods
2.1. Preparation of HEC Pellets
2.2. Carbonization of the HEC Pellets
2.3. Characterizations
2.3.1. Measurement of Surface Area and Pore Size by N2 Adsorption Analysis
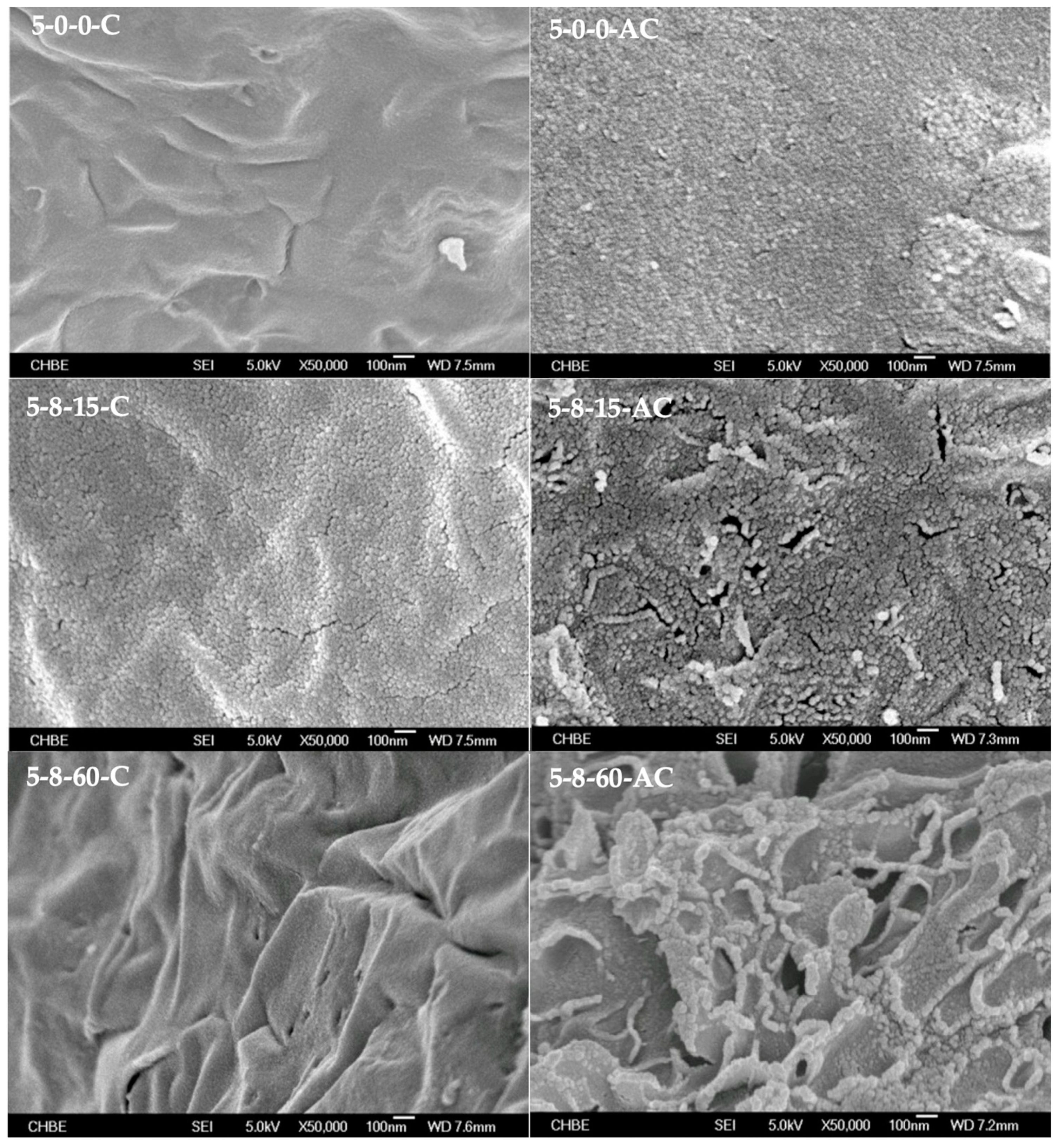
2.3.2. Surface Morphology Recorded by FE-SEM
2.3.3. Analysis of the Pendant Functional Groups on Carbonaceous Substances
2.3.4. Determination of Surface Basicity
2.3.5. Evaluation of the Adsorption Capacities
3. Results and Discussion
3.1. Impact of HEC Pellet Compression on the Evolution of Carbonaceous Intermediates as Templates for Constructing Porous AC Media
- The effect of applied load: 5-0-0-AC < 5-2-15-AC < 5-8-15-AC or 2.5-0-0-AC < 2.5-2-15-AC < 2.5-8-15-AC.
- The impact of the load holding duration: 5-2-15-AC < 5-2-60-AC and 5-8-15-AC < 5-8-60-AC. Conversely, this trend is reversed with the use of half the amount of HEC to form pellets, as demonstrated by the comparisons: 2.5-8-15-AC > 2.5-8-60-AC, respectively. This reversal is attributed to a significant creep-recovery extent in 2.5-8-15-P, which will be discussed in detail later.
