1. Introduction
microRNAs (miRNAs) are small non-coding RNAs that post-transcriptionally regulate up to 60% of all cellular proteins [
1]. Consequently, miRNAs are poised to control dynamic signaling cascades by adjusting the absolute amount of proteins available required to accomplish multiple physiological processes, thereby allowing cells to rapidly adapt to changing internal and external stimuli. Aging can be considered a chronic stimulus that elicits a complex array of cellular changes, many of which are the direct result of reduced levels of proteins that protect the cell against age-related damage [
2]. The ovarian hormone 17β-estradiol (E
2) also regulates the amount of proteins that can contribute to cellular protection in many tissues, especially in the brain; however, this E
2-mediated protection is lost following the menopausal transition and advanced age [
3]. The “Timing Hypothesis” speculates that E
2 is neuroprotective within a critically narrow window of time following menopause, suggesting that there is a biological switch in E
2 action that is temporally dependent [
4]. Our previously published work raised the possibility that miRNAs could underlie this temporal switch in E
2 action in the aging female brain. For instance, miR-9-5p and miR-9-3p were differentially regulated by E
2 in the hypothalamus and hippocampus of aged female rats [
5,
6]. This finding was particularly relevant due to the extensively characterized roles of miR-9-5p and miR-9-3p in maintaining healthy neuronal function through their regulation of neuronal differentiation, dendritic branching, and apoptosis [
7,
8,
9,
10,
11,
12]. Notably, our previous data demonstrated that the ability of E
2 to regulate these miRNAs in aged rats was dependent on the length of time between ovarian hormone depletion (via ovariectomy) and the subsequent timing of replacement E
2 treatment [
6]. Those data further supported the notion that differential regulation of miR-9-5p and miR-9-3p could, in part, explain the disparate functional outcomes of E
2 treatment in women closer to the menopausal transition compared to those given E
2 many years past menopause.
Regulation of miRNAs by E
2 can theoretically be achieved at multiple steps in the miRNA biogenesis pathway. Briefly, miRNA biogenesis is initiated by the transcription of a long primary transcript (pri-miRNA) which is subsequently cleaved by the RNase III enzyme (DROSHA) to form the precursor (pre-miRNA); after nuclear export, pre-miRNA is cleaved by another RNase III enzyme (DICER) to form the miRNA duplex which contains the 20–23 nt mature miRNA products [
13]. To date, the literature investigating the role of E
2 in the regulation of miRNAs has primarily focused on regulation of miRNA transcription (i.e., formation of the pri-miRNA). The ability for E
2 to regulate miRNAs at the level of transcription is conceptually reasonable, and somewhat expected, due to the canonical E
2 signaling pathways which are mediated primarily by nuclear estrogen receptors (ER)s that act as transcription factors to initiate or inhibit gene expression. Indeed, there have been multiple reports suggesting that E
2 can have transcriptional effects on miRNAs. For example, E
2-bound ERβ inhibited the transcription of pri-miR-30a in a breast cancer cell line (MCF7) by binding to two proximal sites near its transcription initiation start site [
14]. Notably, E
2 broadly decreased total miRNA expression in the MCF7 cell line, suggesting that the ERβ-mediated inhibition of transcription could be a shared mechanism to globally downregulate miRNA expression in ER positive cancer cells [
15]. Contrary to data from cancer cell models, E
2 regulation of miRNAs in the brain exhibits distinct molecular patterns; specifically, a global decrease in miRNA expression has not been observed. In fact, our data showed that only a subset of miRNAs were altered by E
2 treatment in the rat brain, and some were increased while others decreased, raising the potential for E
2 to selectively target miRNAs in the brain.
In this study we focused on two of our previously identified E
2-regulated miRNAs: miR-9-5p and miR-9-3p. These miRNAs are both derived from the same primary transcript (pri-miR-9) and share the same precursor (pre-miR-9), but the mature duplex is unwound to form individual miRNAs with unique mRNA targets [
13]. Our data showed that E
2 treatment increased both miR-9-5p and miR-9-3p in the aged rat hypothalamus, but the pri- and pre-miR-9 levels were unaffected, suggesting that E
2 regulation of miR-9-5p and miR-9-3p was not at the level of transcription (i.e., pri-) or DROSHA processing (i.e., pre-) [
6]. Therefore, the objective of this study was to determine a mechanism for E
2 regulation of mature miR-9-5p and miR-9-3p and investigate the potential consequences of E
2 on their ability to effectively repress mRNA translation using polysome profiling. We hypothesized that E
2 regulates miR-9-5p and miR-9-3p through stabilization of the mature miRNA in an age-dependent manner. We tested this hypothesis using a biochemical approach with neuronal cell lines and hypothalamic tissue isolated from aged female rats. Collectively, we show that E
2 treatment stabilized miR-9-5p and miR-9-3p in the rat hypothalamus and this stabilization was dependent on age of treatment. Moreover, our data suggest that E
2 might differentially modulate the ability of miR-9-5p and miR-9-3p to repress the translation of their mRNA targets by shifting their association with polysomes. Together, these data provide a putative mechanistic explanation underlying the “Timing Hypothesis” by highlighting a novel role for E
2 on mature miRNA degradation.
