

Epigenetic Regulation of HIV-1 Sense and Antisense Transcription in Response to Latency-Reversing Agents


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
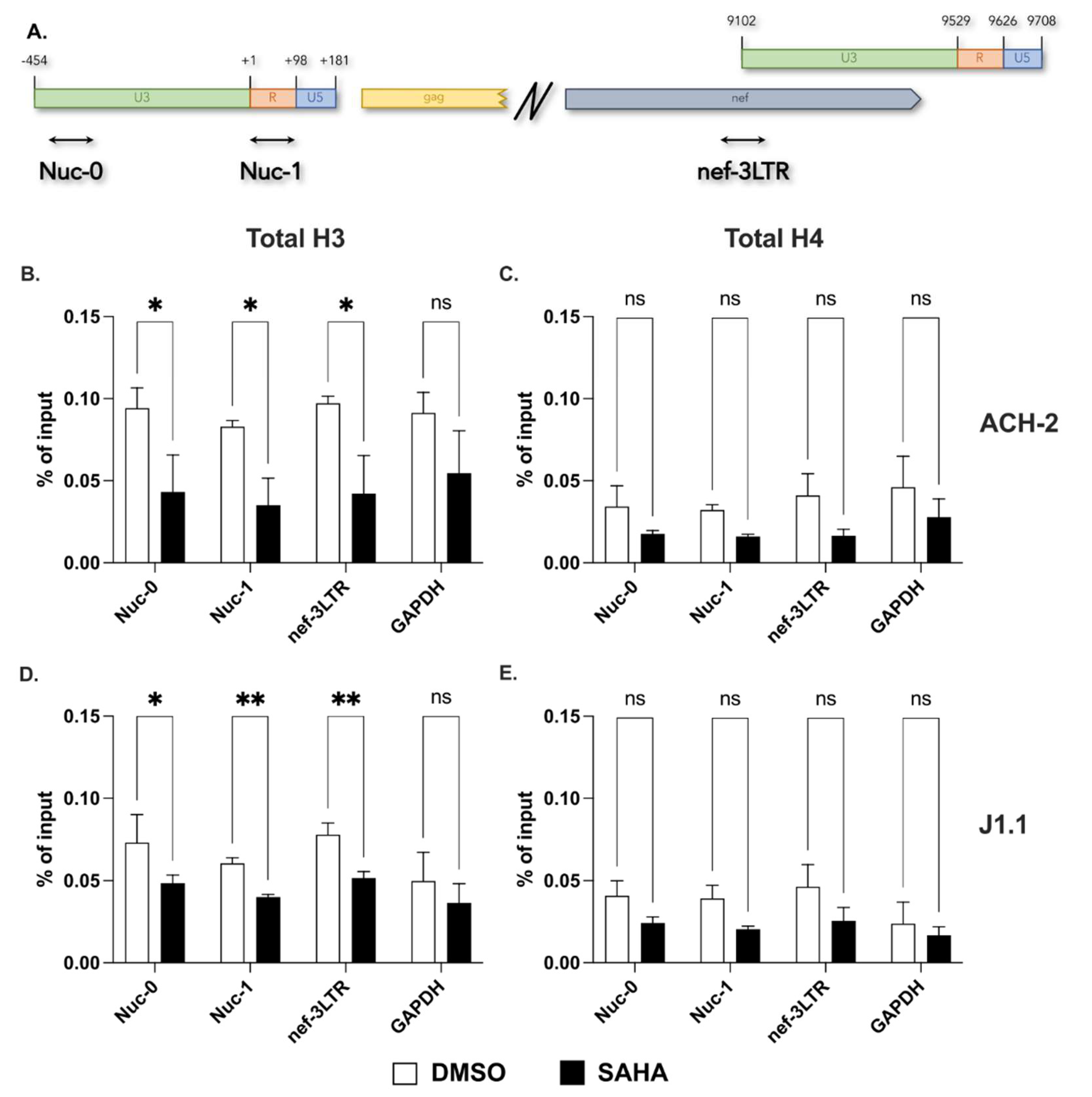
2.1. Presence of a Nucleosome on the Proviral 3′ LTR
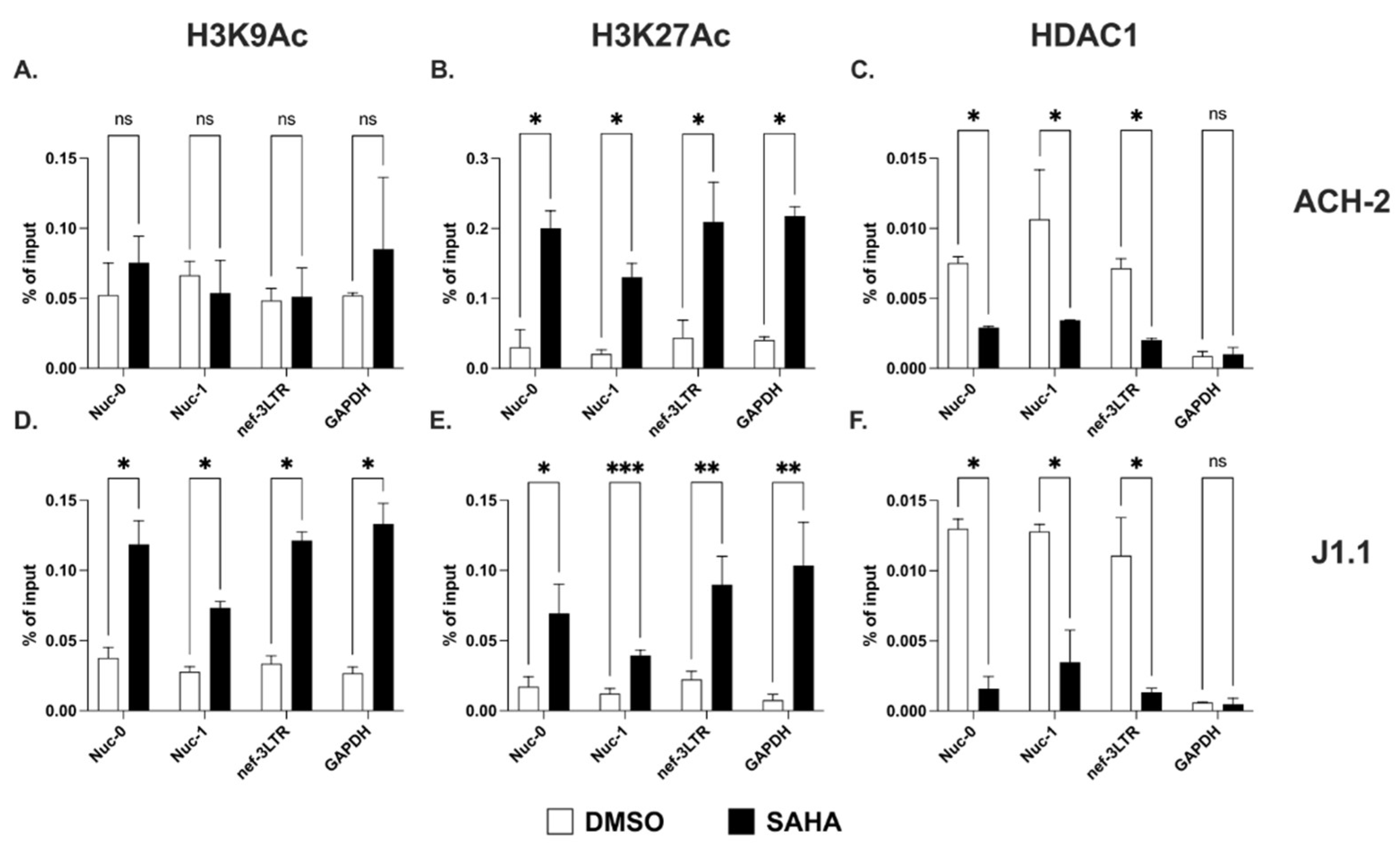
2.2. Acetylation of Lysines on Histone H3 at the 3′ LTR
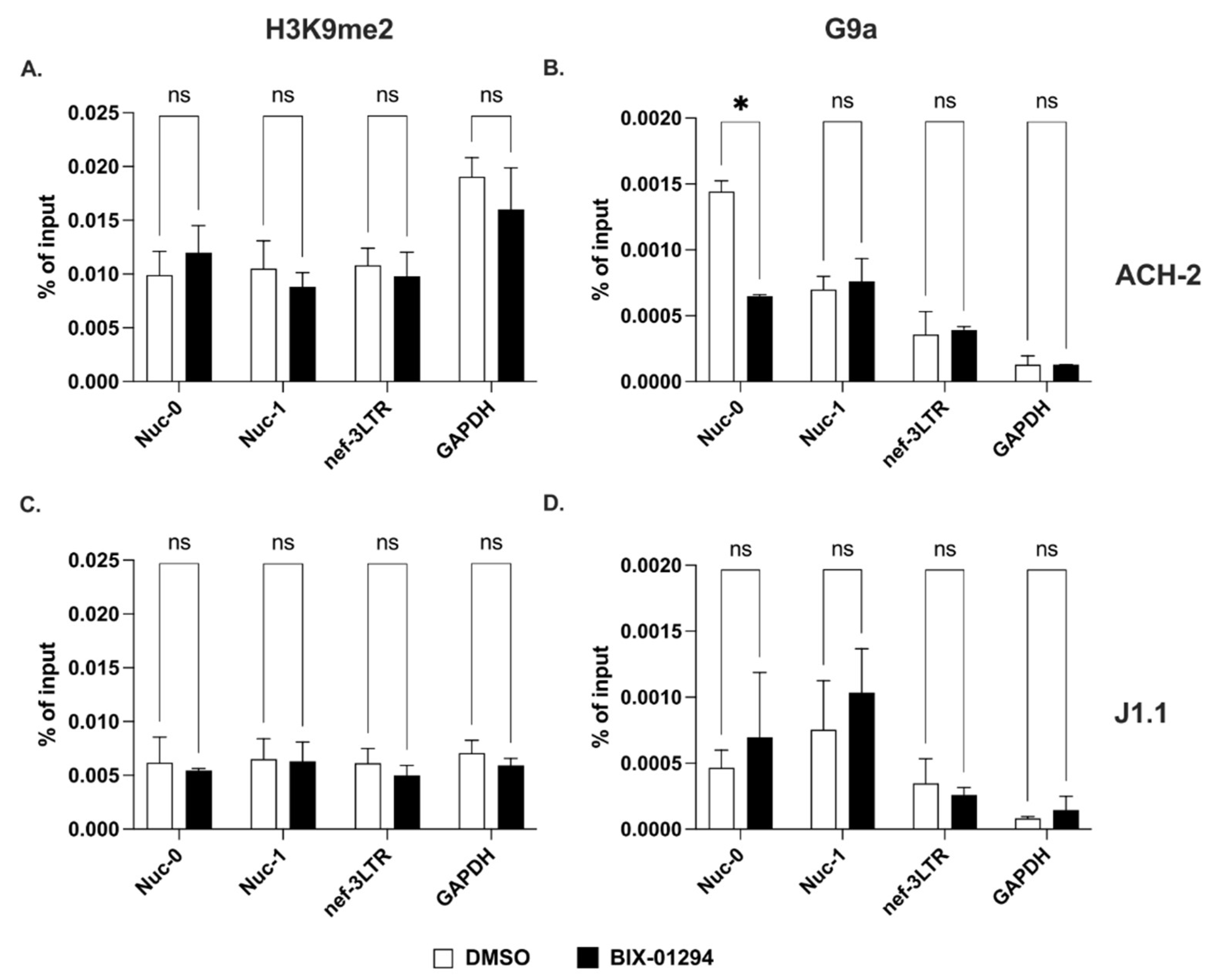
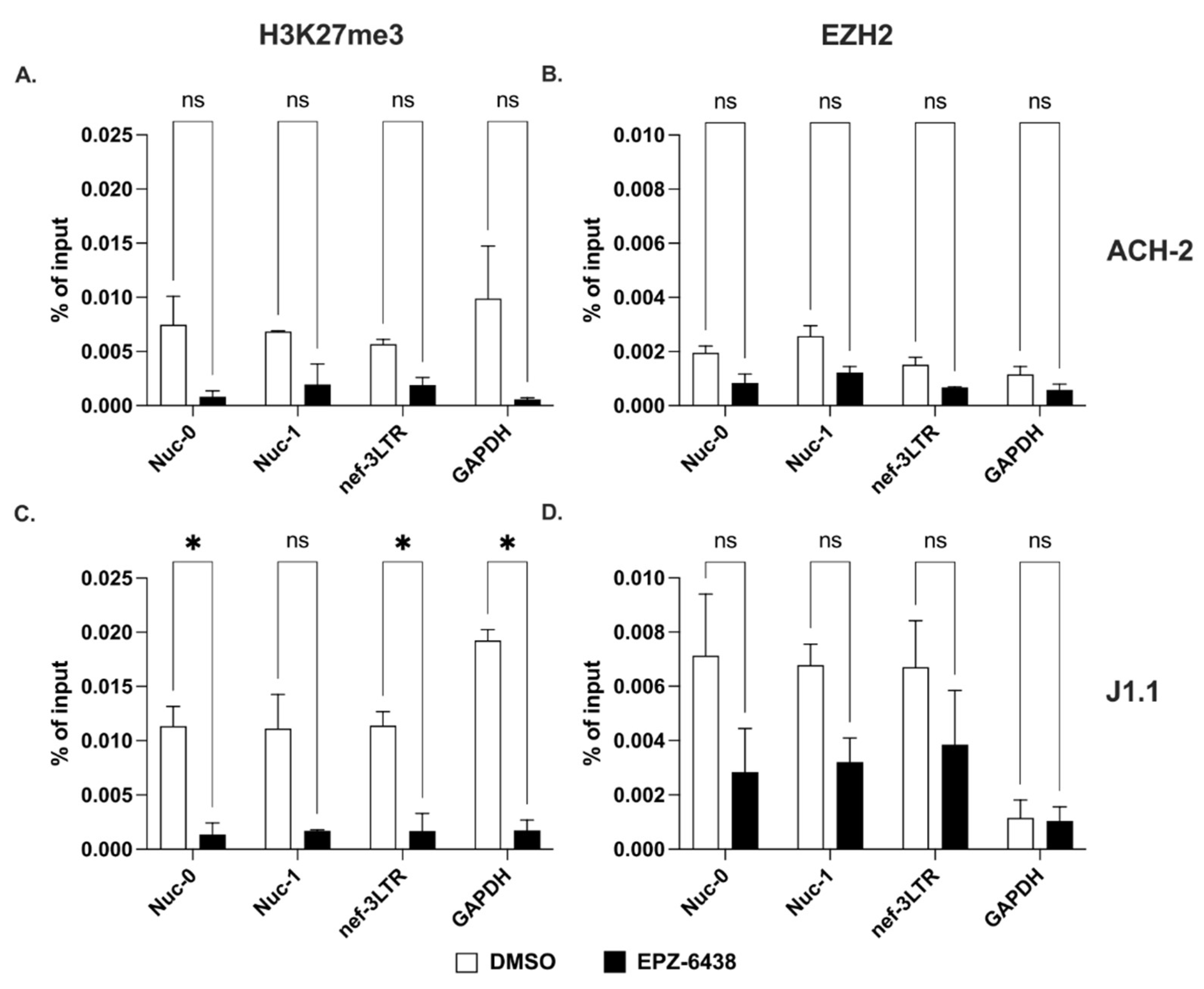
2.3. Methylation of Lysines on Histone H3 at the 3′ LTR
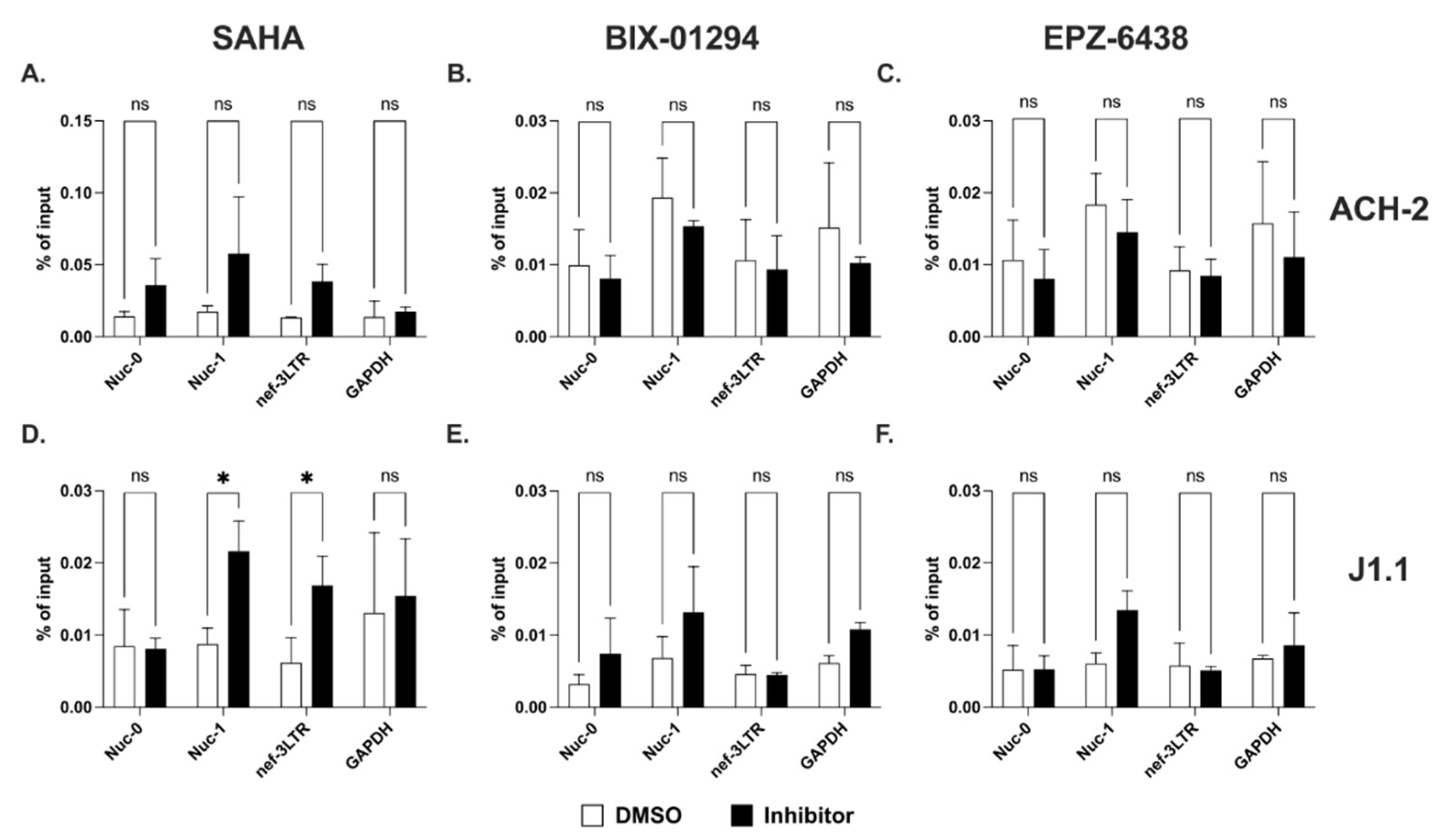
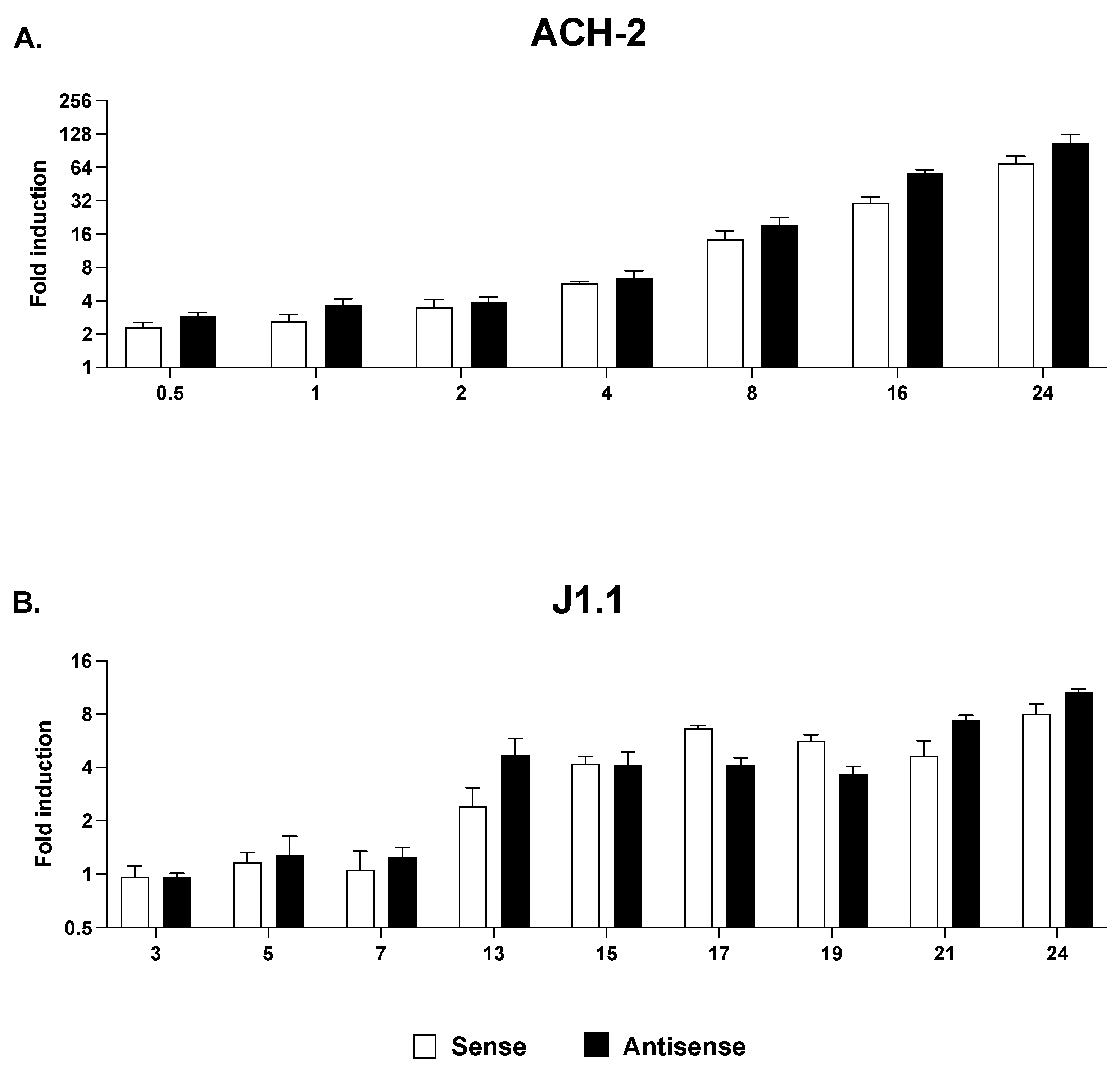
2.4. Sense and Antisense Transcription in Response to HDAC and HKMT Inhibitors
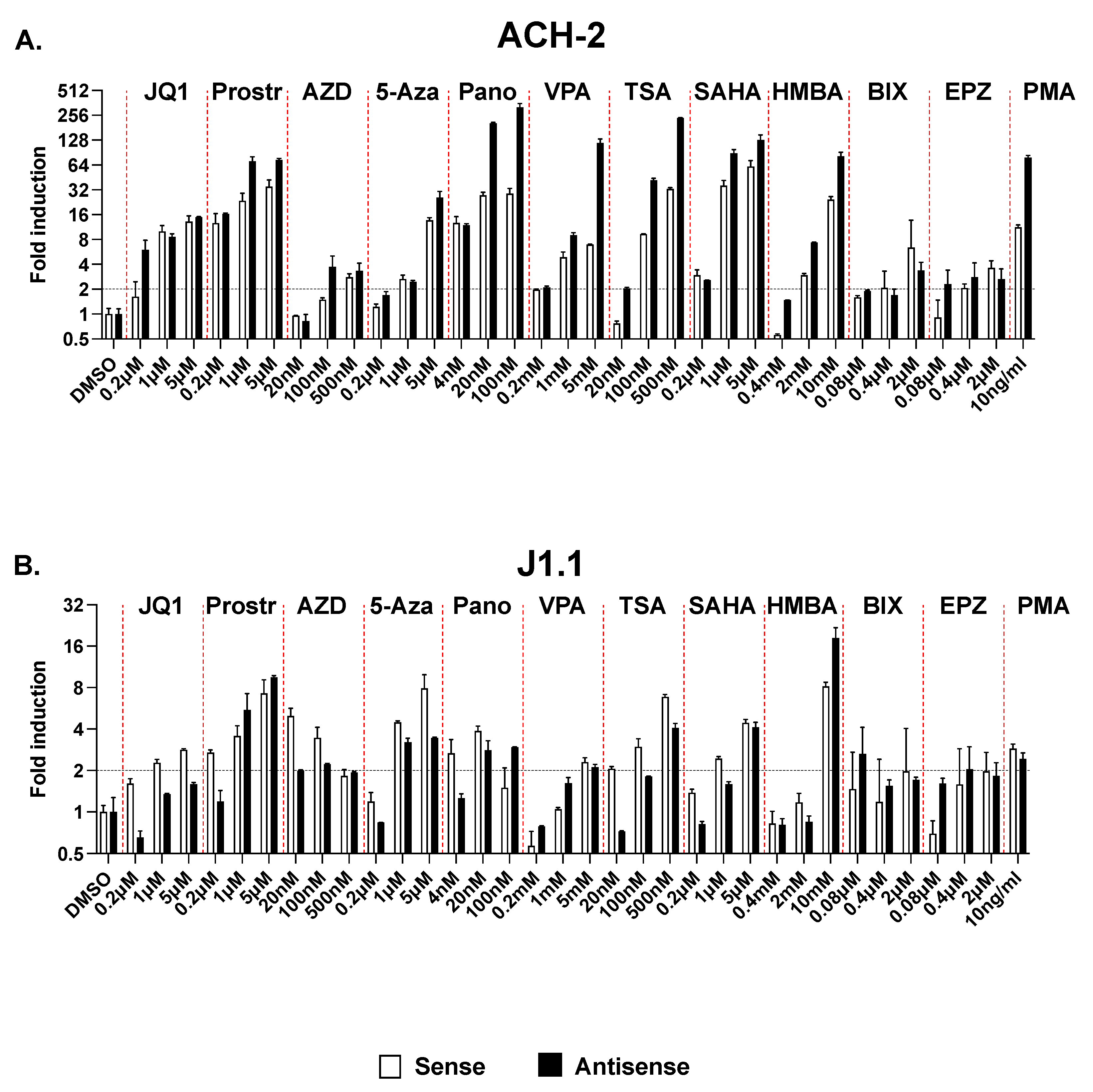
2.5. Effect of Multiple LRAs on Sense and Antisense Transcription
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Reagents
4.2. Reverse Transcription and Real-Time PCR (RT-qPCR) Assay
4.3. ChIP Analysis
4.4. Latency Reversal
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mbonye, U.; Karn, J. The Molecular Basis for Human Immunodeficiency Virus Latency. Annu. Rev. Virol. 2017, 4, 261–285. [Google Scholar]
- Mbonye, U.; Karn, J. Transcriptional control of HIV latency: Cellular signaling pathways, epigenetics, happenstance and the hope for a cure. Virology 2014, 454–455, 328–339. [Google Scholar]
- Kinoshita, S.; Su, L.; Amano, M.; Timmerman, L.A.; Kaneshima, H.; Nolan, G.P. The T cell activation factor NF-ATc positively regulates HIV-1 replication and gene expression in T cells. Immunity 1997, 6, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Nabel, G.; Baltimore, D.A. An inducible transcription factor activates expression of human immunodeficiency virus in T cells. Nature 1987, 326, 711–713. [Google Scholar] [CrossRef]
- Selliah, N.; Zhang, M.; DeSimone, D.; Kim, H.; Brunner, M.; Ittenbach, R.F.; Rui, H.; Cron, R.Q.; Finkel, T.H. The gamma-cytokine regulated transcription factor, STAT5, increases HIV-1 production in primary CD4 T cells. Virology 2006, 344, 283–291. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yang, X.; Chen, Y.; Gabuzda, D. ERK MAP kinase links cytokine signals to activation of latent HIV-1 infection by stimulating a cooperative interaction of AP-1 and NF-kB. J. Biol. Chem. 1999, 274, 27981–27988. [Google Scholar]
