
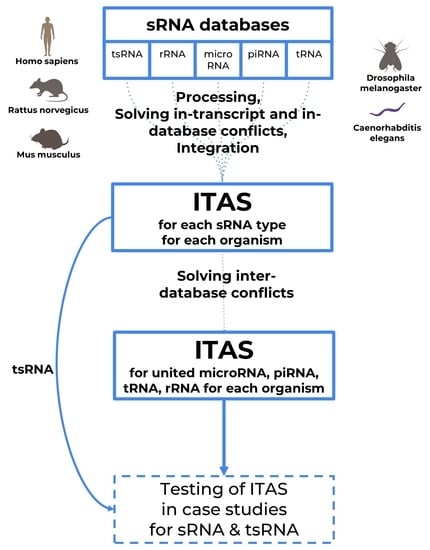
ITAS: Integrated Transcript Annotation for Small RNA

 , , , , , and
, , , , , and
Abstract
:
1. Introduction
1.1. sRNAs in Biology and Medicine
1.2. Expression and Fragment Analysis of RNA-seq Data
2. Results
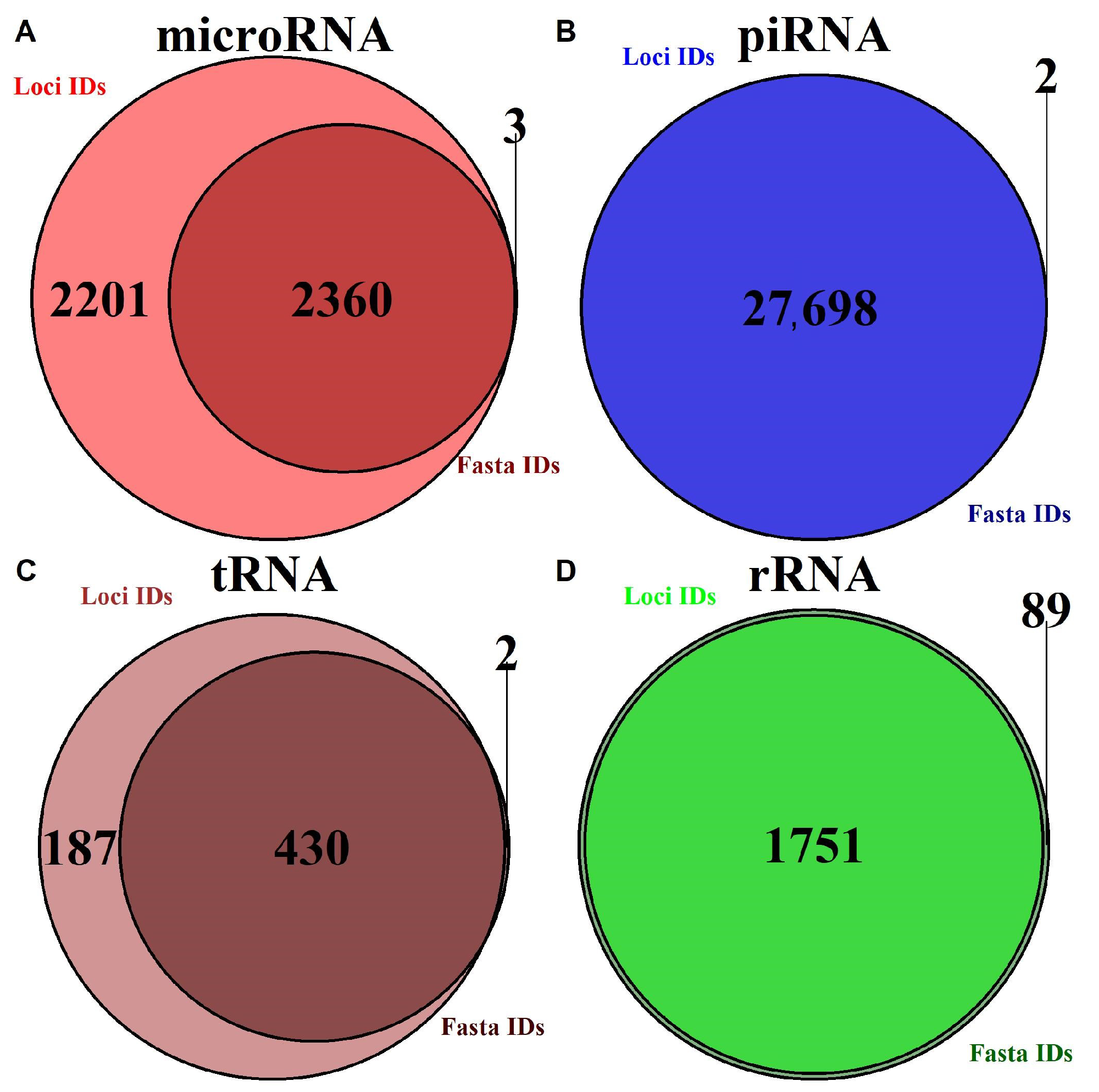
2.1. Database Summary
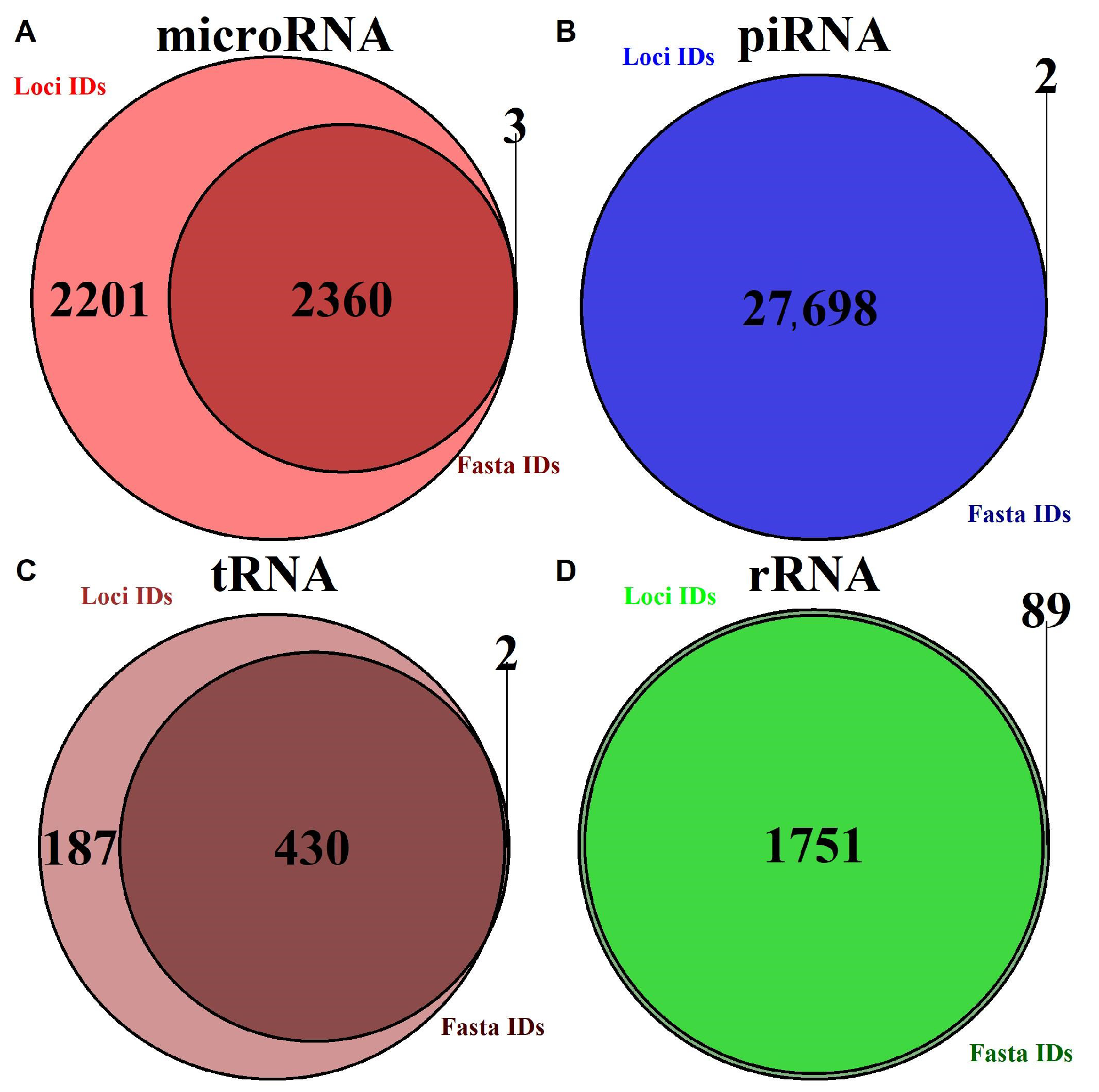
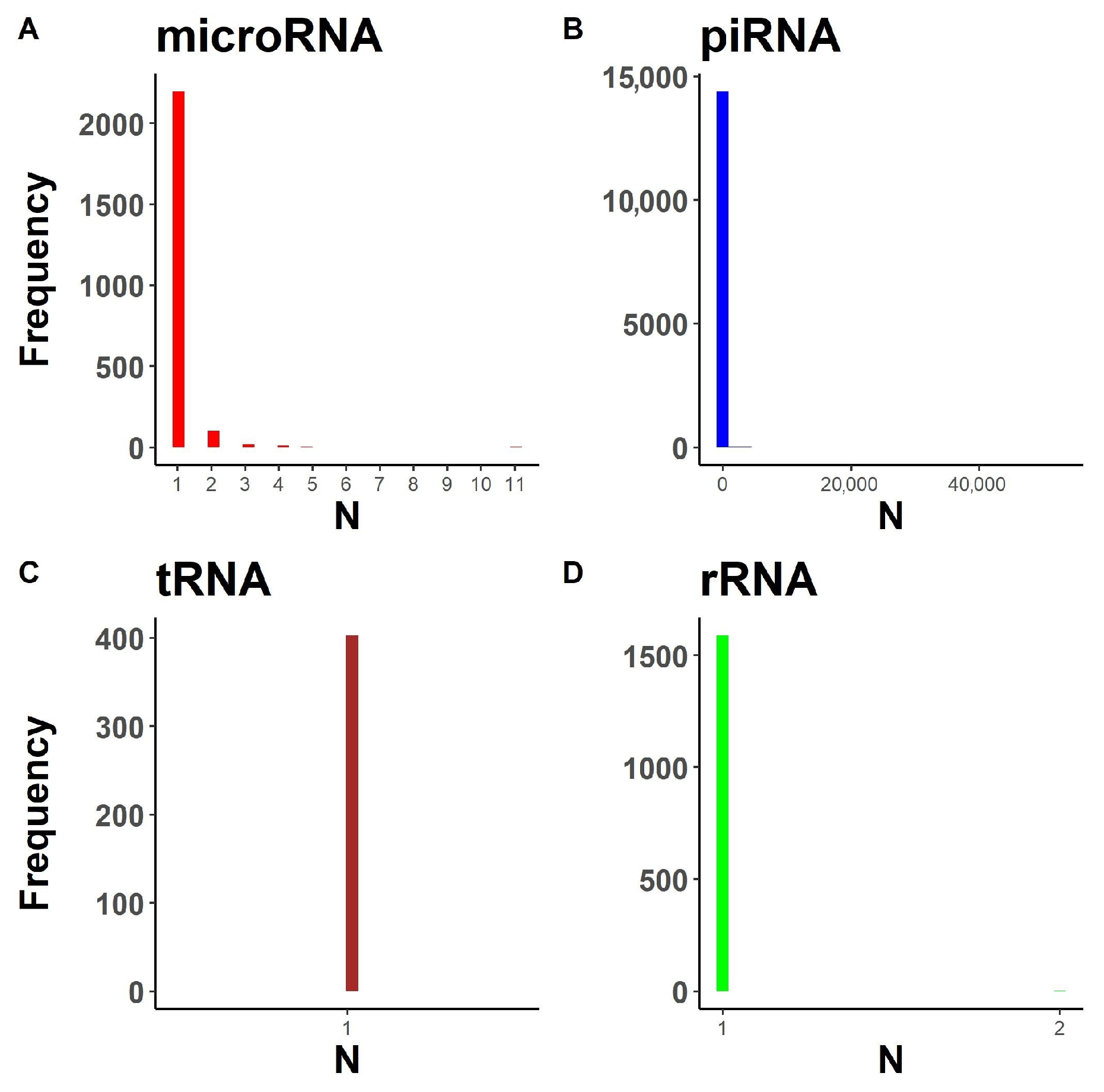