3. Discussion
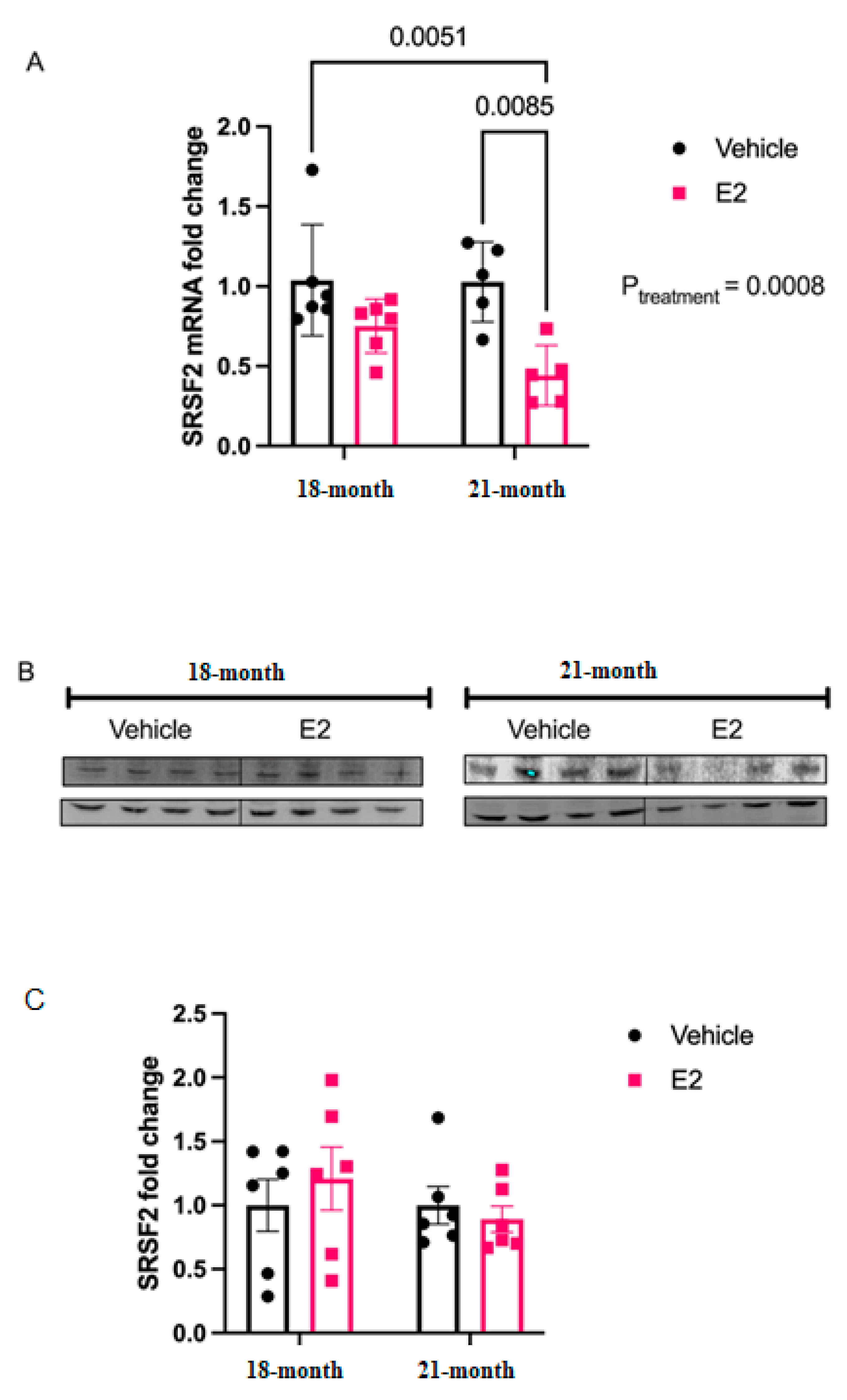
The molecular mechanisms underlying the disparate effects of hormone replacement therapy as postulated by the Timing Hypothesis are still being explored. In the present study, we describe the novel finding that E2 treatment can regulate mature miRNAs in an age-dependent manner independent of its effects on the pri- and precursor forms of miRNA. The predominant view of how E2 regulates miRNAs tends to focus on E2-induced changes in miRNA transcription, as mediated by the genomic actions of estrogen receptors (ER)s. Here, we used a biochemical approach to reveal that E2 treatment can post-transcriptionally regulate the stability of mature miRNAs in both hypothalamic-derived neuronal cell lines and in the paraventricular nucleus (PVN), a hypothalamic subregion, of aged female rats. Specifically, E2 stabilized both strands of the miR-9 duplex (-5p and -3p), which collectively have been shown to be important for driving not only neuronal differentiation, but also for regulating synaptic plasticity in post-mitotic neurons. We also demonstrated the ability of E2 to affect mature miRNA activity using polysome profiling, revealing a novel mechanism of miRNA regulation that could contribute to the molecular mechanisms underlying the Timing Hypothesis.