- The impact of HEC mass under the same pressure and the load-holding duration: 5-2-15-AC < 2.5-2-15-AC, 5-8-15-AC < 2.5-8-15-AC, and 5-8-60-AC < 2.5-8-60-AC. This trend can be related to the relative creep-relaxation rate (∆x/xref %) of their corresponding HEC pellets (Table 1). A lower rate, as explained earlier, suggests a greater extent of creep recovery, thus favoring formation of a larger BET surface area.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AC Sample | SBET/m2/g | Specific Pore Vtotal/cc·g−1 | Oxygrp to C Ratio * |
|---|---|---|---|
| 2.5-0-0-AC | 950 | 0.49 | 0.31 |
| 2.5-2-15-AC | 1580 | 1.07 | 0.56 |
| 2.5-8-15-AC | 2080 | 1.43 | 0.64 |
| 2.5-8-60-AC | 1790 | 1.30 | 0.58 |
| 5-0-0-AC | 820 | 0.35 | 0.31 |
| 5-2-15-AC | 1170 | 0.59 | 0.85 |
| 5-8-15-AC | 1370 | 0.86 | 0.52 |
| 5-8-60-AC | 1710 | 1.17 | 0.56 |
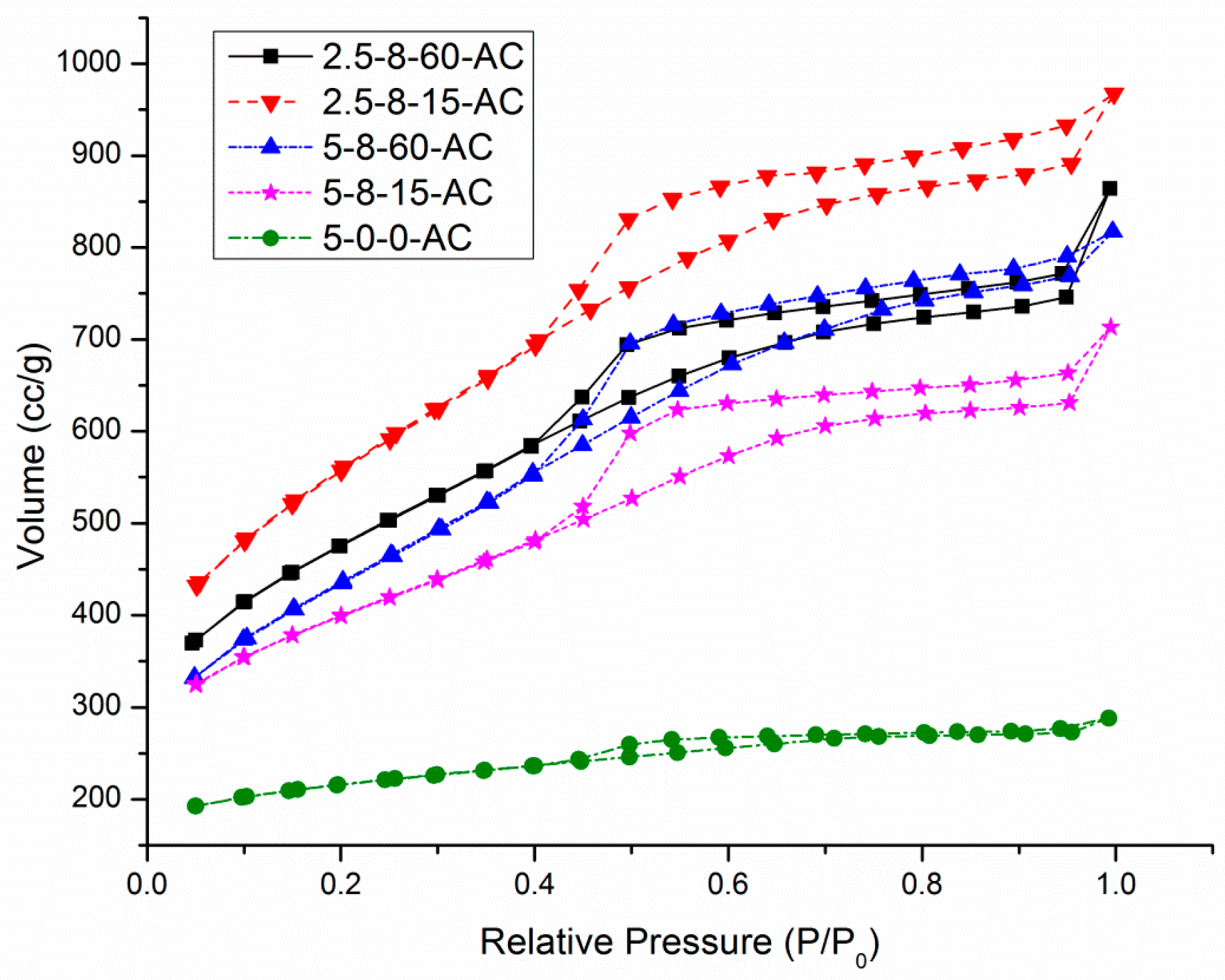
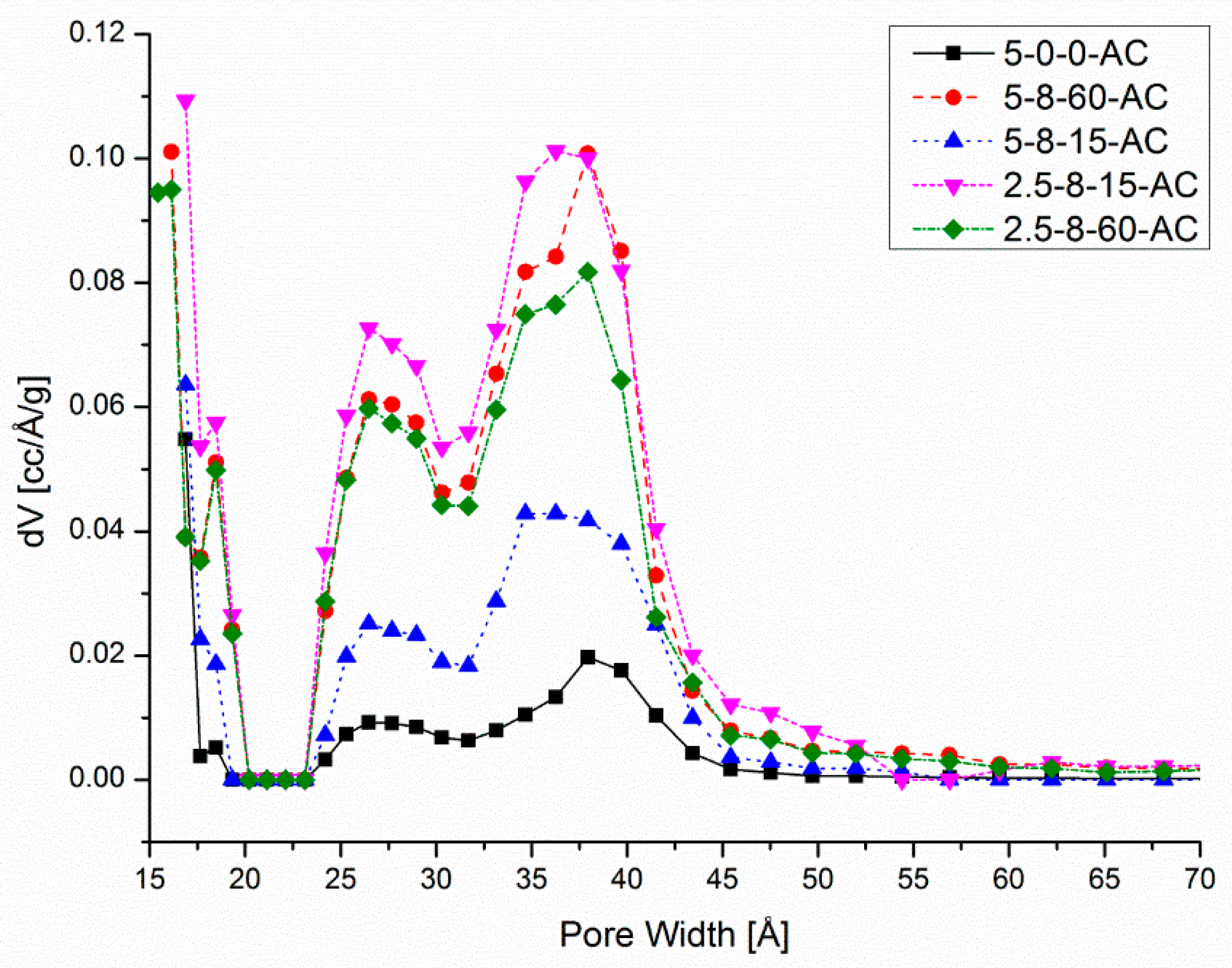
3.2. The Porous Properties of AC Samples Derived from HEC Pellets Made by Using the Compression Load of 8 mt
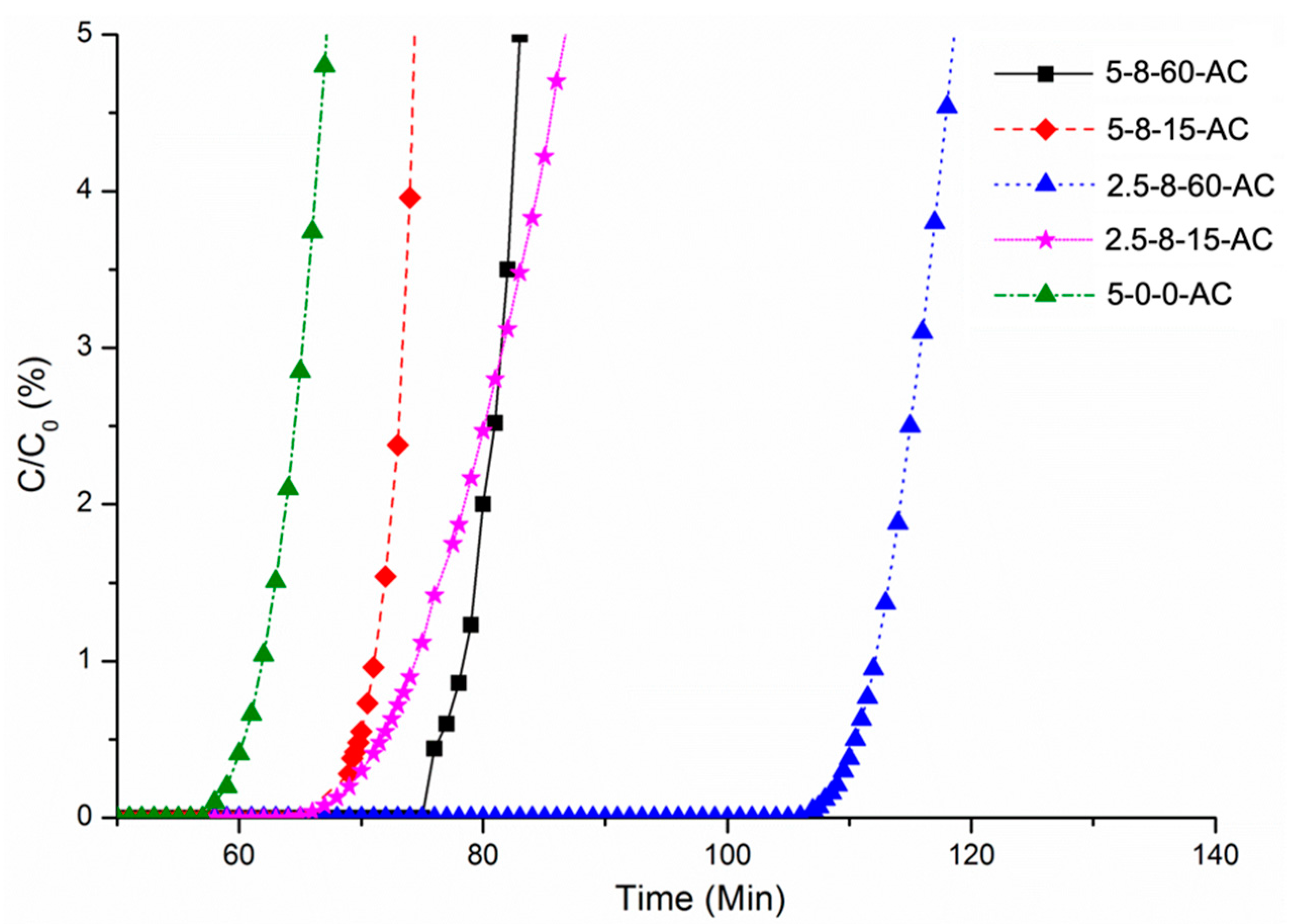
3.3. Assessment of the H2S Removal Efficacies of the AC Samples from a Gas Stream
4. Conclusions
- HEC fine powder was compressed in a cylindrical die set, yielding compaction in both the radial and axial directions. By keeping the load at 8 mt with a load-holding duration, it was observed that a greater polymer compaction, i.e., the relative creep-relaxation extent, was realized in a 5 g pellet than in a 2.5 g pellet. Conversely, extending the duration of load (8 mt) application resulted in compaction for either the 2.5 g or the 5.0 g pellet.