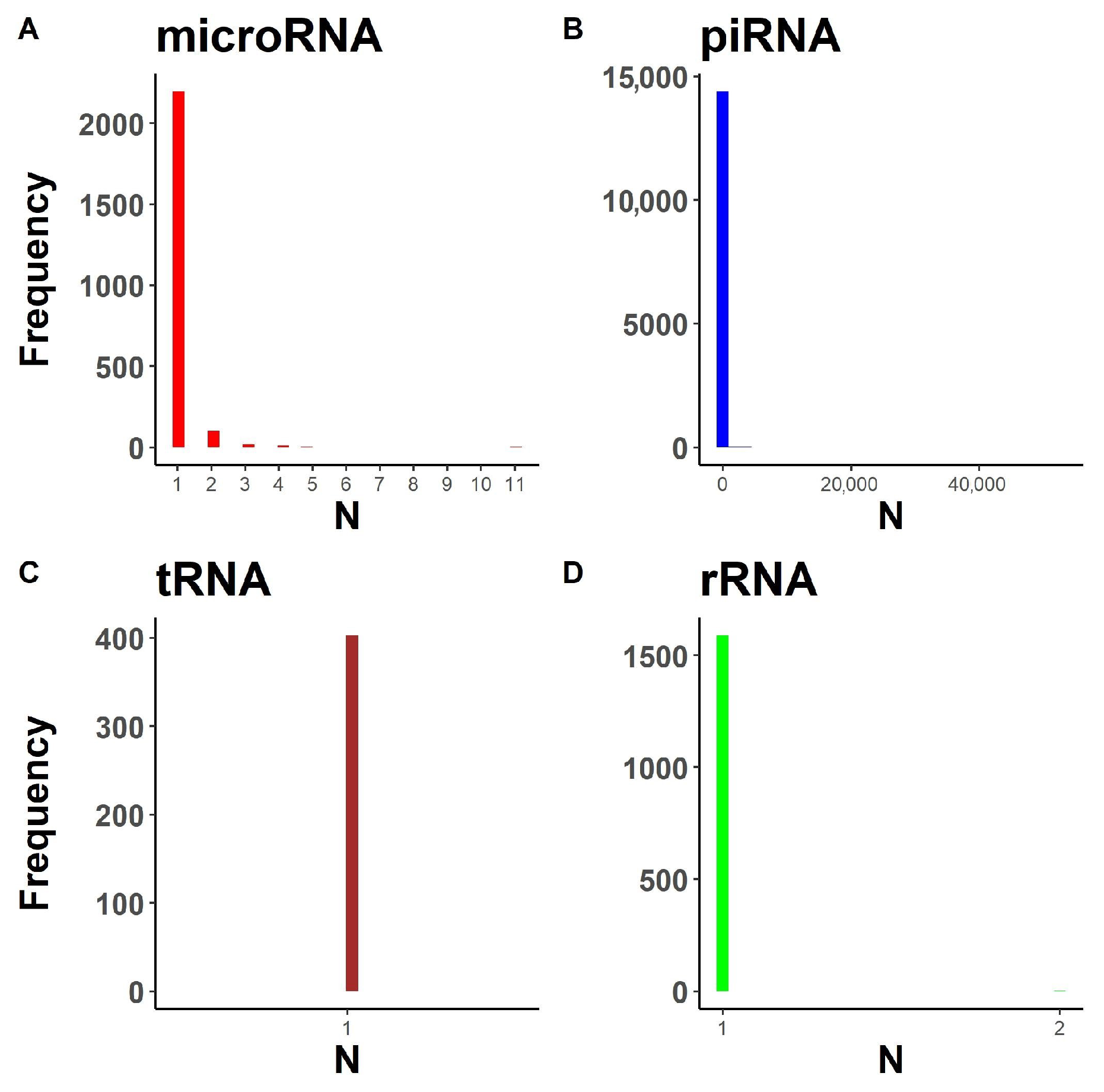
2.1.1. Annotation Entry Data Are Incomplete
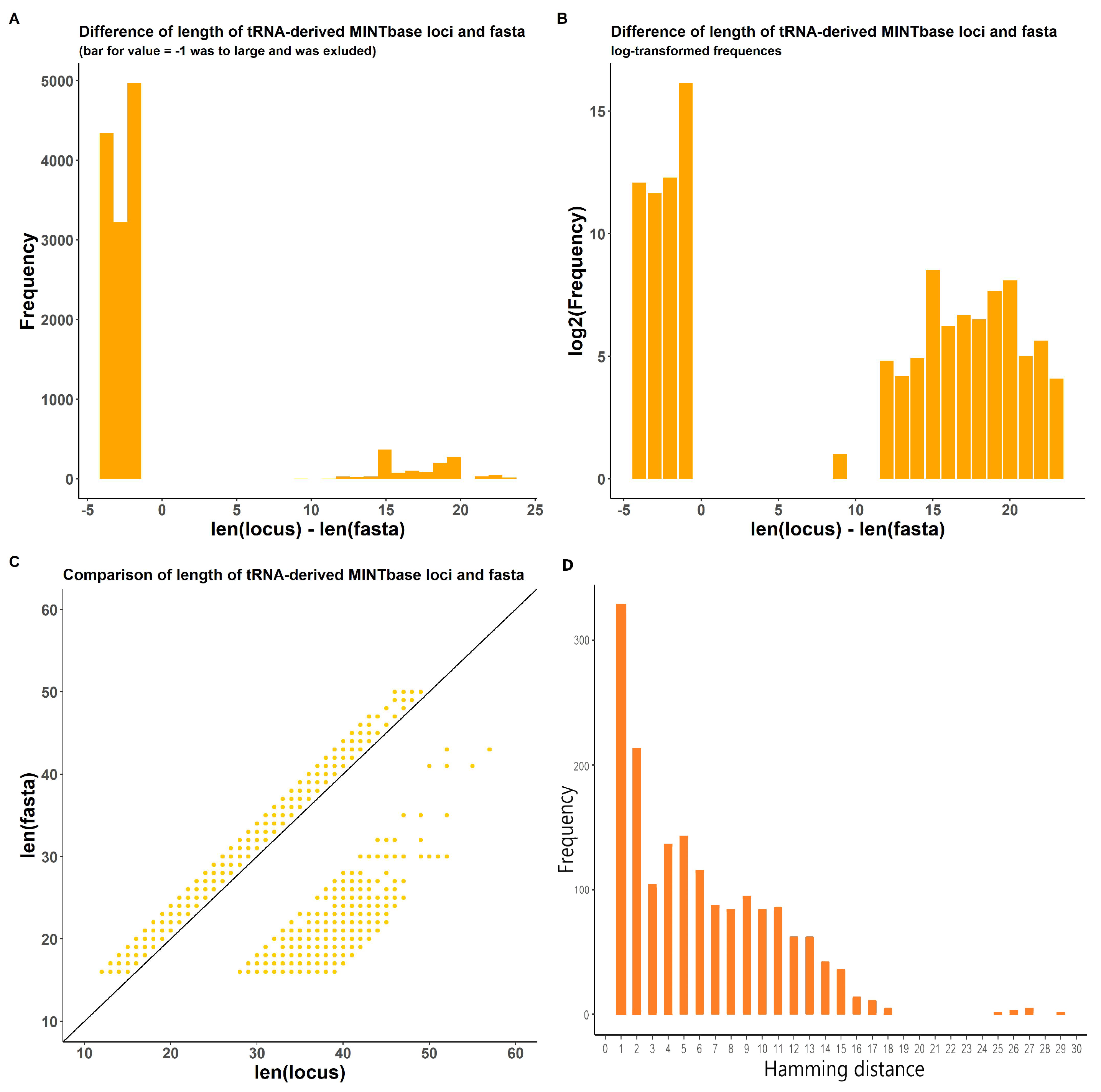
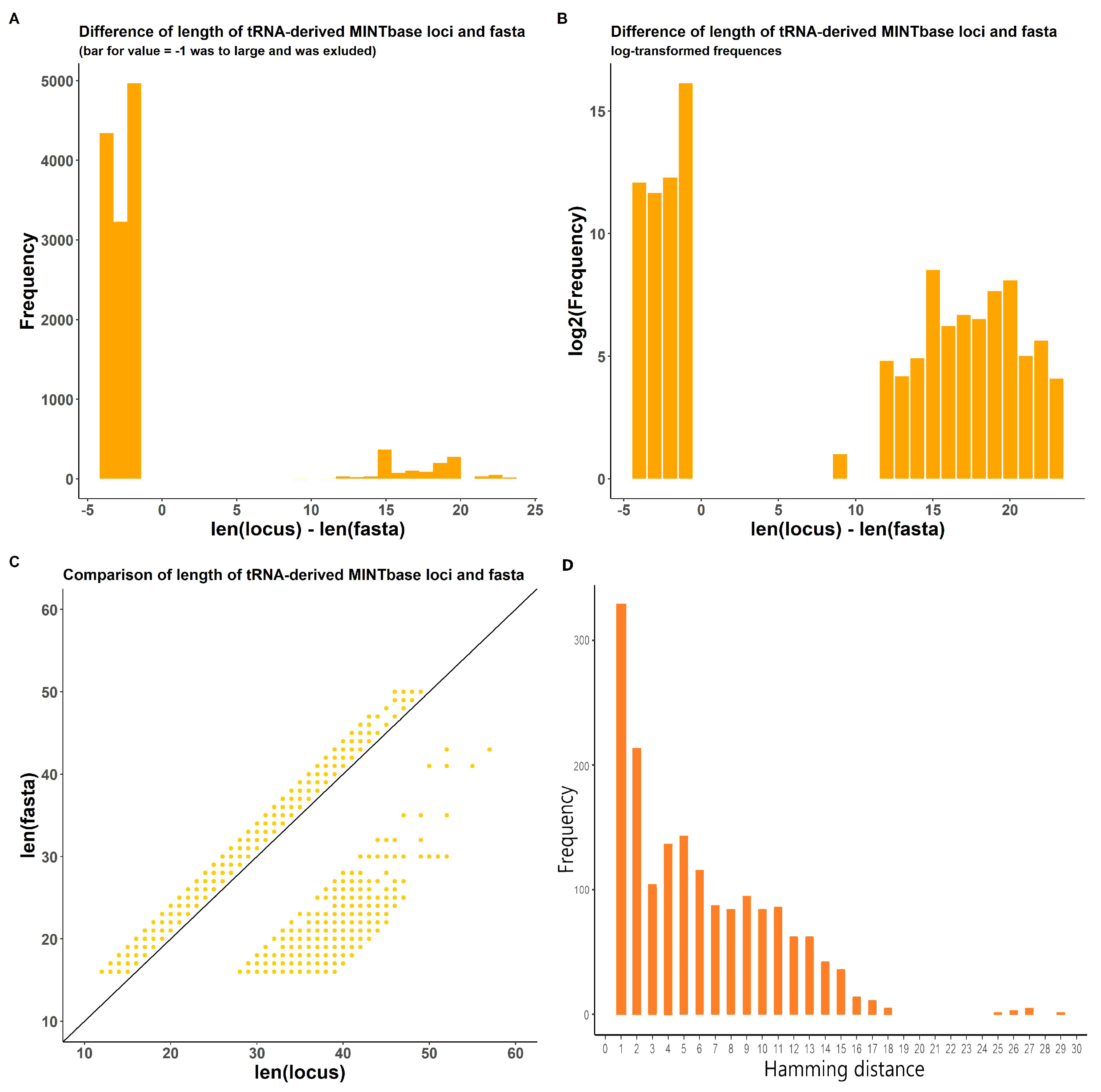
2.1.2. In-Transcript Data Conflicts
2.1.3. In-Database Transcript Data Conflicts
2.1.4. Inter-Database Transcript Data Conflicts
2.2. Integrated Transcript Annotation for Small RNA (ITAS)
- Retrieve and fill in missing data for the incomplete entries;
- Approach the problem of multiple loci per transcript;
- Identify, correct if possible and filter out otherwise entries for transcripts with conflicting fasta-delivered data and locus-delivered data;
- Identify and filter out in-database loci-wise intersecting entries;
- Identify and filter out inter-database loci-wise intersecting entries.