- Romerio, F.; Gabriel, M.N.; Margolis, D.M. Repression of human immunodeficiency virus type 1 through the novel cooperation of human factors YY1 and LSF. J. Virol. 1997, 71, 9375–9382. [Google Scholar] [CrossRef]
- Coull, J.J.; Romerio, F.; Sun, J.M.; Volker, J.L.; Galvin, K.M.; Davie, J.R.; Shi, Y.; Hansen, U.; Margolis, D.M. The human factors YY1 and LSF repress the human immunodeficiency virus type 1 long terminal repeat via recruitment of histone deacetylase 1. J. Virol. 2000, 74, 6790–6799. [Google Scholar]
- Tyagi, M.; Karn, J. CBF-1 promotes transcriptional silencing during the establishment of HIV-1 latency. EMBO J. 2007, 26, 4985–4995. [Google Scholar] [CrossRef]
- Tyagi, M.; Pearson, R.J.; Karn, J. Establishment of HIV latency in primary CD4+ cells is due to epigenetic transcriptional silencing and P-TEFb restriction. J. Virol. 2010, 84, 6425–6437. [Google Scholar] [CrossRef]
- Marban, C.; Suzanne, S.; Dequiedt, F.; de Walque, S.; Redel, L.; Van Lint, C.; Aunis, D.; Rohr, O. Recruitment of chromatin-modifying enzymes by CTIP2 promotes HIV-1 transcriptional silencing. EMBO J. 2007, 26, 412–423. [Google Scholar] [CrossRef] [PubMed]
- Boehm, D.; Calvanese, V.; Dar, R.D.; Xing, S.; Schroeder, S.; Martins, L.; Aull, K.; Li, P.C.; Planelles, V.; Bradner, J.E.; et al. BET bromodomain-targeting compounds reactivate HIV from latency via a Tat-independent mechanism. Cell Cycle 2013, 12, 452–462. [Google Scholar] [CrossRef] [PubMed]
- Dingwall, C.; Ernberg, I.; Gait, M.J.; Green, S.M.; Heaphy, S.; Karn, J.; Lowe, A.D.; Singh, M.; Skinner, M.A. HIV-1 Tat protein stimulates transcription by binding to a U-rich bulge in the stem of the TAR RNA structure. EMBO J. 1990, 9, 4145–4153. [Google Scholar] [CrossRef] [PubMed]
- Kao, S.Y.; Calman, A.F.; Luciw, P.A.; Peterlin, B.M. Anti-termination of transcription within the long terminal repeat of HIV-1 by tat gene product. Nature 1987, 330, 489–493. [Google Scholar] [CrossRef]
- Ghose, R.; Liou, L.Y.; Herrmann, C.H.; Rice, A.P. Induction of TAK (cyclin T1/P-TEFb) in purified resting CD4(+) T lymphocytes by combination of cytokines. J. Virol. 2001, 75, 11336–11343. [Google Scholar] [CrossRef] [PubMed]
- Verdin, E.; Paras, P., Jr.; Van Lint, C. Chromatin disruption in the promoter of human immunodeficiency virus type 1 during transcriptional activation. EMBO J. 1993, 12, 3249–3259. [Google Scholar] [CrossRef] [PubMed]