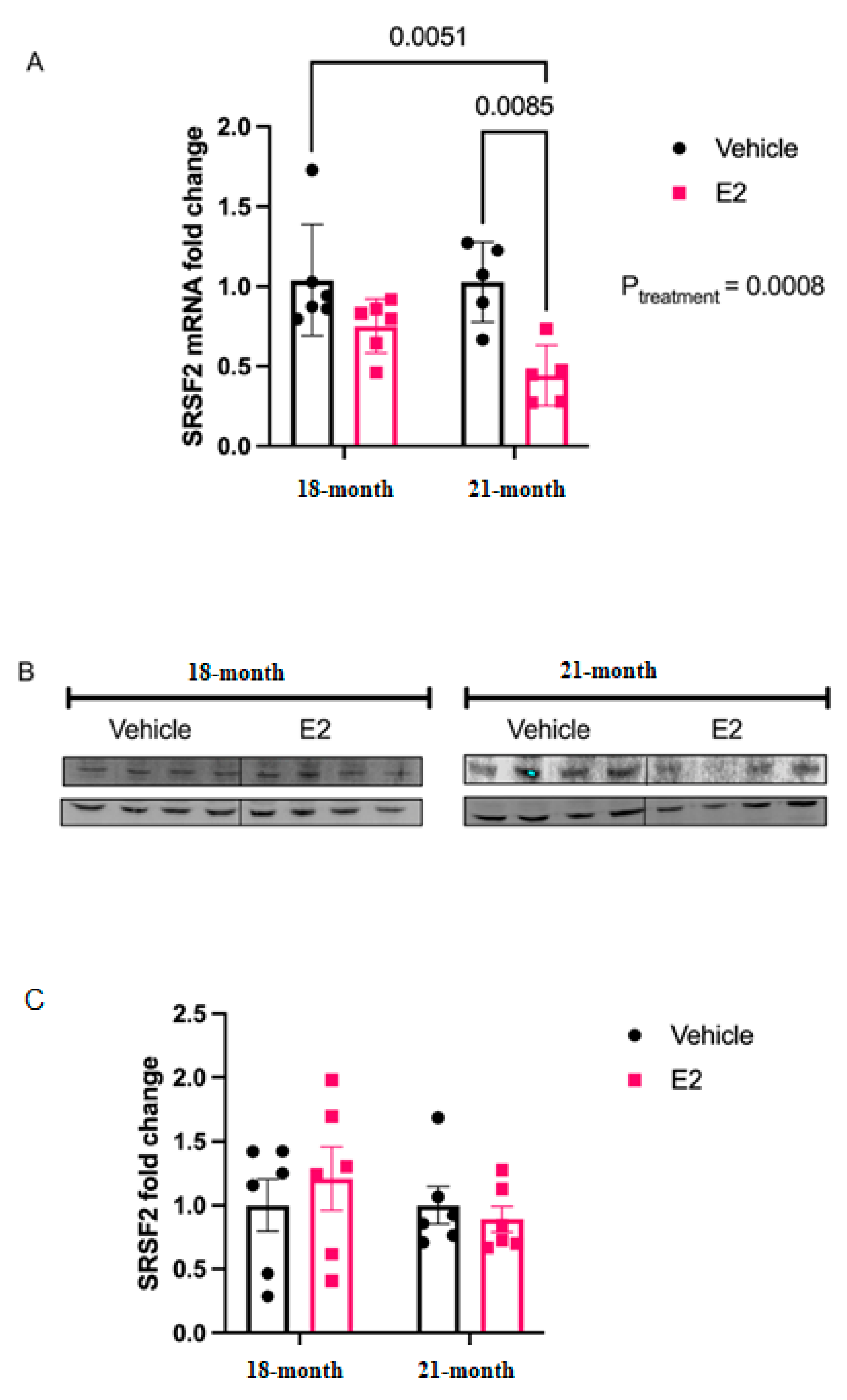
Our data provide evidence that the E
2-mediated regulation of miR-9-5p and miR-9-3p is executed at the mature level and not at the level of transcription in the aged brain. These results are novel, as other reports have shown that E
2 regulates various aspects of miRNA biogenesis in other tissue types. For instance, ERs have been shown to bind directly to the promotors of miRNA genes to regulate the transcription of their primary miRNAs [
14,
20]. Indirect regulation of miRNA transcription has also been reported whereby steroid signaling mechanisms have been shown to recruit other transcription factors to miRNA promoter sites; specifically, c-MYC was recruited to the promotor site of miR-17-92 [
21]. In general, E
2 treatment has been associated with the transcriptional repression of miRNAs, especially when the precursor strand harbors a G-rich terminal loop [
15,
22]. However, it is important to consider that most of these studies were performed using reproductive tissues, and E
2 regulation of miRNAs in the central nervous system is notably distinct from peripheral tissues. While E
2 treatment upregulated levels of mature miR-9-5p and miR-9-3p in the rat hypothalamus, the primary and precursor levels remained unchanged [
6], suggesting that this upregulation was not mediated by an increase in transcription.