- Integrated annotation data for different biotypes of sRNA with removed inter-database conflicts (statistics presented in Table 4);
- Separate annotations for specific small RNA types with no inter-database conflict filtering applied (statistics presented in Table 5);
- Separate annotation for tsRNA analysis (statistics presented in Table 5, last column).
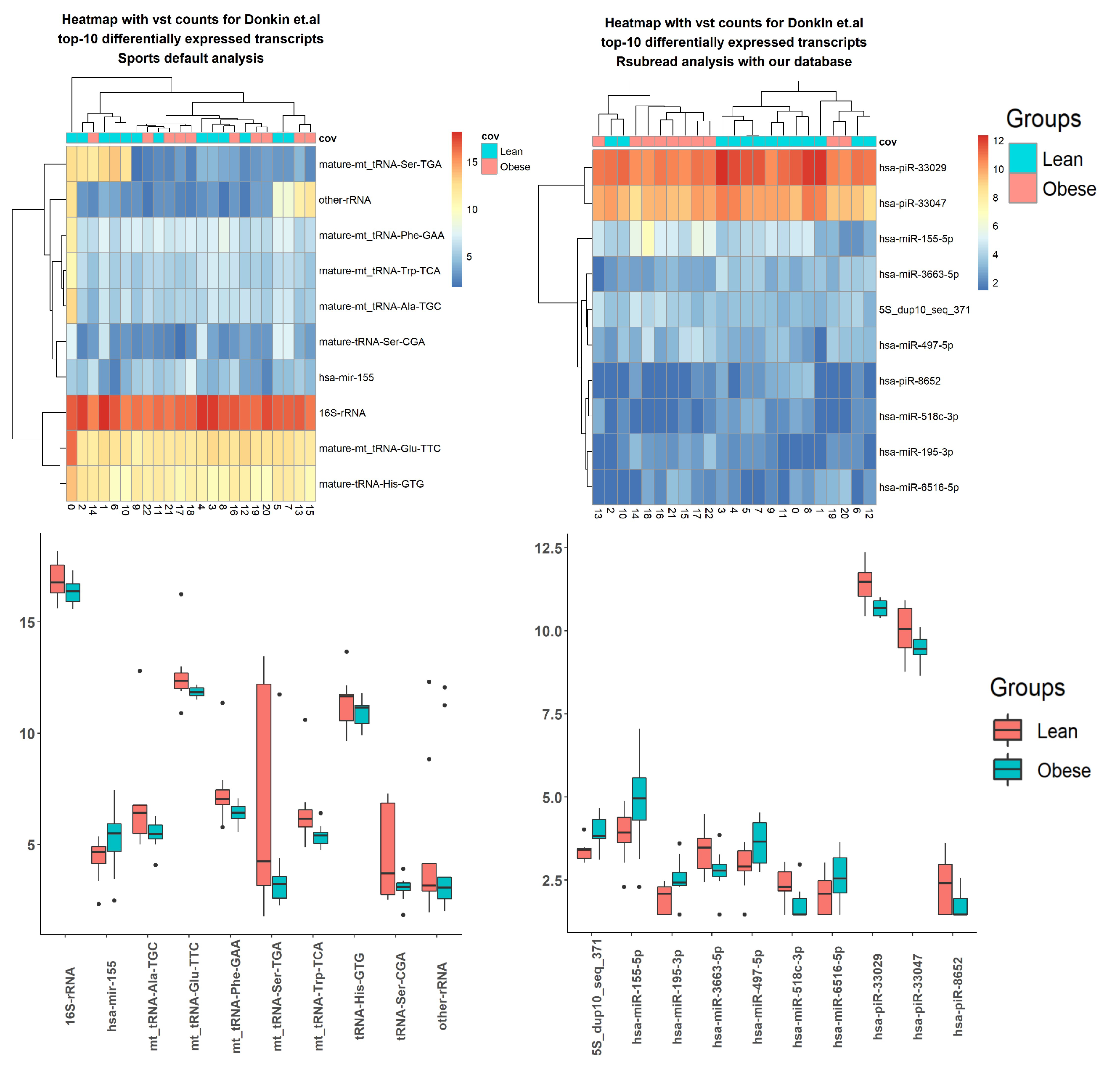
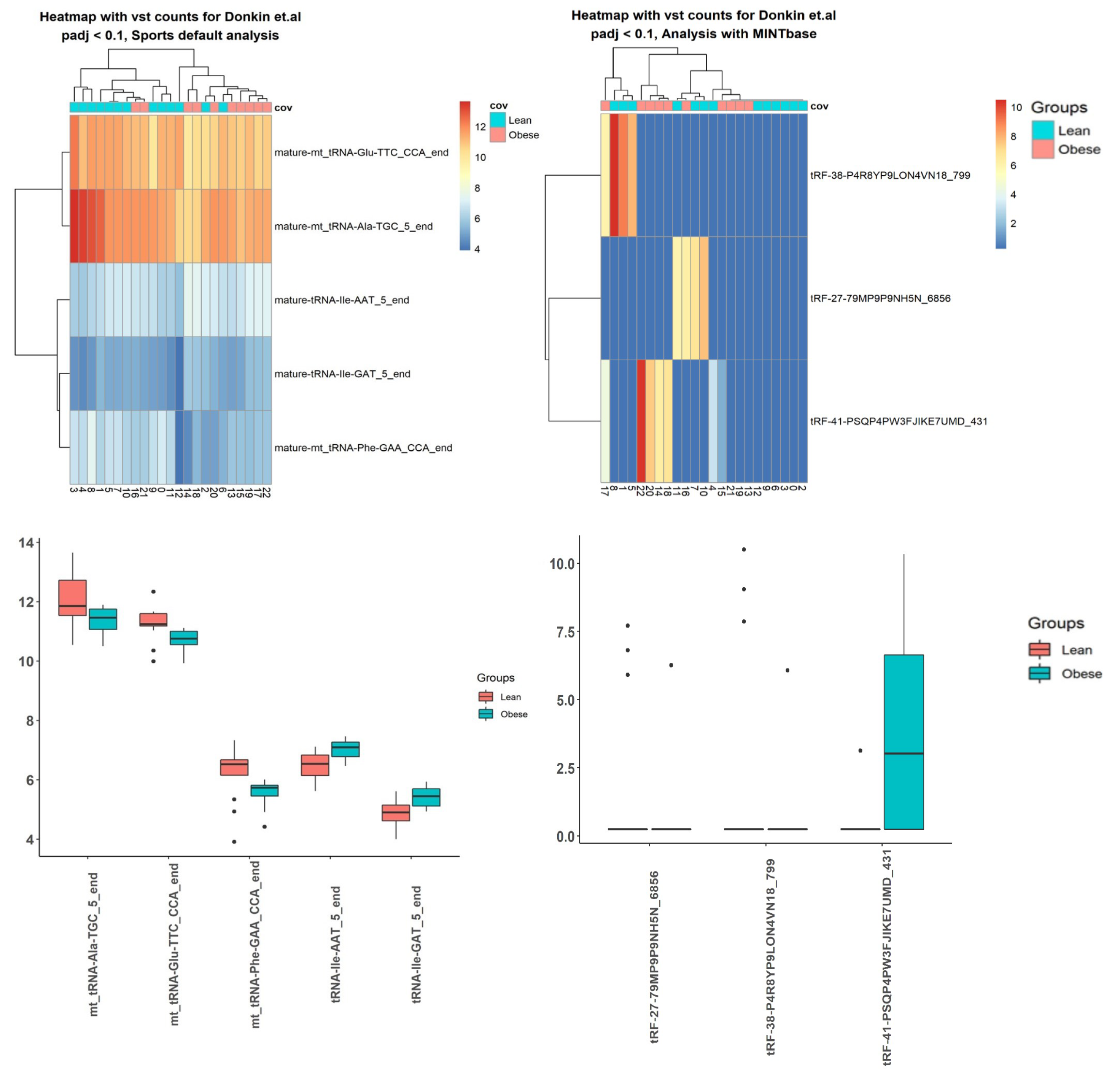
2.3. Case Studies
3. Materials and Methods
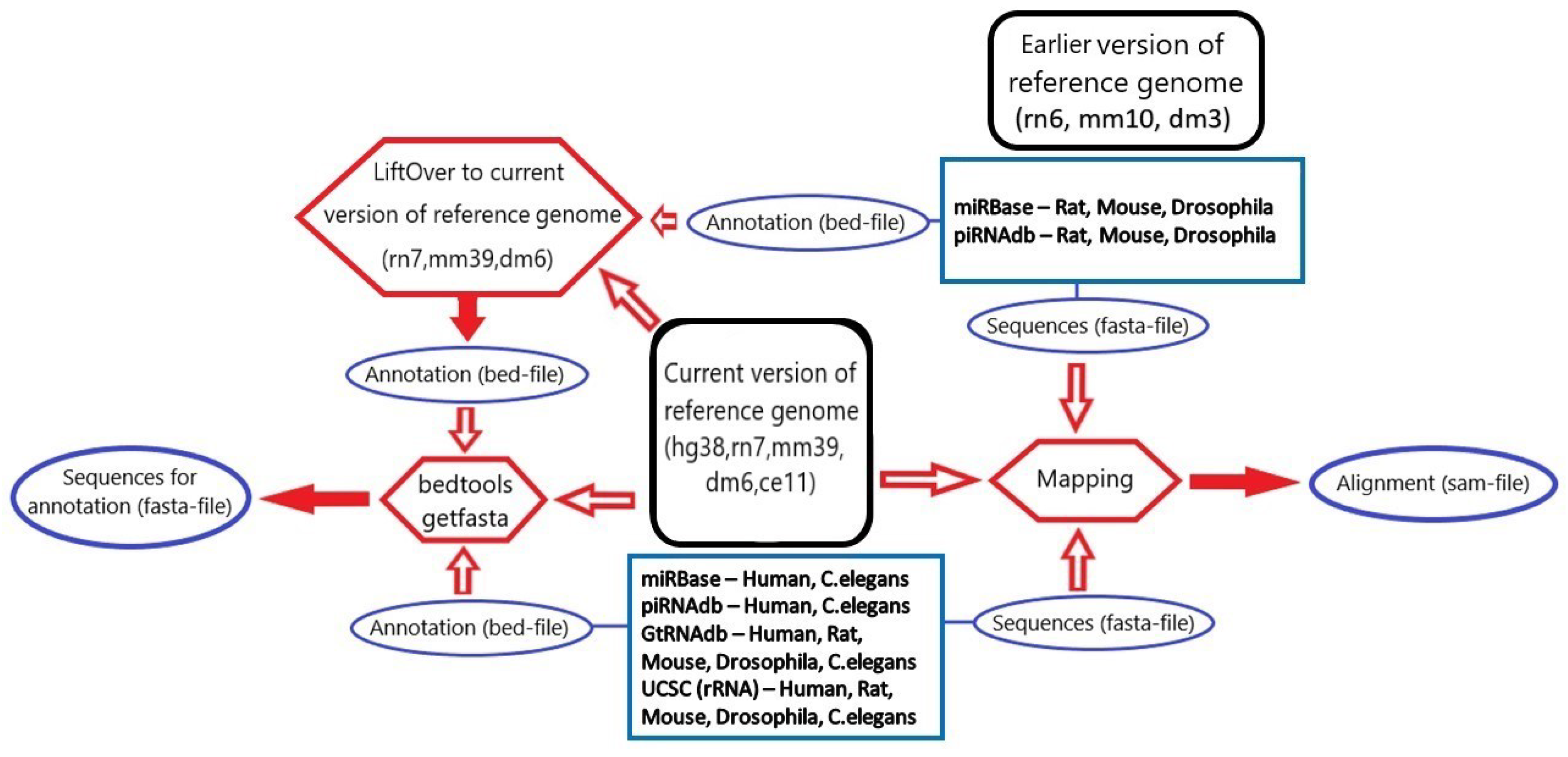
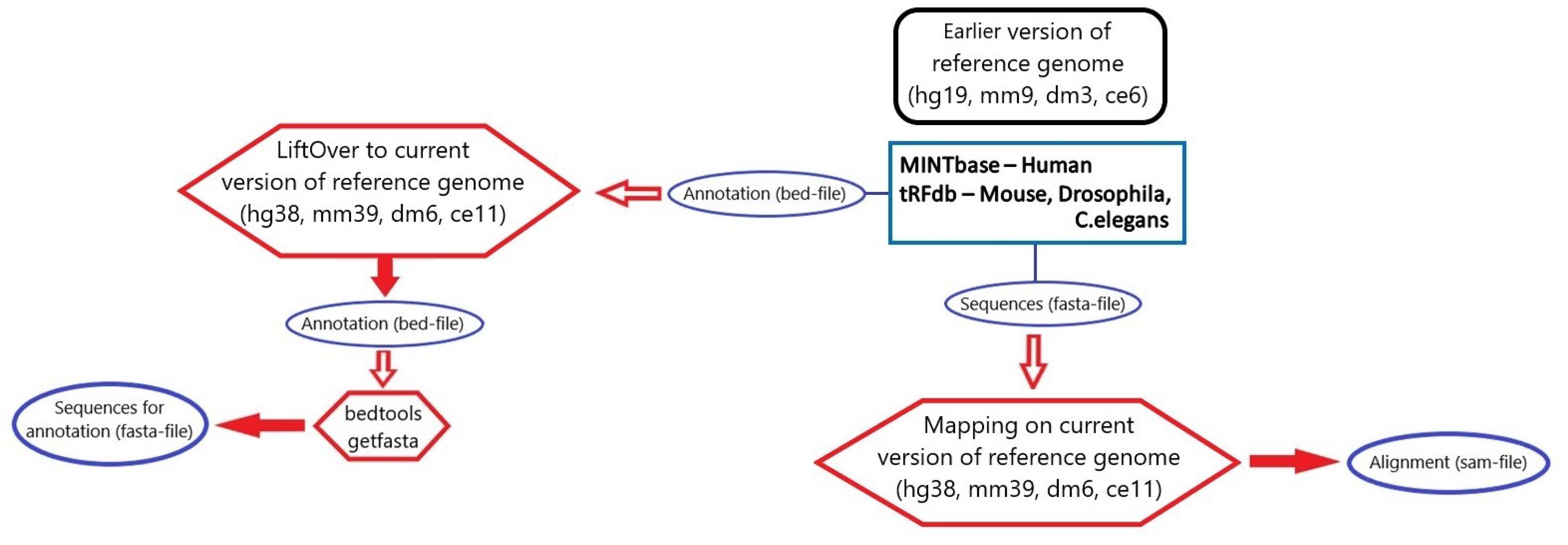
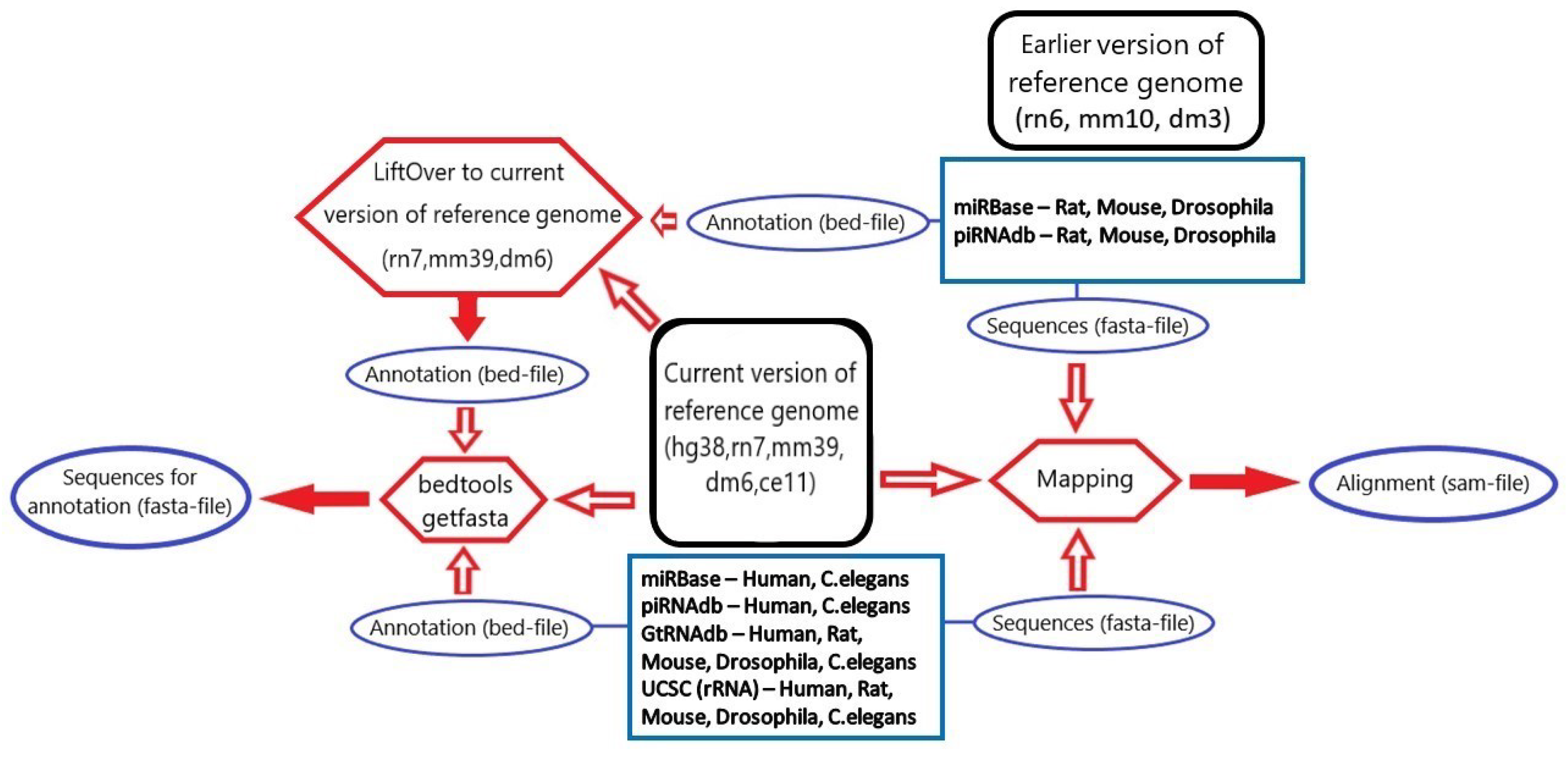
3.1. microRNA, piRNA and tRNA Processing
3.2. rRNA Processing
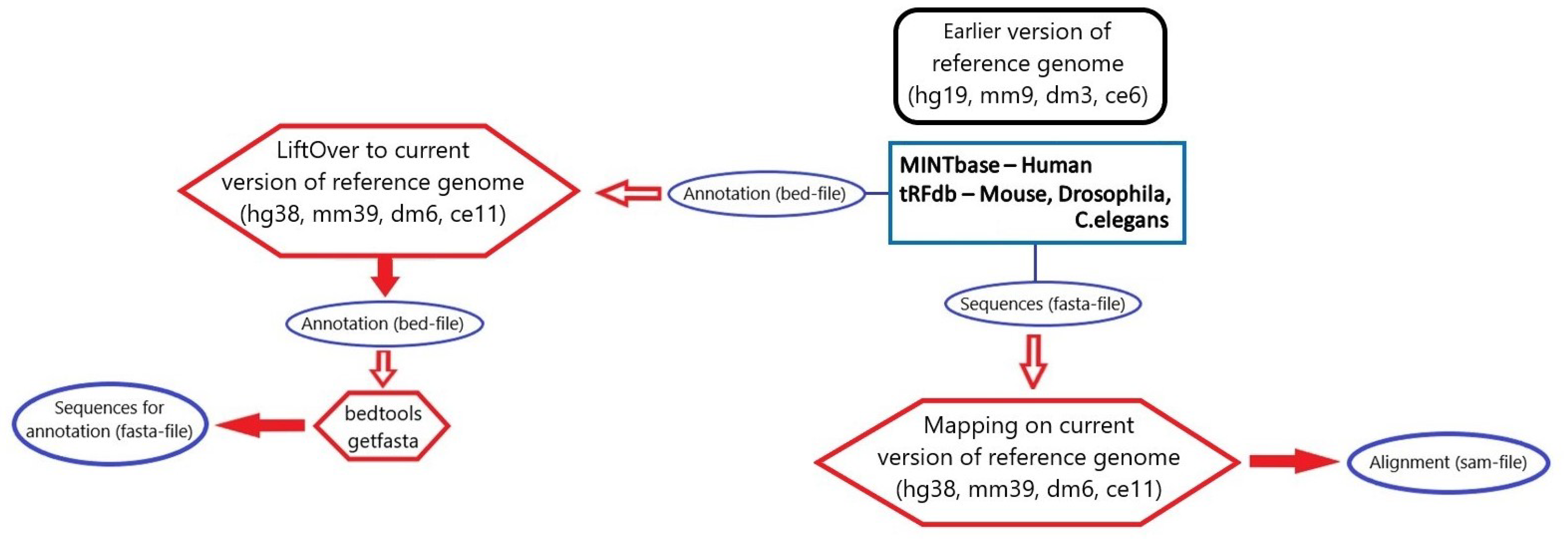
3.3. tsRNA Processing
3.4. Database Integration
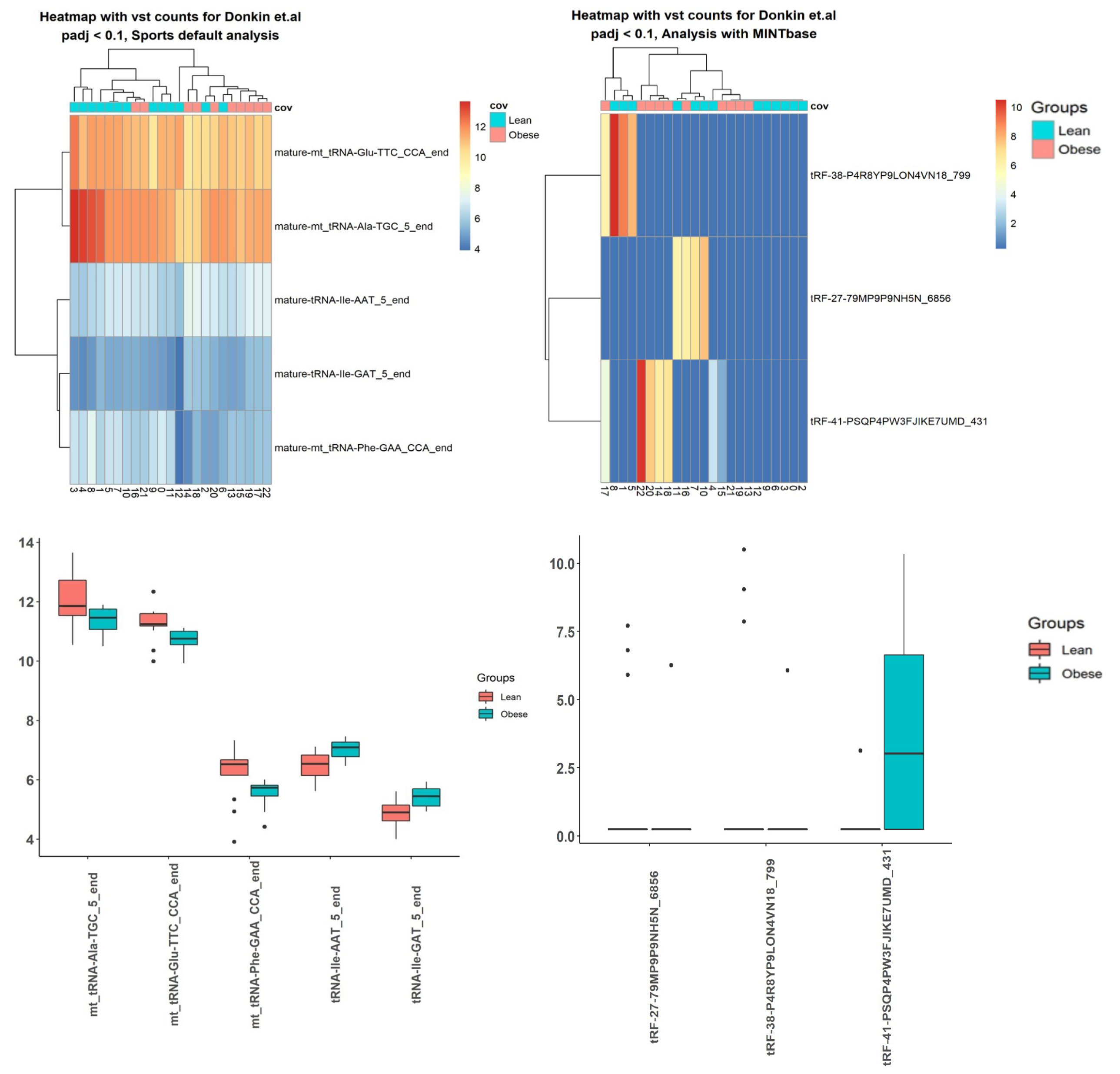
3.5. Case Studies— sRNA
3.6. Case Studies— Fragments
3.7. Program Code Availability
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Storz, G. An expanding universe of noncoding RNAs. Science 2002, 296, 1260–1263. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Peng, J.; Yi, C. The epitranscriptome of small non-coding RNAs. Non-Coding RNA Res. 2021, 6, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Holoch, D.; Moazed, D. RNA-mediated epigenetic regulation of gene expression. Nat. Rev. Genet. 2015, 16, 71–84. [Google Scholar] [CrossRef]
- Penner-Goeke, S.; Binder, E.B. Epigenetics and depression. Dialogues Clin. Neurosci. 2019, 21, 397. [Google Scholar] [CrossRef]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef] [PubMed]
- Santiago, J.; Silva, J.V.; Howl, J.; Santos, M.A.; Fardilha, M. All you need to know about sperm RNAs. Hum. Reprod. Update 2022, 28, 67–91. [Google Scholar] [CrossRef]
- Krawetz, S.A.; Kruger, A.; Lalancette, C.; Tagett, R.; Anton, E.; Draghici, S.; Diamond, M.P. A survey of small RNAs in human sperm. Hum. Reprod. 2011, 26, 3401–3412. [Google Scholar] [CrossRef] [Green Version]
- Marcho, C.; Oluwayiose, O.A.; Pilsner, J.R. The preconception environment and sperm epigenetics. Andrology 2020, 8, 924–942. [Google Scholar] [CrossRef]
- Kotsyfakis, M.; Patelarou, E. MicroRNAs as biomarkers of harmful environmental and occupational exposures: A systematic review. Biomarkers 2019, 24, 623–630. [Google Scholar] [CrossRef]
- Zhang, Y.; Shi, J.; Rassoulzadegan, M.; Tuorto, F.; Chen, Q. Sperm RNA code programmes the metabolic health of offspring. Nat. Rev. Endocrinol. 2019, 15, 489–498. [Google Scholar] [CrossRef] [Green Version]
- Cecere, G. Small RNAs in epigenetic inheritance: From mechanisms to trait transmission. FEBS Lett. 2021, 595, 2953–2977. [Google Scholar] [CrossRef] [PubMed]