- Rafati, H.; Parra, M.; Hakre, S.; Moshkin, Y.; Verdin, E.; Mahmoudi, T. Repressive LTR nucleosome positioning by the BAF complex is required for HIV latency. PLoS Biol. 2011, 9, e1001206. [Google Scholar] [CrossRef]
- Friedman, J.; Cho, W.K.; Chu, C.K.; Keedy, K.S.; Archin, N.M.; Margolis, D.M.; Karn, J. Epigenetic silencing of HIV-1 by the histone H3 lysine 27 methyltransferase enhancer of Zeste 2. J. Virol. 2011, 85, 9078–9089. [Google Scholar] [CrossRef]
- Keedy, K.S.; Archin, N.M.; Gates, A.T.; Espeseth, A.; Hazuda, D.J.; Margolis, D.M. A limited group of class I histone deacetylases act to repress human immunodeficiency virus type-1 expression. J. Virol. 2009, 83, 4749–4756. [Google Scholar] [CrossRef]
- Vire, E.; Brenner, C.; Deplus, R.; Blanchon, L.; Fraga, M.; Didelot, C.; Morey, L.; Van Eynde, A.; Bernard, D.; Vanderwinden, J.M.; et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature 2006, 439, 871–874. [Google Scholar] [CrossRef]
- Cheng, A.S.; Lau, S.S.; Chen, Y.; Kondo, Y.; Li, M.S.; Feng, H.; Ching, A.K.; Cheung, K.F.; Wong, H.K.; Tong, J.H.; et al. EZH2-mediated concordant repression of Wnt antagonists promotes beta-catenin-dependent hepatocarcinogenesis. Cancer Res. 2011, 71, 4028–4039. [Google Scholar] [CrossRef]
- Tae, S.; Karkhanis, V.; Velasco, K.; Yaneva, M.; Erdjument-Bromage, H.; Tempst, P.; Sif, S. Bromodomain protein 7 interacts with PRMT5 and PRC2, and is involved in transcriptional repression of their target genes. Nucleic Acids Res. 2011, 39, 5424–5438. [Google Scholar] [CrossRef] [PubMed]
- Imai, K.; Togami, H.; Okamoto, T. Involvement of histone H3 lysine 9 (H3K9) methyltransferase G9a in the maintenance of HIV-1 latency and its reactivation by BIX01294. J. Biol. Chem. 2010, 285, 16538–16545. [Google Scholar] [CrossRef] [PubMed]
- Jadlowsky, J.K.; Wong, J.Y.; Graham, A.C.; Dobrowolski, C.; Devor, R.L.; Adams, M.D.; Fujinaga, K.; Karn, J. Negative elongation factor is required for the maintenance of proviral latency but does not induce promoter-proximal pausing of RNA polymerase II on the HIV long terminal repeat. Mol. Cell. Biol. 2014, 34, 1911–1928. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Inukai, N.; Narita, T.; Wada, T.; Handa, H. Evidence that negative elongation factor represses transcription elongation through binding to a DRB sensitivity-inducing factor/RNA polymerase II complex and RNA. Mol. Cell. Biol. 2002, 22, 2918–2927. [Google Scholar] [CrossRef]