- The pyrolysis of HEC pellets yielded carbonaceous substances comprising polycyclic aromatic hydrocarbons (PAHs) and a minor fraction of aliphatic species. Both types of species carried oxygenated groups. The atomic ratio of the unsubstituted carbon species to the oxygenated groups, according to XPS analysis, affected the evolution of the porous structure of AC during the subsequent calcination activation process. The optimal ratio range, resulting in high surface areas, originated from the tap densities (g · cm−3) observed in the HEC pellets, ranging from 1.22 to 1.27 compared to 1.14 for the HEC powder used.
- By referencing the HEC powder chosen as the control, the HEC pellets demonstrated evidently larger volumes of total pores and mesopores after their conversion into corresponding AC samples. Moreover, extending the initial load-holding duration from 15 to 60 min resulted in the formation of a wrinkled AC medium embedded with interpenetrating mesopores in addition to micropores. These two distinct porous characteristics worked together to enhance the removal of H2S from a gas stream compared to microporous AC adsorbents with particulate morphology.
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Reza, M.S.; Yun, C.S.; Afroze, S.; Radenahmad, M.S.; Bakar, A.; Saidur, R.; Taweekun, J.; Azad, A.K. Preparation of activated carbon from biomass and its’ applications in water and gas purification, a review. Arab. J. basic Appl. Sci. 2020, 27, 208–238. [Google Scholar] [CrossRef]
- Jin, P.; Jin, X.; Wang, X.; Feng, Y.C.X. Biological activated carbon treatment process for advanced water and wastewater treatment. In Biomass Now-Cultivation and Utilization; Matovic, M.D., Ed.; InTech: London, UK, 2013; pp. 153–192. [Google Scholar] [CrossRef]
- De Mello, R.; Motheo, A.J.; Sáez, C.; Rodrigo, M.A. Recent progress in the combination of activated carbon adsorption and electrolysis for the treatment of wastes. Curr. Opin. Electrochem. 2022, 36, 101167. [Google Scholar] [CrossRef]
- Naji, S.Z.; Tye, C.T. A review of the synthesis of activated carbon for biodiesel production: Precursor, preparation, and modification. Energy Convers. Manag. 2022, X13, 100152. [Google Scholar] [CrossRef]
- Guo, Z.; Zhu, G.; Gao, B.; Zhang, D.; Tian, G.; Chen, Y.; Zhang, W.; Qiu, S. Adsorption of vitamin B12 on ordered mesoporous carbons coated with PMMA. Carbon 2005, 43, 2344–2351. [Google Scholar] [CrossRef]
- Shen, W.; Wang, H.; Guan, R.; Li, Z. Surface modification of AC fiber and its adsorption for vitamin B1 and folic acid. Colloids Surf. A Physicochem. Eng. Asp. 2008, 331, 263–267. [Google Scholar] [CrossRef]
- Slocum, A.; Santora, S.; Ly, M.; Zhang, J.; Castano, J.; Becerra-Arteaga, A. Development of an activated carbon filtration step and high throughput screening method to remove host cell proteins from a recombinant enzyme process. Biotechnol. Prog. 2021, 37, e3151. [Google Scholar] [CrossRef] [PubMed]