- Micheel, J.; Safrastyan, A.; Wollny, D. Advances in Non-Coding RNA Sequencing. Non-Coding RNA 2021, 7, 70. [Google Scholar] [CrossRef] [PubMed]
- Benesova, S.; Kubista, M.; Valihrach, L. Small RNA-Sequencing: Approaches and Considerations for miRNA Analysis. Diagnostics 2021, 11, 964. [Google Scholar] [CrossRef] [PubMed]
- Han, B.W.; Wang, W.; Zamore, P.D.; Weng, Z. piPipes: A set of pipelines for piRNA and transposon analysis via small RNA-seq, RNA-seq, degradome-and CAGE-seq, ChIP-seq and genomic DNA sequencing. Bioinformatics 2015, 31, 593–595. [Google Scholar] [CrossRef] [Green Version]
- Ray, R.; Pandey, P. piRNA analysis framework from small RNA-Seq data by a novel cluster prediction tool-PILFER. Genomics 2018, 110, 355–365. [Google Scholar] [CrossRef]
- Huang, P.J.; Liu, Y.C.; Lee, C.C.; Lin, W.C.; Gan, R.R.C.; Lyu, P.C.; Tang, P. DSAP: Deep-sequencing small RNA analysis pipeline. Nucleic Acids Res. 2010, 38, W385–W391. [Google Scholar] [CrossRef] [Green Version]
- Hackenberg, M.; Rodríguez-Ezpeleta, N.; Aransay, A.M. miRanalyzer: An update on the detection and analysis of microRNAs in high-throughput sequencing experiments. Nucleic Acids Res. 2011, 39, W132–W138. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Nair, A.; Chen, X.; Prodduturi, N.; Wang, J.; Kocher, J.P. UClncR: Ultrafast and comprehensive long non-coding RNA detection from RNA-seq. Sci. Rep. 2017, 7, 1–10. [Google Scholar]
- Shi, J.; Ko, E.A.; Sanders, K.M.; Chen, Q.; Zhou, T. SPORTS1. 0: A tool for annotating and profiling non-coding RNAs optimized for rRNA-and tRNA-derived small RNAs. Genom. Proteom. Bioinform. 2018, 16, 144–151. [Google Scholar] [CrossRef]
- Rahman, R.U.; Gautam, A.; Bethune, J.; Sattar, A.; Fiosins, M.; Magruder, D.S.; Capece, V.; Shomroni, O.; Bonn, S. Oasis 2: Improved online analysis of small RNA-seq data. BMC Bioinform. 2018, 19, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Kim, T.K.; Baxter, D.; Scherler, K.; Gordon, A.; Fong, O.; Etheridge, A.; Galas, D.J.; Wang, K. sRNAnalyzer—A flexible and customizable small RNA sequencing data analysis pipeline. Nucleic Acids Res. 2017, 45, 12140–12151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pogorelcnik, R.; Vaury, C.; Pouchin, P.; Jensen, S.; Brasset, E. sRNAPipe: A Galaxy-based pipeline for bioinformatic in-depth exploration of small RNAseq data. Mob. DNA 2018, 9, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Oshlack, A.; Robinson, M.D.; Young, M.D. From RNA-seq reads to differential expression results. Genome Biol. 2010, 11, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupnikov, A.; Tripathi, S.; de Matos Simoes, R.; McArt, D.; Salto-Tellez, M.; Glazko, G.; Dehmer, M.; Emmert-Streib, F. samExploreR: Exploring reproducibility and robustness of RNA-seq results based on SAM files. Bioinformatics 2016, 32, 3345–3347. [Google Scholar] [CrossRef]