- Pagano, J.M.; Kwak, H.; Waters, C.T.; Sprouse, R.O.; White, B.S.; Ozer, A.; Szeto, K.; Shalloway, D.; Craighead, H.G.; Lis, J.T. Defining NELF-E RNA binding in HIV-1 and promoter-proximal pause regions. PLoS Genet. 2014, 10, e1004090. [Google Scholar] [CrossRef]
- Sedore, S.C.; Byers, S.A.; Biglione, S.; Price, J.P.; Maury, W.J.; Price, D.H. Manipulation of P-TEFb control machinery by HIV: Recruitment of P-TEFb from the large form by Tat and binding of HEXIM1 to TAR. Nucleic Acids Res. 2007, 35, 4347–4358. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Mbonye, U.; Hokello, J.; Karn, J. T-cell receptor signaling enhances transcriptional elongation from latent HIV proviruses by activating P-TEFb through an ERK-dependent pathway. J. Mol. Biol. 2011, 410, 896–916. [Google Scholar] [CrossRef] [PubMed]
- Bukrinsky, M.I.; Etkin, A.F. Plus strand of the HIV provirus DNA is expressed at early stages of infection. AIDS Res. Hum. Retrovir. 1990, 6, 425–426. [Google Scholar] [CrossRef] [PubMed]
- Vanhee-Brossollet, C.; Thoreau, H.; Serpente, N.; D’Auriol, L.; Levy, J.P.; Vaquero, C. A natural antisense RNA derived from the HIV-1 env gene encodes a protein which is recognized by circulating antibodies of HIV+ individuals. Virology 1995, 206, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Michael, N.L.; Vahey, M.T.; d’Arcy, L.; Ehrenberg, P.K.; Mosca, J.D.; Rappaport, J.; Redfield, R.R. Negative-strand RNA transcripts are produced in human immunodeficiency virus type 1-infected cells and patients by a novel promoter downregulated by Tat. J. Virol. 1994, 68, 979–987. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Sklutuis, R.; Groebner, J.L.; Romerio, F. HIV-1 Natural Antisense Transcription and Its Role in Viral Persistence. Viruses 2021, 13, 795. [Google Scholar] [CrossRef] [PubMed]
- Zapata, J.C.; Campilongo, F.; Barclay, R.A.; DeMarino, C.; Iglesias-Ussel, M.D.; Kashanchi, F.; Romerio, F. The Human Immunodeficiency Virus 1 ASP RNA promotes viral latency by recruiting the Polycomb Repressor Complex 2 and promoting nucleosome assembly. Virology 2017, 506, 34–44. [Google Scholar] [CrossRef]
- Haddad, F.; Qin, A.X.; Giger, J.M.; Guo, H.; Baldwin, K.M. Potential pitfalls in the accuracy of analysis of natural sense-antisense RNA pairs by reverse transcription-PCR. BMC Biotechnol. 2007, 7, 21. [Google Scholar] [CrossRef]
