
Knockdown of lncRNA TP53TG1 Enhances the Efficacy of Sorafenib in Human Hepatocellular Carcinoma Cells



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Sorafenib Induces lncRNA TP53TG1 Expression
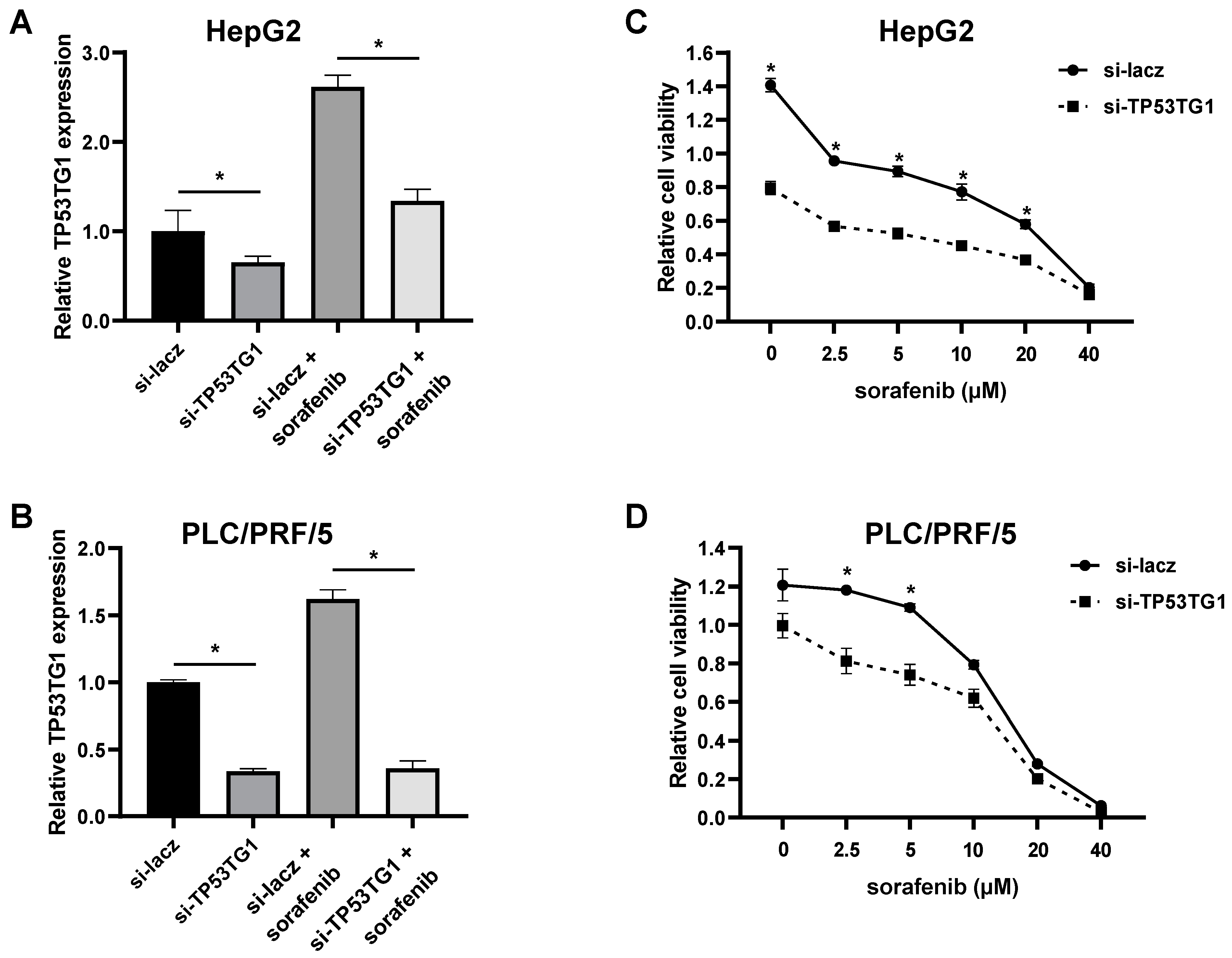
2.2. TP53TG1 Knockdown Increases Sorafenib Sensitivity
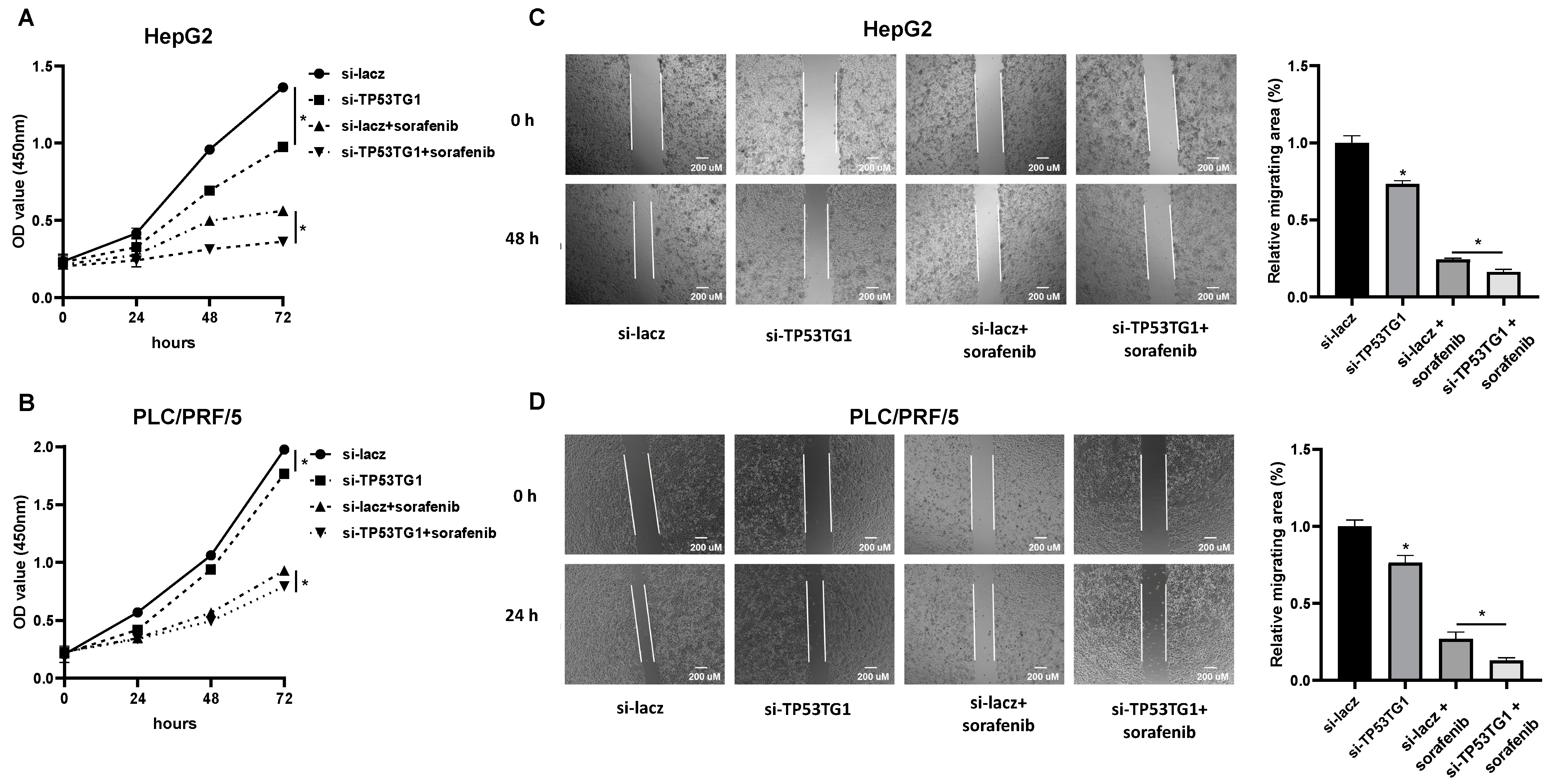
2.3. TP53TG1 Knockdown Has a Synergetic Effect with Sorafenib on Cell Proliferation and Migration
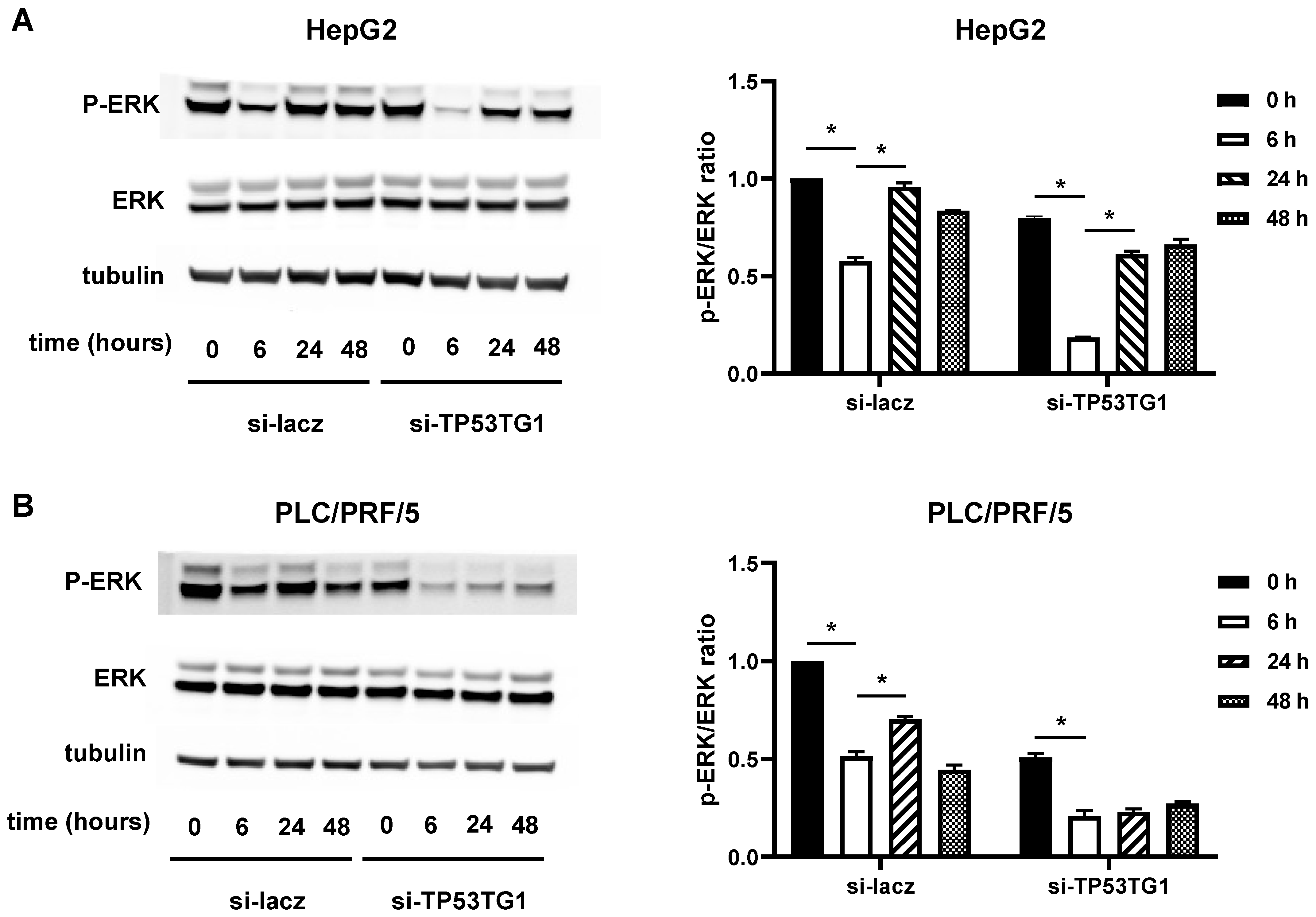
2.4. TP53TG1 May Affect Sorafenib Efficacy through ERK Signaling
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Transfection
4.2. Cell Counting kit-8 (CCK-8) Assay
4.3. RNA Extraction, Reverse Transcription, and qRT-PCR
4.4. Western Blotting
4.5. Wound-Healing Assay
4.6. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yan, L.; Lin, J.; Ke, K.; Wu, Z.; Huang, J.; Huang, N.; Yang, W. A meta-analysis comparing hepatic arterial infusion chemotherapy and sorafenib for advanced hepatocellular carcinoma. Transl. Cancer Res. 2022, 11, 99–112. [Google Scholar] [CrossRef] [PubMed]
- Raoul, J.-L.; Adhoute, X.; Penaranda, G.; Perrier, H.; Castellani, P.; Oules, V.; Bourlière, M. Sorafenib: Experience and Better Manage-ment of Side Effects Improve Overall Survival in Hepatocellular Carcinoma Patients: A Real-Life Retrospective Analysis. Liver Cancer 2019, 8, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Gao, Z.-H.; Qu, X.-J. The adverse effects of sorafenib in patients with advanced cancers. Basic Clin. Pharmacol. Toxicol. 2015, 116, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Boudou-Rouquette, P.; Ropert, S.; Mir, O.; Coriat, R.; Billemont, B.; Tod, M.; Cabanes, L.; Franck, N.; Blanchet, B.; Goldwasser, F. Variability of sorafenib toxicity and exposure over time: A pharmacokinetic/pharmacodynamic analysis. Oncologist 2012, 17, 1204–1212. [Google Scholar] [CrossRef]