- Schurch, N.J.; Schofield, P.; Gierliński, M.; Cole, C.; Sherstnev, A.; Singh, V.; Wrobel, N.; Gharbi, K.; Simpson, G.G.; Owen-Hughes, T.; et al. How many biological replicates are needed in an RNA-seq experiment and which differential expression tool should you use? RNA 2016, 22, 839–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupnikov, A.; McInerney, C.; Savage, K.; McIntosh, S.; Emmert-Streib, F.; Kennedy, R.; Salto-Tellez, M.; Prise, K.; McArt, D. Robustness of differential gene expression analysis of RNA-seq. Comput. Struct. Biotechnol. J. 2021, 19, 3470–3481. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Di Bella, S.; La Ferlita, A.; Carapezza, G.; Alaimo, S.; Isacchi, A.; Ferro, A.; Pulvirenti, A.; Bosotti, R. A benchmarking of pipelines for detecting ncRNAs from RNA-Seq data. Briefings Bioinform. 2020, 21, 1987–1998. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piuco, R.; Galante, P.A. piRNAdb: A piwi-interacting RNA database. bioRxiv 2021. [Google Scholar] [CrossRef]
- Chan, P.P.; Lowe, T.M. GtRNAdb 2.0: An expanded database of transfer RNA genes identified in complete and draft genomes. Nucleic Acids Res. 2016, 44, D184–D189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karolchik, D.; Baertsch, R.; Diekhans, M.; Furey, T.S.; Hinrichs, A.; Lu, Y.; Roskin, K.M.; Schwartz, M.; Sugnet, C.W.; Thomas, D.J.; et al. The UCSC genome browser database. Nucleic Acids Res. 2003, 31, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Mudunuri, S.B.; Anaya, J.; Dutta, A. tRFdb: A database for transfer RNA fragments. Nucleic Acids Res. 2015, 43, D141–D145. [Google Scholar] [CrossRef] [PubMed]
- Pliatsika, V.; Loher, P.; Telonis, A.G.; Rigoutsos, I. MINTbase: A framework for the interactive exploration of mitochondrial and nuclear tRNA fragments. Bioinformatics 2016, 32, 2481–2489. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Donkin, I.; Versteyhe, S.; Ingerslev, L.R.; Qian, K.; Mechta, M.; Nordkap, L.; Mortensen, B.; Appel, E.V.R.; Jørgensen, N.; Kristiansen, V.B.; et al. Obesity and bariatric surgery drive epigenetic variation of spermatozoa in humans. Cell Metab. 2016, 23, 369–378. [Google Scholar] [CrossRef] [Green Version]