- Landry, S.; Halin, M.; Lefort, S.; Audet, B.; Vaquero, C.; Mesnard, J.M.; Barbeau, B. Detection, characterization and regulation of antisense transcripts in HIV-1. Retrovirology 2007, 4, 71. [Google Scholar] [CrossRef]
- Lefebvre, G.; Desfarges, S.; Uyttebroeck, F.; Munoz, M.; Beerenwinkel, N.; Rougemont, J.; Telenti, A.; Ciuffi, A. Analysis of HIV-1 expression level and sense of transcription by high-throughput sequencing of the infected cell. J. Virol. 2011, 85, 6205–6211. [Google Scholar] [CrossRef]
- Kobayashi-Ishihara, M.; Yamagishi, M.; Hara, T.; Matsuda, Y.; Takahashi, R.; Miyake, A.; Nakano, K.; Yamochi, T.; Ishida, T.; Watanabe, T. HIV-1-encoded antisense RNA suppresses viral replication for a prolonged period. Retrovirology 2012, 9, 38. [Google Scholar] [CrossRef] [PubMed]
- Champiat, S.; Raposo, R.A.; Maness, N.J.; Lehman, J.L.; Purtell, S.E.; Hasenkrug, A.M.; Miller, J.C.; Dean, H.; Koff, W.C.; Hong, M.A.; et al. Influence of HAART on alternative reading frame immune responses over the course of HIV-1 infection. PLoS ONE 2012, 7, e39311. [Google Scholar] [CrossRef] [PubMed]
- Laverdure, S.; Gross, A.; Arpin-Andre, C.; Clerc, I.; Beaumelle, B.; Barbeau, B.; Mesnard, J.M. HIV-1 antisense transcription is preferentially activated in primary monocyte-derived cells. J. Virol. 2012, 86, 13785–13789. [Google Scholar] [CrossRef]
- Saayman, S.; Ackley, A.; Turner, A.M.; Famiglietti, M.; Bosque, A.; Clemson, M.; Planelles, V.; Morris, K.V. An HIV-encoded antisense long noncoding RNA epigenetically regulates viral transcription. Mol. Ther. J. Am. Soc. Gene Ther. 2014, 22, 1164–1175. [Google Scholar] [CrossRef]
- Berger, C.T.; Llano, A.; Carlson, J.M.; Brumme, Z.L.; Brockman, M.A.; Cedeno, S.; Harrigan, P.R.; Kaufmann, D.E.; Heckerman, D.; Meyerhans, A.; et al. Immune screening identifies novel T cell targets encoded by antisense reading frames of HIV-1. J. Virol. 2015, 89, 4015–4019. [Google Scholar] [CrossRef] [PubMed]
- Bet, A.; Maze, E.A.; Bansal, A.; Sterrett, S.; Gross, A.; Graff-Dubois, S.; Samri, A.; Guihot, A.; Katlama, C.; Theodorou, I.; et al. The HIV-1 antisense protein (ASP) induces CD8 T cell responses during chronic infection. Retrovirology 2015, 12, 15. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi-Ishihara, M.; Terahara, K.; Martinez, J.P.; Yamagishi, M.; Iwabuchi, R.; Brander, C.; Ato, M.; Watanabe, T.; Meyerhans, A.; Tsunetsugu-Yokota, Y. HIV LTR-Driven Antisense RNA by Itself Has Regulatory Function and May Curtail Virus Reactivation From Latency. Front. Microbiol. 2018, 9, 1066. [Google Scholar] [CrossRef] [PubMed]