- Fuertes, A.B.; Lota, G.; Centeno, T.A.; Frackowiak, E. Templated mesoporous carbons for supercapacitor application. Electrochimica Acta 2005, 50, 2799–2805. [Google Scholar] [CrossRef]
- Ahmad, A.; Gondal, M.A.; Hassan, M.; Rashid, I.; Ullah, S.; Alzahrani, A.S.; Memon, W.A.; Mabood, F.; Melhi, S. Preparation and Characterization of Physically Activated Carbon and Its Energetic Application for All-Solid-State Supercapacitors: A Case Study. ACS Omega 2023, 8, 21653–21663. [Google Scholar] [CrossRef] [PubMed]
- Strizhenov, E.M.; Shkolin, A.V.; Chugaev, S.S.; Men’shchikov, I.E.; Solovtsova, O.V.; Shiryaev, A.A.; Nickolsky, M.S. Adsorbed natural gas storage facility based on activated carbon of wood waste origin. Adsorption 2023, 29, 291–307. [Google Scholar] [CrossRef]
- Guan, C.; Loo, S.; Wang, K.; Yang, C. Methane storage in carbon pellets prepared via a binderless method. Energy. Convers. Manag. 2011, 52, 1258–1262. [Google Scholar] [CrossRef]
- Zhang, T.; Walawender, W.P.; Fan, L.T. Grain-based AC for natural gas storage. Bioresour. Technol. 2010, 101, 1983–1991. [Google Scholar] [CrossRef] [PubMed]
- Inomata, K.; Kanazawa, K.; Urabe, Y.; Hosono, H.; Araki, T. Natural gas storage in AC pellets without a binder. Carbon 2002, 40, 87–93. [Google Scholar] [CrossRef]
- Zhu, R.; Yu, Q.; Li, M.; Zhao, H.; Jin, S.; Huang, Y.; Fan, J.; Chen, J. Analysis of factors influencing pore structure development of agricultural and forestry waste-derived activated carbon for adsorption application in gas and liquid phases: A review. J. Environ. Chem. Eng. 2021, 9, 105905. [Google Scholar] [CrossRef]
- Li, L.; Liu, S.; Liu, J. Surface modification of coconut shell-based AC for the improvement of hydrophobic VOC removal. J. Hazard. Mater. 2011, 192, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Zhi, Y.; Liu, J. Surface modification of activated carbon for enhanced adsorption of perfluoroalkyl acids from aqueous solutions. Chemosphere 2016, 144, 1224–1232. [Google Scholar] [CrossRef] [PubMed]
- Jha, M.K.; Joshi, S.; Sharma, R.K.; Kim, A.A.; Pant, B.; Park, M.; Pant, H.R. Surface Modified Activated Carbons: Sustainable Bio-Based Materials for Environmental Remediation. Nanomaterials 2021, 11, 3140. [Google Scholar] [CrossRef] [PubMed]
- Lingjun Kong, L.; Liu, M.; Diao, Z.; Chen, D.; Chang, X.; Xiong, Y. Coupling template nanocasting and self-activation for fabrication of nanoporous carbon. Sci. Rep. 2016, 6, 38176. [Google Scholar] [CrossRef] [PubMed]
- Rambau, K.M.; Musyoka, N.M.; Manyala, N.; Ren, J.; Langmi, H.W. Mechanochemical approach in the synthesis of activated carbons from waste tyres and its hydrogen storage applications. Mater. Today Proc. 2018, 5, 10505–10513. [Google Scholar] [CrossRef]
- Alcañiz-Monge, J.; Trautwein, G.; Pérez-Cadenas, M.; Román-Martínez, M.C. Effects of compression on the textural properties of porous solids. Microporous Mesoporous Mater. 2009, 126, 291–301. [Google Scholar] [CrossRef]