- Ingerslev, L.R.; Donkin, I.; Fabre, O.; Versteyhe, S.; Mechta, M.; Pattamaprapanont, P.; Mortensen, B.; Krarup, N.T.; Barrès, R. Endurance training remodels sperm-borne small RNA expression and methylation at neurological gene hotspots. Clin. Epigenetics 2018, 10, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, M.; Liu, W.; Chen, Y.; Zhang, F.; Xu, B.; Liu, S.; Chen, G.; Shi, H.; Wu, L. Identification of small non-coding RNAs as sperm quality biomarkers for in vitro fertilization. Cell Discov. 2019, 5, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, A.R. BEDTools: The Swiss-army tool for genome feature analysis. Curr. Protoc. Bioinform. 2014, 47, 11–12. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Fromm, B.; Høye, E.; Domanska, D.; Zhong, X.; Aparicio-Puerta, E.; Ovchinnikov, V.; Umu, S.; Chabot, P.; Kang, W.; Aslanzadeh, M.; et al. MirGeneDB 2.1: Toward a complete sampling of all major animal phyla. Nucleic Acids Res. 2021, 50, D204–D210. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RNA Type | Database | Loci and Sequence | Loci Only | Sequence Only | Genome Version |
|---|---|---|---|---|---|
| precursor microRNA | miRBase | 1002 | 913 | 2 | hg38 |
| mature microRNA | miRBase | 1477 | 1405 | 1 | hg38 |
| piRNA | piRNAdb | 812,343 | 0 | 2 | hg38 |
| tRNA | GtRNAdb | 430 | 187 | 2 | hg38 |
| rRNA | UCSC | 1752 | 0 | 186 | hg38 |
| tsRNA | MINTbase | 125,285 | 0 | 0 | hg19 |
| RNA Type | Database | Loci & Seq | Loci Only | Seq Only | Ext Loci & Seq | Loci & Ext Seq | Ext Loci & Ext Seq | Inbase Conflict | Interbase Conflict |
|---|---|---|---|---|---|---|---|---|---|
| mature | miRBase | 1291 | 1227 | 1 | 0 | 0 | 0 | 140 | 21 |
| microRNA | |||||||||
| piRNA | piRNAdb | 422,017 | 0 | 1 | 0 | 0 | 0 | 388,826 | 1268 |
| tRNA | GtRNAdb | 225 | 176 | 2 | 0 | 0 | 0 | 0 | 214 |
| rRNA | UCSC | 1591 | 0 | 186 | 0 | 0 | 0 | 37 | 126 |
| tRNA-derived | MINTbase | 0 | 0 | 8120 | 115,040 | 33 | 8 | * | * |
| Species | Database | Loci & Seq | Loci Only | Seq Only | Ext Loci & Seq | Loci & Ext Seq | Ext Loci & Ext Seq |
|---|---|---|---|---|---|---|---|
| Homo sapiens | MINTBase | 0 | 0 | 8120 | 115,040 | 33 | 8 |
| Mus musculus | tRFdb | 335 | 0 | 0 | 0 | 0 | 8 |
| Drosophila melanogaster | tRFdb | 147 | 0 | 0 | 0 | 0 | 0 |
| Caenorhabditis elegans | tRFdb | 247 | 0 | 0 | 0 | 0 | 0 |
| Mature | ||||||
|---|---|---|---|---|---|---|
| Species | Genome Version | microRNA | piRNA | tRNA | rRNA | tsRNA |
| Homo sapiens | hg38 | 2330 | 14,439 | 403 | 1776 | 18,948 |
| Mus musculus | mm39 | 1870 | 9715 | 1044 | 1376 | 13,105 |
| Rattus norvegicus | rn6 | 616 | 7976 | 966 | 239 | 9797 |
| Caenorhabditis elegans | ce11 | 397 | 8376 | 633 | 5 | 9411 |
| Drosophila melanogaster | dm6 | 435 | 8296 | 154 | 93 | 8978 |