Moreover, we generally observed no effect of E
2 treatment to the miRNA processing machinery in both cell lines and in the PVN of aged female rats. One exception was a significant reduction in
Drosha mRNA levels in our aged animals. These results from brain tissue contrast with the well-characterized effects of E
2 on the various aspects of miRNA processing in reproductive tissues. For instance, E
2 and progesterone signaling concomitantly increased
Xpo5 mRNA expression in mouse uterine tissue, and
Dicer expression was observed to be increased with E
2 treatment in MCF7 cells [
23,
24]. Furthermore, E
2 positive cell lines had increased
Dicer and decreased
Ago2 expression compared to ER negative cell lines [
25]. In our system, by contrast, we did not observe broad E
2-mediated changes to the miRNA processing machinery, suggesting that global miRNA processing was not altered by E
2 treatment. Therefore, E
2 regulation of miR-9, at least in the aged rat hypothalamus, likely exerts its actions directly at the level of mature miR-9 specific duplex strands.
The subcellular localization of mature miRNAs in the aged rat brain has not been well studied and, to our knowledge, this is the first report showing that E
2 shifts specific miRNA polysome occupancy. Under cellular stress, active translation shifts towards the formation of processing bodies or stress granules and mRNA translation is stalled [
26,
27]. On the other hand, miRNA association with cytosolic polysomes is indicative of active translational repression. Therefore, we hypothesized that E
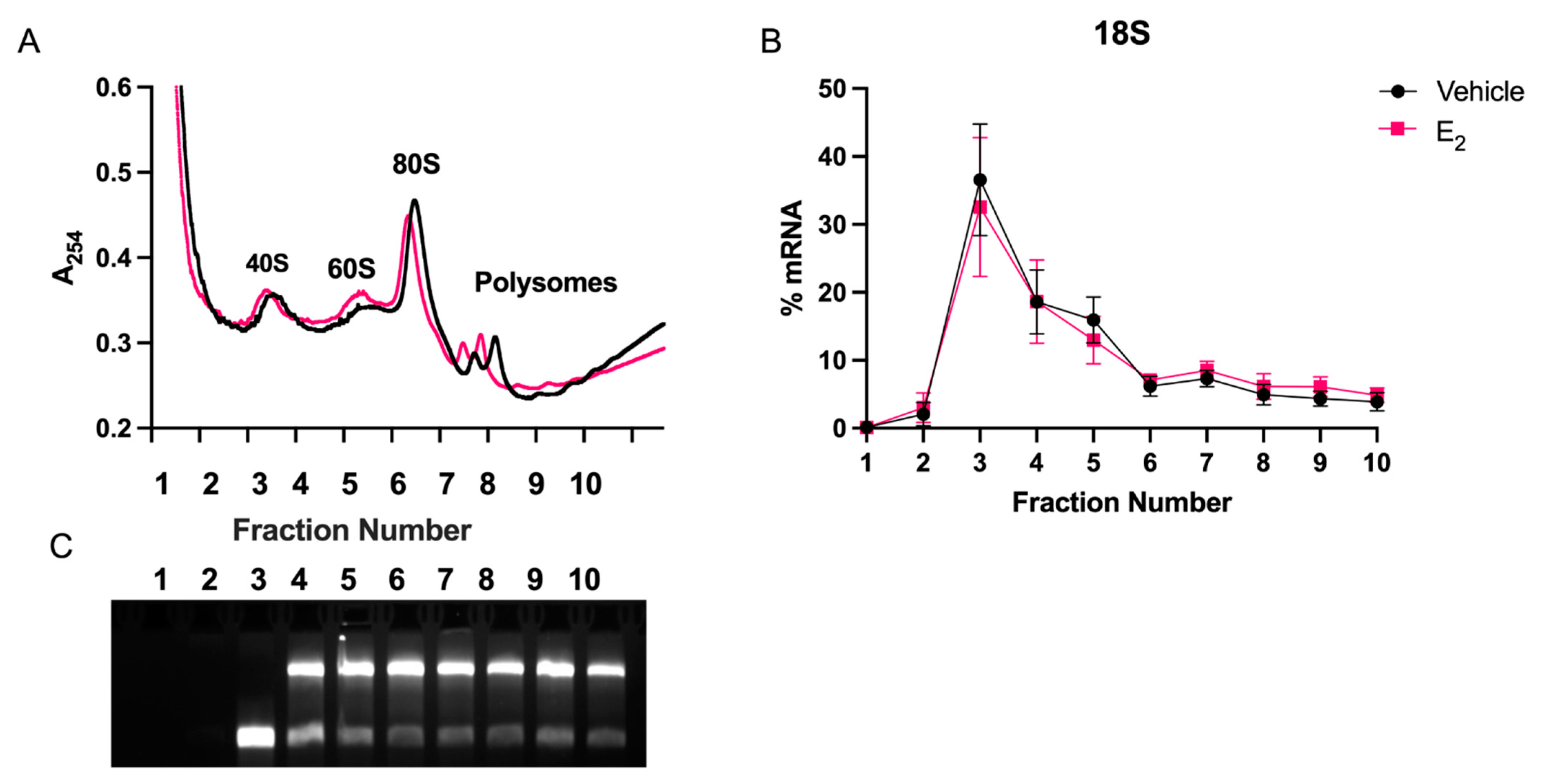
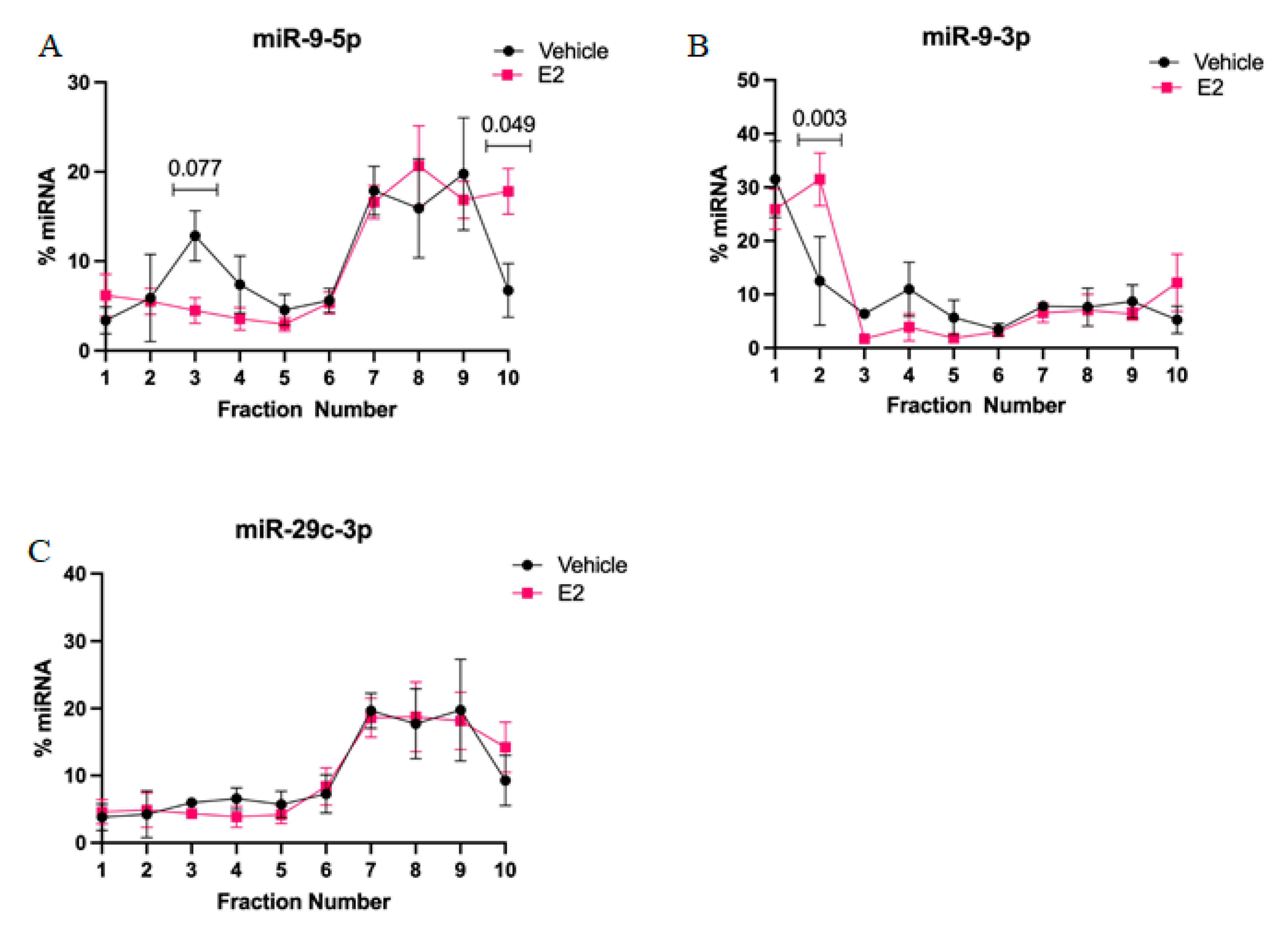
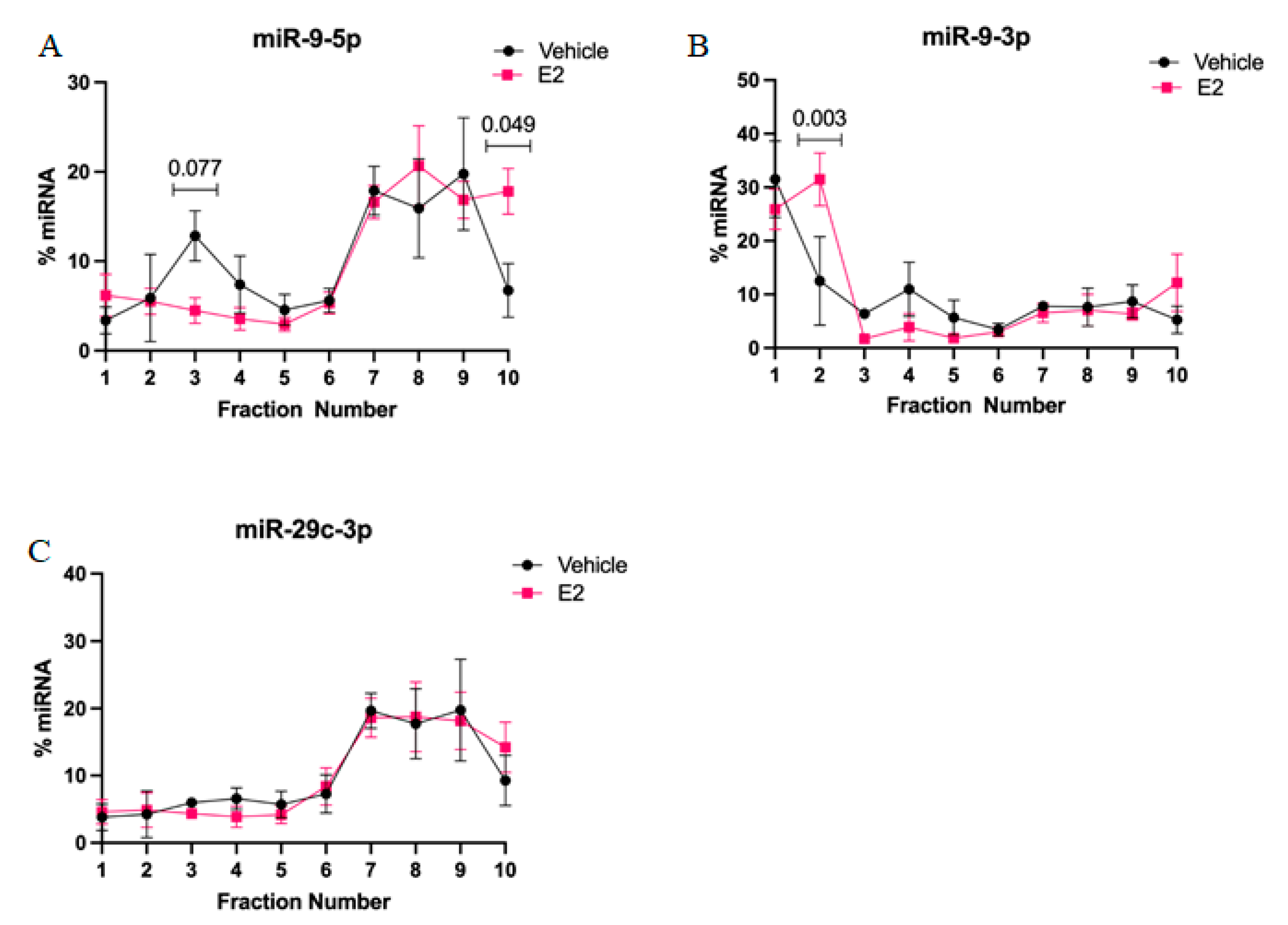
2-mediated stabilization of miRNAs could subsequently alter the activity and cellular location of miRNA in actively translating polysomes. First, we found that miR-9-5p and miR-9-3p display very different polysome profiles, which likely reflects their guide and passenger strand status, respectively. While miR-9-5p primarily occupies the heavier polysome fractions (~60% total miR-9-5p), miR-9-3p occupies the lightest polysome fractions (~40% total miR-9-3p). Treatment with E
2 showed a trend, but was not statistically significant, towards decreasing miR-9-5p in the 40S ribosomal complex (i.e., fraction 3), which may partly account for its significantly increased shift in the heavier polysome fractions (i.e., fraction 10). This indicates that E
2 increased the ability of miR-9-5p to translationally repress its target mRNAs. In contrast, E
2 significantly shifted miR-9-3p towards the 40S ribosomal subunit (i.e., fraction 2), indicating a role for translational stalling. Moreover, previous studies demonstrated that the 40S ribosome is a marker of stress granules, a cellular compartment that functions to stall translation of mRNAs under stress conditions [
27,
28].