- Savoret, J.; Chazal, N.; Moles, J.P.; Tuaillon, E.; Boufassa, F.; Meyer, L.; Lecuroux, C.; Lambotte, O.; Van De Perre, P.; Mesnard, J.M.; et al. A Pilot Study of the Humoral Response Against the AntiSense Protein (ASP) in HIV-1-Infected Patients. Front. Microbiol. 2020, 11, 20. [Google Scholar] [CrossRef]
- Bentley, K.; Deacon, N.; Sonza, S.; Zeichner, S.; Churchill, M. Mutational analysis of the HIV-1 LTR as a promoter of negative sense transcription. Arch. Virol. 2004, 149, 2277–2294. [Google Scholar] [CrossRef]
- Affram, Y.; Zapata, J.C.; Gholizadeh, Z.; Tolbert, W.D.; Zhou, W.; Iglesias-Ussel, M.D.; Pazgier, M.; Ray, K.; Latinovic, O.S.; Romerio, F. The HIV-1 Antisense Protein ASP Is a Transmembrane Protein of the Cell Surface and an Integral Protein of the Viral Envelope. J. Virol. 2019, 93, e00574-19. [Google Scholar] [CrossRef]
- Arpin-Andre, C.; Laverdure, S.; Barbeau, B.; Gross, A.; Mesnard, J.M. Construction of a reporter vector for analysis of bidirectional transcriptional activity of retrovirus LTR. Plasmid 2014, 74, 45–51. [Google Scholar] [CrossRef]
- Padeken, J.; Methot, S.P.; Gasser, S.M. Establishment of H3K9-methylated heterochromatin and its functions in tissue differentiation and maintenance. Nat. Rev. Mol. Cell Biol. 2022, 23, 623–640. [Google Scholar] [CrossRef]
- Wiles, E.T.; Selker, E.U. H3K27 methylation: A promiscuous repressive chromatin mark. Curr. Opin. Genet. Dev. 2017, 43, 31–37. [Google Scholar] [CrossRef]
- Chen, Y.C.; Koutelou, E.; Dent, S.Y.R. Now open: Evolving insights to the roles of lysine acetylation in chromatin organization and function. Mol. Cell 2022, 82, 716–727. [Google Scholar] [CrossRef]
- Colin, L.; Van Lint, C. Molecular control of HIV-1 postintegration latency: Implications for the development of new therapeutic strategies. Retrovirology 2009, 6, 111. [Google Scholar] [CrossRef]