- Nikanjam, M.; Liu, S.; Yang, J.; Kurzrock, R. Dosing Three-Drug Combinations That Include Targeted Anti-Cancer Agents: Analysis of 37,763 Patients. Oncologist 2017, 22, 576–584. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, G.; Liu, X.; Song, Y.; Xie, J.; Li, G.; Ren, J.; Wang, H.; Mou, J.; Dai, J. Sorafenib inhibited cell growth through the MEK/ERK signaling pathway in acute promyelocytic leukemia cells. Oncol. Lett. 2018, 15, 5620–5626. [Google Scholar] [CrossRef]
- Guo, Y.-J.; Pan, W.-W.; Liu, S.-B.; Shen, Z.-F.; Xu, Y.; Hu, L.-L. ERK/MAPK signalling pathway and tumorigenesis. Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef]
- Sun, Y.; Liu, W.-Z.; Liu, T.; Feng, X.; Yang, N.; Zhou, H.-F. Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. J. Recept. Signal Transduct. 2015, 35, 600–604. [Google Scholar] [CrossRef]
- Tayama, H.; Karasawa, H.; Yamamura, A.; Okamura, Y.; Katsuoka, F.; Suzuki, H.; Kajiwara, T.; Kobayashi, M.; Hatsuzawa, Y.; Shiihara, M.; et al. The association between ERK inhibitor sensitivity and molecular characteristics in colorectal cancer. Biochem. Biophys. Res. Commun. 2021, 560, 59–65. [Google Scholar] [CrossRef]
- Zimmer, L.; Livingstone, E.; Krackhardt, A.; Schultz, E.S.; Göppner, D.; Assaf, C.; Trebing, D.; Stelter, K.; Windemuth-Kieselbach, C.; Ugurel, S.; et al. Encorafenib, binimetinib plus pembrolizumab triplet therapy in patients with advanced BRAF(V600) mutant melanoma: Safety and tolerability results from the phase I IMMU-TARGET trial. Eur. J. Cancer 2021, 158, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Kane, R.C.; Farrell, A.T.; Saber, H.; Tang, S.; Williams, G.; Jee, J.M.; Liang, C.; Booth, B.; Chidambaram, N.; Morse, D.; et al. Sorafenib for the treatment of advanced renal cell carcinoma. Clin. Cancer Res. 2006, 12, 7271–7278. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Gholipour, M.; Hussen, B.M.; Taheri, M. The Impact of Long Non-Coding RNAs in the Pathogenesis of Hepatocellular Carcinoma. Front. Oncol. 2021, 11, 649107. [Google Scholar] [CrossRef]
- Zhang, Y.; Tang, L. The Application of lncRNAs in Cancer Treatment and Diagnosis. Recent Pat. Anti-Cancer Drug Discov. 2018, 13, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.; Feng, B.; Abudoureyimu, M.; Zhi, Y.; Zhou, H.; Wang, T.; Chu, X.; Chen, P.; Wang, R. Non-coding RNAs: Emerging Regulators of Sorafenib Resistance in Hepatocellular Carcinoma. Front. Oncol. 2019, 9, 1156. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Dong, X.; He, C.; Tan, G.; Li, Z.; Zhai, B.; Feng, J.; Jiang, X.; Liu, C.; Jiang, H.; et al. LncRNA SNHG1 contributes to sorafenib resistance by activating the Akt pathway and is positively regulated by miR-21 in hepatocellular carcinoma cells. J. Exp. Clin. Cancer Res. 2019, 38, 183. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhou, Y.; Yang, L.; Ma, Y.; Peng, X.; Yang, S.; Li, H.; Liu, J. LncRNA NEAT1 promotes autophagy via regulating miR-204/ATG3 and enhanced cell resistance to sorafenib in hepatocellular carcinoma. J. Cell. Physiol. 2020, 235, 3402–3413. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, H.; Du, Y.; Liu, P.; Zhang, J.; Li, Y.; Shen, H.; Xing, L.; Xue, X.; Chen, J.; et al. Long noncoding RNA TP53TG1 promotes pancreatic ductal adenocarcinoma development by acting as a molecular sponge of microRNA-96. Cancer Sci. 2019, 110, 2760–2772. [Google Scholar] [CrossRef]