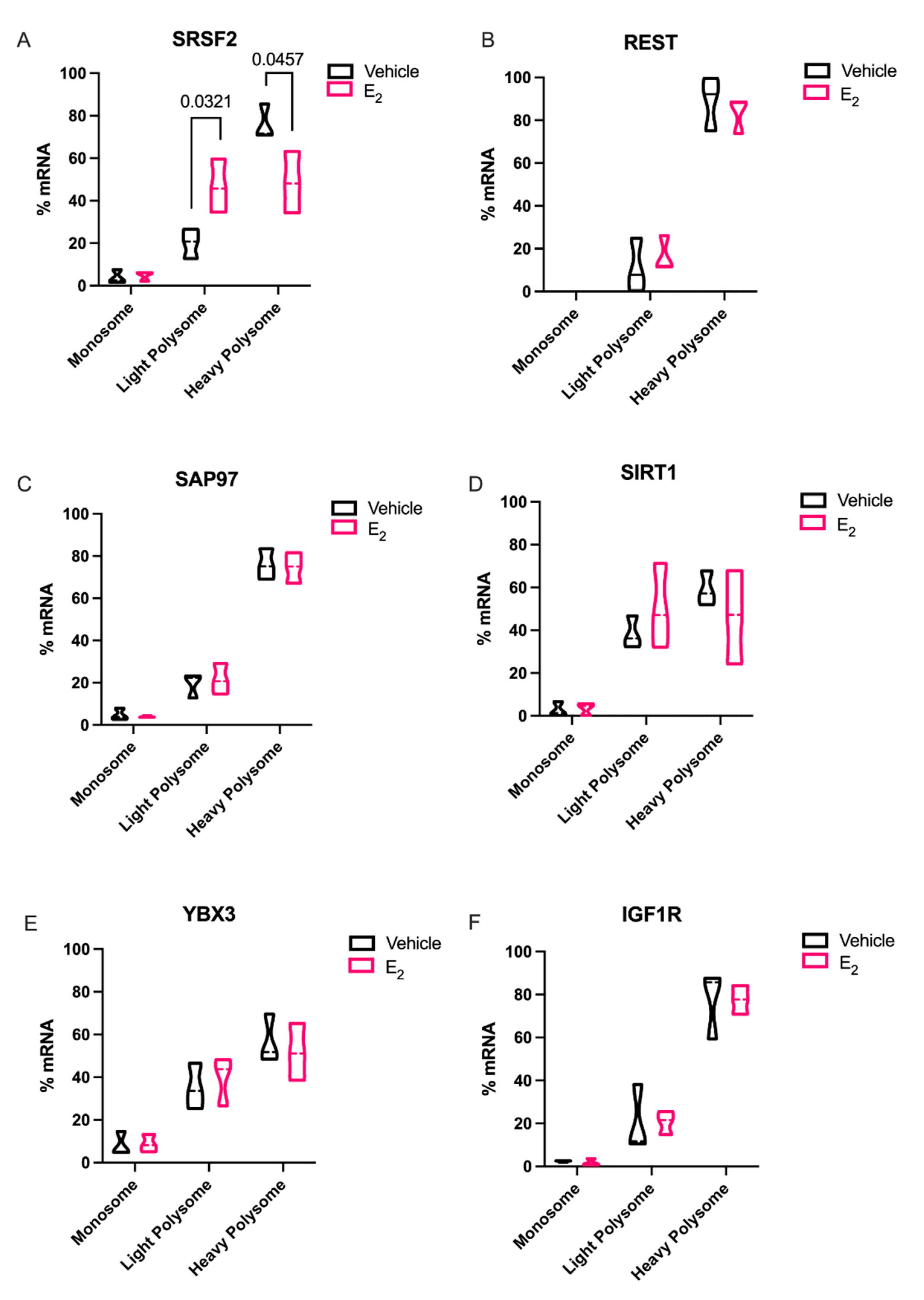
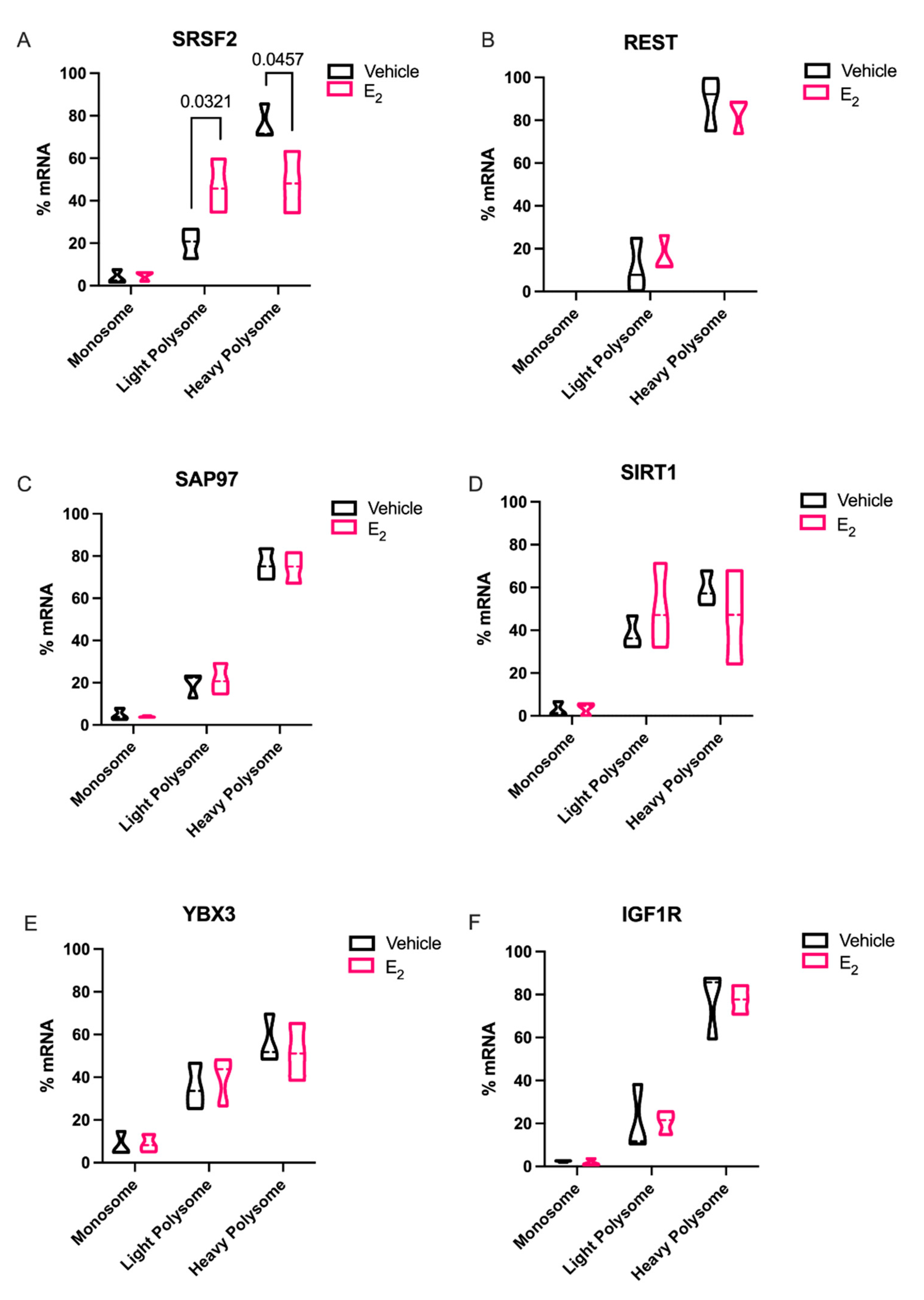
Elongating the half-life of these rapidly degraded miRNAs, as demonstrated with E
2 treatment, would theoretically afford it more time to repress their mRNA targets. Therefore, we investigated the mRNA targets of both miR-9-5p and miR-9-3p with the expectation that miR-9-5p and/or miR-9-3p targets would be significantly downregulated with E
2 treatment in polysome fractions. Indeed,
Srsf2, a predicted target of miR-9-3p was translationally stalled with E
2 treatment. SRSF2 is a ubiquitous RNA binding protein and is part of the core family of proteins in the spliceosome. Notably, SRSF2 is increased during physiological stress and participates in the alternative splicing of acetylcholinesterase thereby altering cholinergic synaptic transmission in the brain [
29,
30]. Our finding that E
2 can inhibit translation of SRSF2 in the aging brain points to a potential mechanism for the protective effects of E
2 on age-related cellular stress.
In sharp contrast to our data for
Srsf2 mRNA in polysomes, other putative miR-9-5p and miR-9-3p mRNA targets that we tested were not altered, despite the fact that REST, SAP97, SIRT1, and YBX3 have all been experimentally validated and are highly conserved targets of these miRNAs. These data underscore the importance of considering other cellular factors that might contribute to their translational regulation. For instance, prediction databases such as miRDB highlight several additional miRNAs that could target these same genes, raising the possibility that several miRNAs could work in concert with miR-9-5p and miR-9-3p to achieve full translational repression of these targets [
31]. Alternatively, a limitation of our polysome data is that we typically obtain low overall yields of total RNA from individual fractions. Therefore, it was necessary to pool several fractions for the measurements of mRNA, which might have masked differences that might have been otherwise observed in a single fraction. Previous studies in breast cancer cell lines found that E