- Münstedt, H. Influence of hydrostatic pressure on rheological properties of polymer melts—A review. J. Rheol. 2020, 64, 751–774. [Google Scholar] [CrossRef]
- Grandes, F.A.; Sakano, V.K.; Rego, A.C.; Rebmann, M.S.; Cardoso, F.A.; Pileggi, R.G. Rrheological behaviour and flow induced microstructural changes of cement-based mortars assessed by pressure mapped squeeze flow. Powder Technol. 2021, 393, 519–538. [Google Scholar] [CrossRef]
- Jeya, R.P.K.; Bouzid, A.-H. Compression Creep and Thermal Ratcheting Behaviour of High Density Polyethylene (HDPE). Polymers 2018, 10, 156. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Nguyen, T.D. Influence of thermoviscoelastic properties and loading conditions on the recovery performance of shape memory polymers. Mech. Mater. 2011, 43, 127–138. [Google Scholar] [CrossRef]
- Sun, M.; Hong, L. Impacts of the pendant functional groups of cellulose precursor on the generation of pore structures of AC. Carbon 2011, 49, 2173–2180. [Google Scholar] [CrossRef]
- Shivanand, P.; Sprockel, O.L. Compaction behaviour of cellulose polymers. Powder Technol. 1992, 69, 177–184. [Google Scholar] [CrossRef]
- Lewis, I.C. Chemistry of carbonization. Carbon 1982, 20, 519–529. [Google Scholar] [CrossRef]
- Norit Activated Carbon. Available online: https://norit.com/applications/biogas-rng/hydrogen-sulfide-removal (accessed on 14 November 2022).
- Sevilla, M.; Fuertes, A.B. The production of carbon materials by hydrothermal carbonization of cellulose. Carbon 2009, 47, 2281–2289. [Google Scholar] [CrossRef]
- Cui, H.; Turn, S.Q.; Reese, M.A. Removal of sulfur compounds from utility pipelined synthetic natural gas using modified AC. Catalysis Today 2009, 139, 274–279. [Google Scholar] [CrossRef]
- Shokri, J.; Adibkia, K. Application of cellulose and cellulose derivatives in pharmaceutical industries. In Cellulose-Medical, Pharmaceutical and Electronic Applications; Van De Ven Theo, G.M., Ed.; InTech: London, UK, 2013. [Google Scholar] [CrossRef]
- Ghori, M.U.; Conway, B. Powder compaction: Compression properties of cellulose ethers. Br. J. Pharm. 2016, 1, 19–29. [Google Scholar] [CrossRef]
- Tang, W.; Wang, C.; Chen, D. Kinetic studies on the pyrolysis of chitin and chitosan. Polym. Degrad. Stab. 2005, 87, 389–394. [Google Scholar] [CrossRef]
- El-Sayed, N.S.; Awad, H.; El-Sayed, G.M. Synthesis and characterization of biocompatible hydrogel based on hydroxyethyl cellulose-g-poly(hydroxyethyl methacrylate). Polym. Bull. 2020, 77, 6333–6347. [Google Scholar] [CrossRef]
- Xing, Z.; Ng, Y.H.; Tay, S.W.; Oon, R.P.H.; Hong, L. Shaping nanofiltration channels in a carbonaceous membrane via controlling the pyrolysis atmosphere. Phys. Chem. Chem. Phys. 2017, 19, 21426–21435. [Google Scholar] [CrossRef] [PubMed]
- Pham, N.K.; Nguyen, T.T. Understanding interaction capacity of CO2 with organic compounds at molecular level: A theoretical approach. In Carbon Dioxide Chemistry, Capture and Oil Recovery; Karame, I., Shaya, J., Srour, H., Eds.; InTech: London, UK, 2017. [Google Scholar] [CrossRef]