- Liao, D.; Liu, X.; Yuan, X.; Feng, P.; Ouyang, Z.; Liu, Y.; Li, C. Long non-coding RNA tumor protein 53 target gene 1 promotes cervical cancer development via regulating microRNA-33a-5p to target forkhead box K2. Cell Cycle 2022, 21, 572–584. [Google Scholar] [CrossRef]
- Sun, J.; Guo, Y.; Chen, T.; Jin, T.; Ma, L.; Ai, L.; Guo, J.; Niu, Z.; Yang, R.; Wang, Q.; et al. Systematic analyses identify the anti-fibrotic role of lncRNA TP53TG1 in IPF. Cell Death Dis. 2022, 13, 525. [Google Scholar] [CrossRef]
- Lu, Q.; Guo, Q.; Xin, M.; Lim, C.; Gamero, A.M.; Gerhard, G.S.; Yang, L. LncRNA TP53TG1 Promotes the Growth and Migration of Hepatocellular Carcinoma Cells via Activation of ERK Signaling. Non-Coding RNA 2021, 7, 52. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Cao, Y.; Chen, C.; Zhang, X.; McNabola, A.; Wilkie, D.; Wilhelm, S.; Lynch, M.; Carter, C. Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res. 2006, 66, 11851–11858. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Liu, Y.; Liang, P.; Wang, B.; Tan, H.; Zhang, Y.; Gao, X.; Gao, J. TP53TG1 enhances cisplatin sensitivity of non-small cell lung cancer cells through regulating miR-18a/PTEN axis. Cell Biosci. 2018, 8, 23. [Google Scholar] [CrossRef]
- Gao, W.; Qiao, M.; Luo, K. Long Noncoding RNA TP53TG1 Contributes to Radioresistance of Glioma Cells Via miR-524–5p/RAB5A Axis. Cancer Biother. Radiopharm. 2021, 36, 600–612. [Google Scholar] [CrossRef]
- Diaz-Lagares, A.; Crujeiras, A.B.; Lopez-Serra, P.; Soler, M.; Setien, F.; Goyal, A.; Sandoval, J.; Hashimoto, Y.; Martinez-Cardús, A.; Gomez, A.; et al. Epigenetic inactivation of the p53-induced long noncoding RNA TP53 target 1 in human cancer. Proc. Natl. Acad. Sci. USA 2016, 113, E7535–E7544. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Jin, R.; Zhao, J.; Liu, J.; Ying, H.; Yan, H.; Zhou, S.; Liang, Y.; Huang, D.; Liang, X.; et al. Potential molecular, cellular and microenvironmental mechanism of sorafenib resistance in hepatocellular carcinoma. Cancer Lett. 2015, 367, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, Y.-C.; Sung, Y.-C.; Ramjiawan, R.R.; Lin, T.-T.; Chang, C.-C.; Jeng, K.-S.; Chang, C.-F.; Liu, C.-H.; Gao, D.-Y.; et al. Overcoming sorafenib evasion in hepatocellular carcinoma using CXCR4-targeted nanoparticles to co-deliver MEK-inhibitors. Sci. Rep. 2017, 7, 44123. [Google Scholar] [CrossRef] [PubMed]
- Galmiche, A.; Chauffert, B.; Barbare, J.-C. New biological perspectives for the improvement of the efficacy of sorafenib in hepatocellular carcinoma. Cancer Lett. 2014, 346, 159–162. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, Z.; Zhang, Y.; Liu, S.; Li, L. Long Non-Coding RNA TP53TG1 Upregulates SHCBP1 to Promote Retinoblastoma Progression by Sponging miR-33b. Cell Transplant. 2021, 30, 9636897211025223. [Google Scholar] [CrossRef]
- Davalos, A.; Goedeke, L.; Smibert, P.; Ramírez, C.M.; Warrier, N.P.; Andreo, U.; Cirera-Salinas, D.; Rayner, K.; Suresh, U.; Pastor-Pareja, J.C.; et al. miR-33a/b contribute to the regulation of fatty acid metabolism and insulin signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 9232–9237. [Google Scholar] [CrossRef]
- Liu, S.M.; Lu, J.; Lee, H.-C.; Chung, F.-H.; Ma, N. miR-524-5p suppresses the growth of oncogenic BRAF melanoma by targeting BRAF and ERK2. Oncotarget 2014, 5, 9444–9459. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Xin, Y.; Huang, M.; Guo, W.-W.; Huang, Q.; Zhang, L.; Jiang, G. Nano-based delivery of RNAi in cancer therapy. Mol. Cancer 2017, 16, 134. [Google Scholar] [CrossRef] [PubMed]