- Nguyen, K.; Das, B.; Dobrowolski, C.; Karn, J. Multiple Histone Lysine Methyltransferases Are Required for the Establishment and Maintenance of HIV-1 Latency. mBio 2017, 8, e00133-17. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, L.B.; Ambrus, J.L., Jr.; Krawczyk, K.A.; Sharma, S.; Brooks, S.; Hsiao, C.B.; Schwartz, S.A. Human Immunodeficiency Virus-Type 1 LTR DNA contains an intrinsic gene producing antisense RNA and protein products. Retrovirology 2006, 3, 80. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.H.; Bracken, A.P.; Pasini, D.; Dietrich, N.; Gehani, S.S.; Monrad, A.; Rappsilber, J.; Lerdrup, M.; Helin, K. A model for transmission of the H3K27me3 epigenetic mark. Nat. Cell Biol. 2008, 10, 1291–1300. [Google Scholar] [CrossRef] [PubMed]
- Hugues, A.; Jacobs, C.S.; Roudier, F. Mitotic Inheritance of PRC2-Mediated Silencing: Mechanistic Insights and Developmental Perspectives. Front. Plant Sci. 2020, 11, 262. [Google Scholar] [CrossRef] [PubMed]
- Folks, T.M.; Clouse, K.A.; Justement, J.; Rabson, A.; Duh, E.; Kehrl, J.H.; Fauci, A.S. Tumor necrosis factor alpha induces expression of human immunodeficiency virus in a chronically infected T-cell clone. Proc. Natl. Acad. Sci. USA 1989, 86, 2365–2368. [Google Scholar] [CrossRef]
- Clouse, K.A.; Powell, D.; Washington, I.; Poli, G.; Strebel, K.; Farrar, W.; Barstad, P.; Kovacs, J.; Fauci, A.S.; Folks, T.M. Monokine regulation of human immunodeficiency virus-1 expression in a chronically infected human T cell clone. J. Immunol. 1989, 142, 431–438. [Google Scholar] [CrossRef]
- Perez, V.L.; Rowe, T.; Justement, J.S.; Butera, S.T.; June, C.H.; Folks, T.M. An HIV-1-infected T cell clone defective in IL-2 production and Ca2+ mobilization after CD3 stimulation. J. Immunol. 1991, 147, 3145–3148. [Google Scholar] [CrossRef]
- Symons, J.; Chopra, A.; Malatinkova, E.; De Spiegelaere, W.; Leary, S.; Cooper, D.; Abana, C.O.; Rhodes, A.; Rezaei, S.D.; Vandekerckhove, L.; et al. HIV integration sites in latently infected cell lines: Evidence of ongoing replication. Retrovirology 2017, 14, 2. [Google Scholar] [CrossRef]
- Schroder, A.R.; Shinn, P.; Chen, H.; Berry, C.; Ecker, J.R.; Bushman, F. HIV-1 integration in the human genome favors active genes and local hotspots. Cell 2002, 110, 521–529. [Google Scholar] [CrossRef]
- Maldarelli, F.; Wu, X.; Su, L.; Simonetti, F.R.; Shao, W.; Hill, S.; Spindler, J.; Ferris, A.L.; Mellors, J.W.; Kearney, M.F.; et al. HIV latency. Specific HIV integration sites are linked to clonal expansion and persistence of infected cells. Science 2014, 345, 179–183. [Google Scholar] [CrossRef]