- Sehaqui, H.; Zhou, Q.; Berglund, L.A. Nanostructured biocomposites of high toughness—A wood cellulose nanofiber network in ductile hydroxyethylcellulose matrix. Soft Matter. 2011, 7, 7342. [Google Scholar] [CrossRef]
- Sandford, S.A.; Bernstein, M.P.; Materese, C.K. The infrared spectra of polycyclic aromatic hydrocarbons with excess peripheral h atoms (Hn-PAHs) and their relation to the 3.4 and 6.9 μm PAH emission features. Astrophys. J. Suppl. Ser. 2013, 205, 30. [Google Scholar] [CrossRef] [PubMed]
- Miao, M.; Zuo, S.; Zhao, Y.; Wang, Y.; Xia, H.; Tan, C.; Gao, H. Selective oxidation rapidly decomposes biomass-based activated carbons into graphite-like crystallites. Carbon 2018, 140, 504–507. [Google Scholar] [CrossRef]
- Wang, T.; Liu, Q.; Shi, L.; Xiang, C.; Liu, Z.; Han, W.; Zhang, L.; Nie, H.; Li, M. Radicals and coking behaviors during thermal cracking of two vacuum resids and their SARA fractions. Fuel 2020, 279, 118374. [Google Scholar] [CrossRef]
- Johansson, K.; Head-Gordon, M.; Schrader, P.; Wilson, K.; Michelsen, H. Resonance-stabilized hydrocarbon-radical chain reactions may explain soot inception and growth. Science 2018, 361, 997–1000. [Google Scholar] [CrossRef] [PubMed]
- Elvati, P.; Violi, A. Homo-dimerization of oxygenated polycyclic aromatic hydrocarbons under flame conditions. Fuel 2018, 222, 307–311. [Google Scholar] [CrossRef]
- Leon, G.; Martin, J.W.; Bringley, E.J.; Akroyd, J.; Kraft, M. The role of oxygenated species in the growth of graphene, fullerenes and carbonaceous particles. Carbon 2021, 182, 203–213. [Google Scholar] [CrossRef]
- Hu, M.; Ye, Z.; Zhang, Q.; Xue, Q.; Li, Z.; Wang, J.; Pan, Z. Towards understanding the chemical reactions between KOH and oxygen-containing groups during KOH-catalyzed pyrolysis of biomass. Fuel 2022, 245, 123286. [Google Scholar] [CrossRef]
- Otte, D.A.L.; Borchmann, D.E.; Lin, C.; Weck, M.; Woerpel, K.A. 13C NMR spectroscopy for the quantitative determination of compound ratios and polymer end groups. Org. Lett. 2014, 16, 1566–1569. [Google Scholar] [CrossRef] [PubMed]
- Eiliazadeh, B.; Briscoe, B.J.; Sheng, Y.; Pitt, K. Investigating density distributions for tablets of different geometry during the compaction of pharmaceuticals. Part. Sci. Technol. 2003, 21, 303–316. [Google Scholar] [CrossRef]
- Tonga, M. Effect of π–conjugation on aggregation–induced emission of α–cyanostilbene incorporated polycyclic aromatic hydrocarbons. J. Photochem. Photobio. A 2021, 412, 113247. [Google Scholar] [CrossRef]
- Khabazipour, M.; Anbia, M. Removal of Hydrogen Sulfide from Gas Streams Using Porous Materials: A Review. Ind. Eng. Chem. Res. 2019, 58, 22133–22164. [Google Scholar] [CrossRef]