- Babu, A.; Munshi, A.; Ramesh, R. Combinatorial therapeutic approaches with RNAi and anticancer drugs using nanodrug delivery systems. Drug Dev. Ind. Pharm. 2017, 43, 1391–1401. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.J.; Erb, H.H.H.; Hobisch, A.; Santer, F.R.; Culi, Z. Sorafenib decreases proliferation and induces apoptosis of prostate cancer cells by inhibition of the androgen receptor and Akt signaling pathways. Endocr. Relat. Cancer 2012, 19, 305–319. [Google Scholar] [CrossRef]
- Sun, W.; Wang, Y.; Cai, M.; Lin, L.; Chen, X.; Cao, Z.; Zhu, K.; Shuai, X. Codelivery of sorafenib and GPC3 siRNA with PEI-modified liposomes for hepatoma therapy. Biomater. Sci. 2017, 5, 2468–2479. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, Q.; Xin, M.; Guo, Q.; Rothberg, B.S.; Gamero, A.M.; Yang, L. Knockdown of lncRNA TP53TG1 Enhances the Efficacy of Sorafenib in Human Hepatocellular Carcinoma Cells. Non-Coding RNA 2022, 8, 61. https://doi.org/10.3390/ncrna8040061
Lu Q, Xin M, Guo Q, Rothberg BS, Gamero AM, Yang L. Knockdown of lncRNA TP53TG1 Enhances the Efficacy of Sorafenib in Human Hepatocellular Carcinoma Cells. Non-Coding RNA. 2022; 8(4):61. https://doi.org/10.3390/ncrna8040061
Chicago/Turabian StyleLu, Qingchun, Mingyang Xin, Qian Guo, Brad S. Rothberg, Ana M. Gamero, and Ling Yang. 2022. "Knockdown of lncRNA TP53TG1 Enhances the Efficacy of Sorafenib in Human Hepatocellular Carcinoma Cells" Non-Coding RNA 8, no. 4: 61. https://doi.org/10.3390/ncrna8040061
APA StyleLu, Q., Xin, M., Guo, Q., Rothberg, B. S., Gamero, A. M., & Yang, L. (2022). Knockdown of lncRNA TP53TG1 Enhances the Efficacy of Sorafenib in Human Hepatocellular Carcinoma Cells. Non-Coding RNA, 8(4), 61. https://doi.org/10.3390/ncrna8040061