- Cohn, L.B.; Silva, I.T.; Oliveira, T.Y.; Rosales, R.A.; Parrish, E.H.; Learn, G.H.; Hahn, B.H.; Czartoski, J.L.; McElrath, M.J.; Lehmann, C.; et al. HIV-1 integration landscape during latent and active infection. Cell 2015, 160, 420–432. [Google Scholar] [CrossRef]
- Einkauf, K.B.; Osborn, M.R.; Gao, C.; Sun, W.; Sun, X.; Lian, X.; Parsons, E.M.; Gladkov, G.T.; Seiger, K.W.; Blackmer, J.E.; et al. Parallel analysis of transcription, integration, and sequence of single HIV-1 proviruses. Cell 2022, 185, 266–282.e215. [Google Scholar] [CrossRef] [PubMed]
- Machida, S.; Depierre, D.; Chen, H.C.; Thenin-Houssier, S.; Petitjean, G.; Doyen, C.M.; Takaku, M.; Cuvier, O.; Benkirane, M. Exploring histone loading on HIV DNA reveals a dynamic nucleosome positioning between unintegrated and integrated viral genome. Proc. Natl. Acad. Sci. USA 2020, 117, 6822–6830. [Google Scholar] [CrossRef] [PubMed]
- Musick, A.; Spindler, J.; Boritz, E.; Perez, L.; Crespo-Velez, D.; Patro, S.C.; Sobolewski, M.D.; Bale, M.J.; Reid, C.; Keele, B.F.; et al. HIV Infected T Cells Can Proliferate in vivo Without Inducing Expression of the Integrated Provirus. Front. Microbiol. 2019, 10, 2204. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Zankharia, U.; Vladimirova, O.; Yi, Y.; Collman, R.G.; Lieberman, P.M. Epigenetic Landscape of HIV-1 Infection in Primary Human Macrophage. J. Virol. 2022, 96, e0016222. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, R.; Caico, I.; Xu, Z.; Iqbal, M.S.; Romerio, F. Epigenetic Regulation of HIV-1 Sense and Antisense Transcription in Response to Latency-Reversing Agents. Non-Coding RNA 2023, 9, 5. https://doi.org/10.3390/ncrna9010005
Li R, Caico I, Xu Z, Iqbal MS, Romerio F. Epigenetic Regulation of HIV-1 Sense and Antisense Transcription in Response to Latency-Reversing Agents. Non-Coding RNA. 2023; 9(1):5. https://doi.org/10.3390/ncrna9010005
Chicago/Turabian StyleLi, Rui, Isabella Caico, Ziyan Xu, Mohammad Shameel Iqbal, and Fabio Romerio. 2023. "Epigenetic Regulation of HIV-1 Sense and Antisense Transcription in Response to Latency-Reversing Agents" Non-Coding RNA 9, no. 1: 5. https://doi.org/10.3390/ncrna9010005
APA StyleLi, R., Caico, I., Xu, Z., Iqbal, M. S., & Romerio, F. (2023). Epigenetic Regulation of HIV-1 Sense and Antisense Transcription in Response to Latency-Reversing Agents. Non-Coding RNA, 9(1), 5. https://doi.org/10.3390/ncrna9010005