| Mature | ||||||
|---|---|---|---|---|---|---|
| Species | Genome Version | microRNA | piRNA | tRNA | rRNA | tsRNA |
| Homo sapiens | hg38 | 2543 | 14,605 | 619 | 1840 | 26,731 |
| Mus musculus | mm39 | 1870 | 9739 | 1135 | 1430 | 65 |
| Rattus norvegicus | rn6 | 647 | 8079 | 1173 | 274 | - |
| Caenorhabditis elegans | ce11 | 424 | 8654 | 721 | 5 | 18 |
| Drosophila melanogaster | dm6 | 481 | 500,536 | 295 | 165 | 22 |
| Data | SPORTS, sRNA | ITAS, sRNA | SPORTS, tsRNA | ITAS, tsRNA |
|---|---|---|---|---|
| Donkin et al. [40] | 11 | 66 | 5 | 3 |
| Ingerslev et al. [41] | 43 | 212 | 12 | 24 |
| Hua et al. [42] | 26 | 242 | 0 | 12 |
| Transcript Name sRNA | SPORTS, p-Value |
|---|---|
| tRNA-Ser-CGA | 3.23 |
| hsa-mir-155 | 0.001 |
| other-rRNA | 0.002 |
| mt-tRNA-Glu-TTC | 0.009 |
| mt-tRNA-Phe-GAA | 0.019 |
| mt-tRNA-Trp-TCA | 0.019 |
| mt-tRNA-Ala-TGC | 0.022 |
| mt-tRNA-Ser-TGA | 0.022 |
| tRNA-His-GTG | 0.028 |
| 16S-rRNA | 0.035 |
| tsRNA | adjusted p-value |
| tRNA-Ile-AAT-5-end | 0.099 |
| mt-tRNA-Glu-TTC-CCA-end | 0.099 |
| mt-tRNA-Ala-TGC-5-end | 0.099 |
| tRNA-Ile-GAT-5-end | 0.099 |
| mt-tRNA-Phe-GAA-CCA-end | 0.099 |
| Transcript name sRNA | ITAS, p-value |
| hsa-piR-33029 | 1.84 |
| hsa-miR-155-5p | 0.002 |
| 5S-dup10-seq-371 | 0.003 |
| hsa-miR-497-5p | 0.006 |
| hsa-piR-8652 | 0.006 |
| hsa-miR-195-3p | 0.007 |
| hsa-piR-33047 | 0.008 |
| hsa-miR-6516-5p | 0.008 |
| hsa-miR-3663-5p | 0.012 |
| hsa-miR-518c-3p | 0.012 |
| tsRNA | adjusted p-value |
| tRF-38-P4R8YP9LON4VN18-799 | 1.09 |
| tRF-27-79MP9P9NH5N-6856 | 1.29 |
| tRF-41-PSQP4PW3FJIKE7UMD-431 | 0.01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stupnikov, A.; Bezuglov, V.; Skakov, I.; Shtratnikova, V.; Pilsner, J.R.; Suvorov, A.; Sergeyev, O. ITAS: Integrated Transcript Annotation for Small RNA. Non-Coding RNA 2022, 8, 30. https://doi.org/10.3390/ncrna8030030
Stupnikov A, Bezuglov V, Skakov I, Shtratnikova V, Pilsner JR, Suvorov A, Sergeyev O. ITAS: Integrated Transcript Annotation for Small RNA. Non-Coding RNA. 2022; 8(3):30. https://doi.org/10.3390/ncrna8030030
Chicago/Turabian StyleStupnikov, Alexey, Vitaly Bezuglov, Ivan Skakov, Victoria Shtratnikova, J. Richard Pilsner, Alexander Suvorov, and Oleg Sergeyev. 2022. "ITAS: Integrated Transcript Annotation for Small RNA" Non-Coding RNA 8, no. 3: 30. https://doi.org/10.3390/ncrna8030030
APA StyleStupnikov, A., Bezuglov, V., Skakov, I., Shtratnikova, V., Pilsner, J. R., Suvorov, A., & Sergeyev, O. (2022). ITAS: Integrated Transcript Annotation for Small RNA. Non-Coding RNA, 8(3), 30. https://doi.org/10.3390/ncrna8030030