- Feng, W.; Kwon, S.; Borguet, E.; Vidic, R. Adsorption of hydrogen sulfide onto activated carbon fibers: Effect of pore structure and surface chemistry. Environ. Sci. Technol. 2005, 39, 9744–9749. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wang, J.; Li, W.; Wang, M.; Qiao, W.; Long, D.; Ling, L. Millimeter-sized mesoporous carbon spheres for highly efficient catalytic oxidation of hydrogen sulfide at room temperature. Carbon 2016, 96, 608–615. [Google Scholar] [CrossRef]
- Morishige, K.; Kawai, T.; Kittaka, S. Capillary condensation of water in mesoporous carbon. J. Phys. Chem. C 2014, 118, 4664–4669. [Google Scholar] [CrossRef]
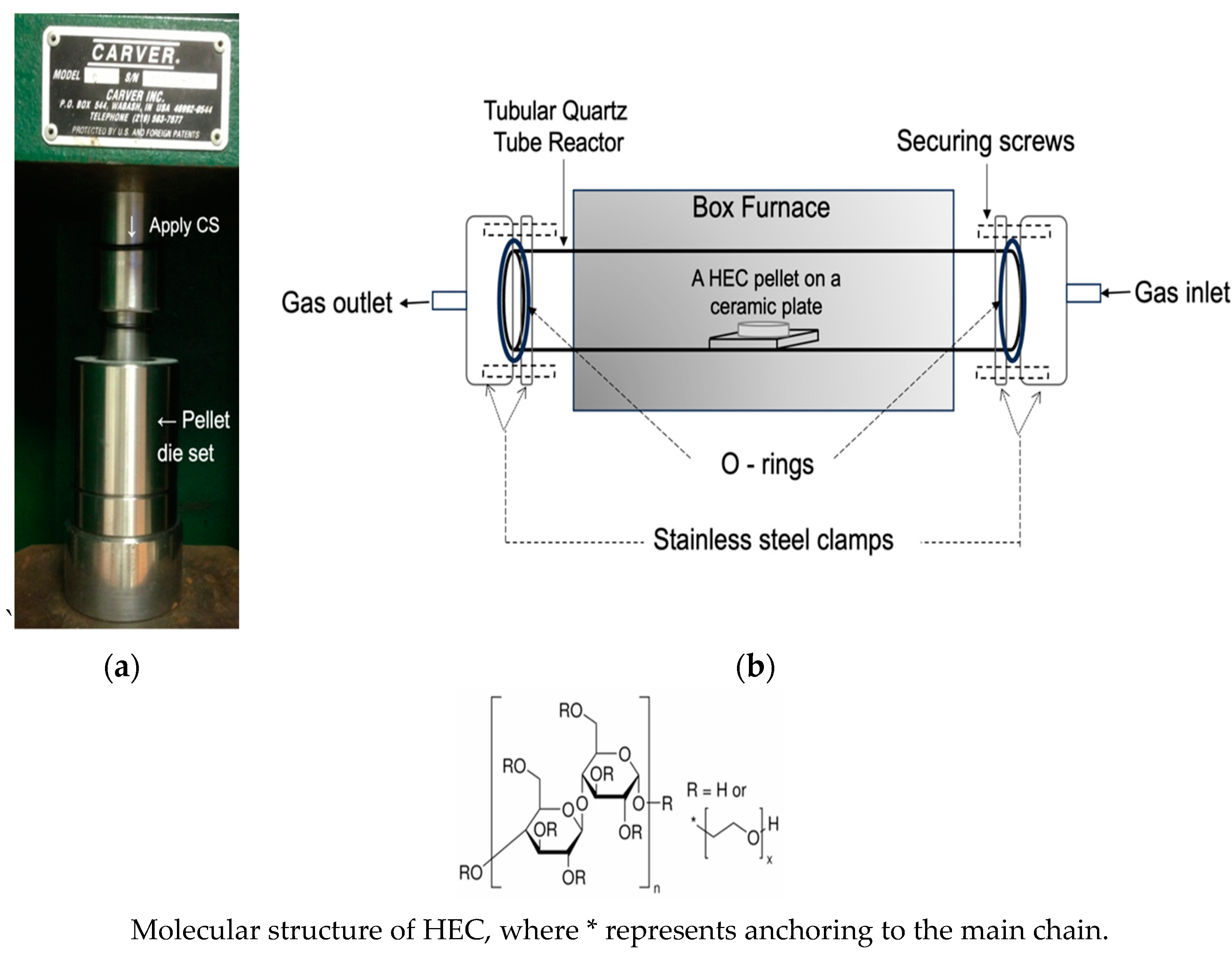

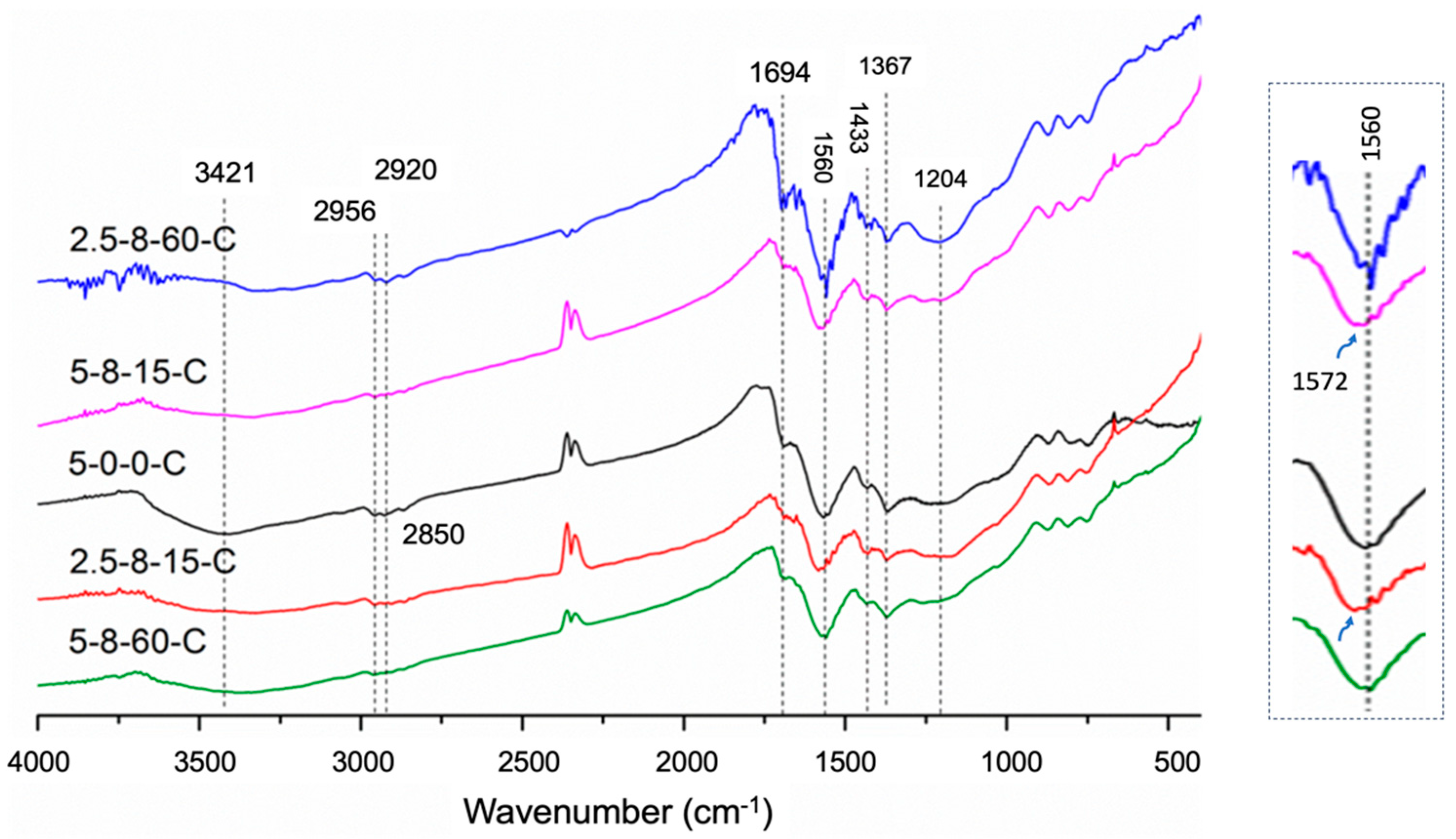

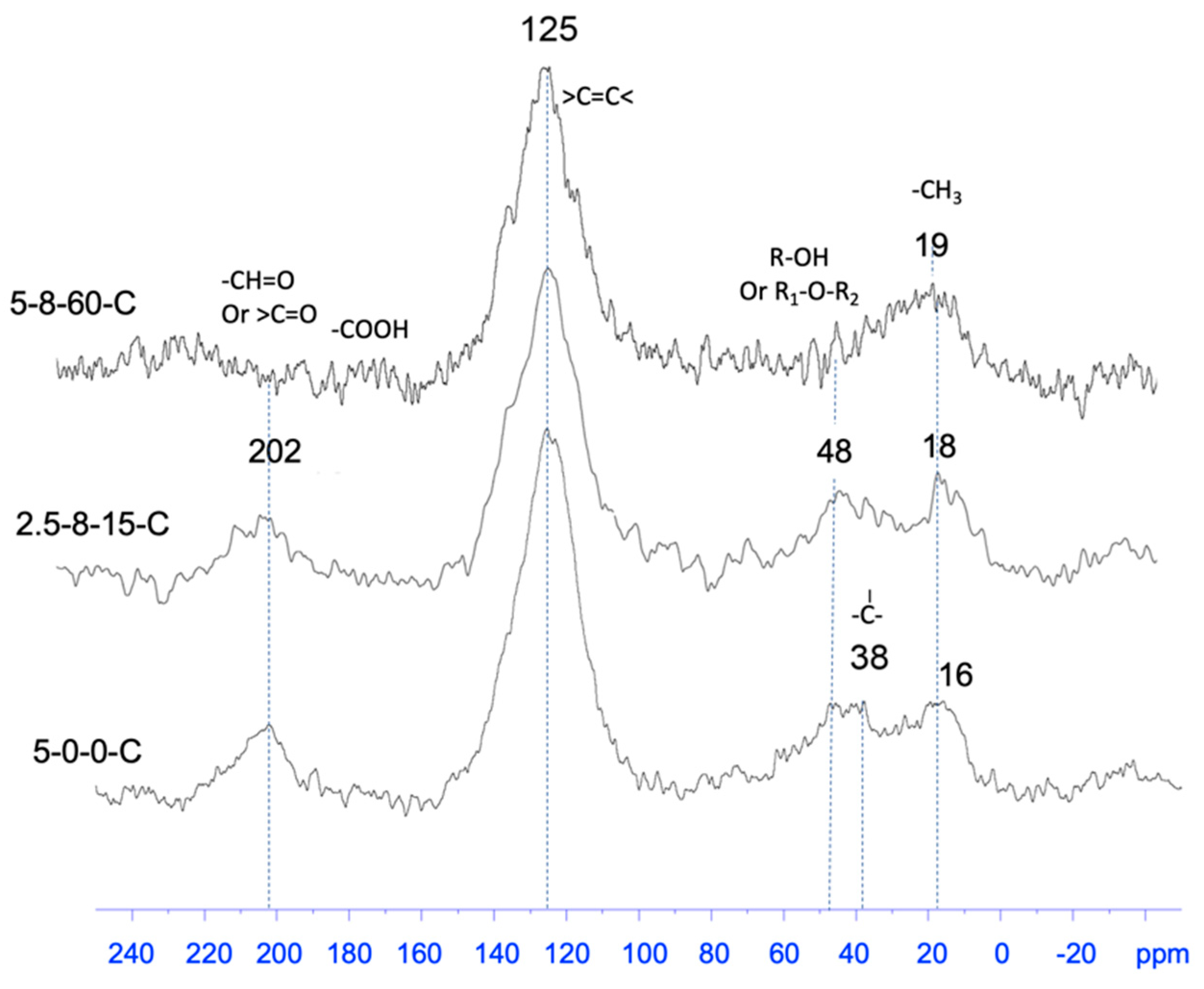

| HEC Sample | Thickness, x/mm | ∆x a | ∆x/xref % | Tap Density (25 °C)/g·cm−3 |
|---|---|---|---|---|
| Powder | - | - | - | 1.14 b |
| 5-2-15-Pref | 5.69 | 0 | 0 | 1.15 |
| 5-8-15-P | 5.25 | 0.44 | 7.73 | 1.24 |
| 5-8-60-P | 5.14 | 0.55 | 9.67 | 1.27 |
| 2.5-2-15-Pref | 2.79 | 0 | 0 | 1.17 |
| 2.5-8-15-P | 2.65 | 0.14 | 5.02 | 1.22 |
| 2.5-8-60-P | 2.56 | 0.23 | 8.24 | 1.27 |
| Carbonaceous Substances | Atomic % | |||
|---|---|---|---|---|
| C-C and C=C | C-O | C=O | COO | |
| 2.5-2-15-C | 63.8 | 24.4 | 6.6 | 4.7 |
| 2.5-8-15-C | 59.7 | 18.5 | 13.6 | 6.1 |
| 2.5-8-60-C | 62.2 | 28.8 | 3.5 | 4.0 |
| 5-2-15-C | 53.4 | 34.0 | 6.3 | 5.1 |
| 5-8-15-C | 64.8 | 25.3 | 3.5 | 5.0 |
| 5-8-60-C | 63.3 | 21.8 | 10.3 | 3.6 |
| 5-0-0-C/2.5-0-0-C | 75.3 | 14.0 | 6.5 | 3.1 |
| AC Sample | Vtotal (cc/g) | Vmicro (cc/g) | Vmeso (cc/g) | Vmeso/Vtotal |
|---|---|---|---|---|
| 5-0-0-AC | 0.35 | 0.24 | 0.11 | 0.32 |
| 5-8-15-AC | 0.86 | 0.51 | 0.35 | 0.41 |
| 5-8-60-AC | 1.17 | 0.60 | 0.57 | 0.49 |
| 2.5-8-15-AC | 1.43 | 1.08 | 0.35 | 0.24 |
| 2.5-8-60-AC | 1.30 | 0.46 | 0.84 | 0.65 |
| AC Sample | Vtotal (cc/g)/Vmeso % | Breakthrough Time (min) | Breakthrough Capacity (mg/g) | pH |
|---|---|---|---|---|
| 5-0-0-AC | 0.35/32 | 61.86 | 13.0 | 10.3 |
| 5-8-15-AC | 0.86/41 | 71.09 | 13.7 | 10.0 |
| 5-8-60-AC | 1.17/49 | 84.55 | 16.1 | 9.7 |
| 2.5-8-15-AC | 1.43/24 | 74.44 | 14.3 | 8.8 |
| 2.5-8-60-AC | 1.30/65 | 112.00 | 21.6 | 9.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, F.; Hong, L. Stimulating Mesoporous Characteristics of Activated Carbon through Pyrolysis of Compacted Hydroxyethyl Cellulose—A Showcase for H2S Removal. C 2024, 10, 43. https://doi.org/10.3390/c10020043
Chen F, Hong L. Stimulating Mesoporous Characteristics of Activated Carbon through Pyrolysis of Compacted Hydroxyethyl Cellulose—A Showcase for H2S Removal. C. 2024; 10(2):43. https://doi.org/10.3390/c10020043
Chicago/Turabian StyleChen, Fuxiang, and Liang Hong. 2024. "Stimulating Mesoporous Characteristics of Activated Carbon through Pyrolysis of Compacted Hydroxyethyl Cellulose—A Showcase for H2S Removal" C 10, no. 2: 43. https://doi.org/10.3390/c10020043
APA StyleChen, F., & Hong, L. (2024). Stimulating Mesoporous Characteristics of Activated Carbon through Pyrolysis of Compacted Hydroxyethyl Cellulose—A Showcase for H2S Removal. C, 10(2), 43. https://doi.org/10.3390/c10020043