2 was able to downregulate REST in polysomes more than total RNA [
32]. While a direct relationship of E
2-mediated miRNA repression of target mRNAs was not established, our data is consistent with the aforementioned study. Our data herein showed that there was a clear trend for E
2 to decrease levels in the heavy polysome fractions, while increasing it in the light polysome fractions, when comparing the statistical median for REST, SIRT1, and IGF1R, similar to what we observed with SRSF2.
Estrogens initiate cellular signaling processes via their interaction with nuclear receptors: ERα and ERβ [
33]. The stabilizing effect of E
2 that was observed for miR-9-5p and miR-9-3p in our cell and tissue models was likely mediated primarily by the actions of ERβ. In the hypothalamic-derived IVB cell lines, ERβ is dominantly expressed, though ERβ expression can still be observed at very low levels [
34,
35]. Additionally, our previous studies showed that an ERβ-, but not ERα-specific agonist, diarylpropionitrile: DPN, significantly increased the expression of miR-9-3p in the rat hypothalamus [
6]. Furthermore, ERβ has recently been shown to indirectly interact with Argonaute 2 (
Ago2) in breast cancer cell lines, suggesting a role for ERβ in RISC loading [
36]. Since miRNA loading to
Ago2 has been positively correlated with stabilization of the miRNA [
37], it remains an intriguing possibility that ERβ is facilitating the loading of miR-9-3p to the RNA-induced silencing complex (RISC). Furthermore, distinct miRNA profiles were observed between ERβ positive and negative breast cancer cell lines [
14], consistent with our previous results in various brain regions. Taken together, these data provide strong evidence that ERβ signaling is critical in the regulation of a specific subset of miRNAs in multiple tissues.
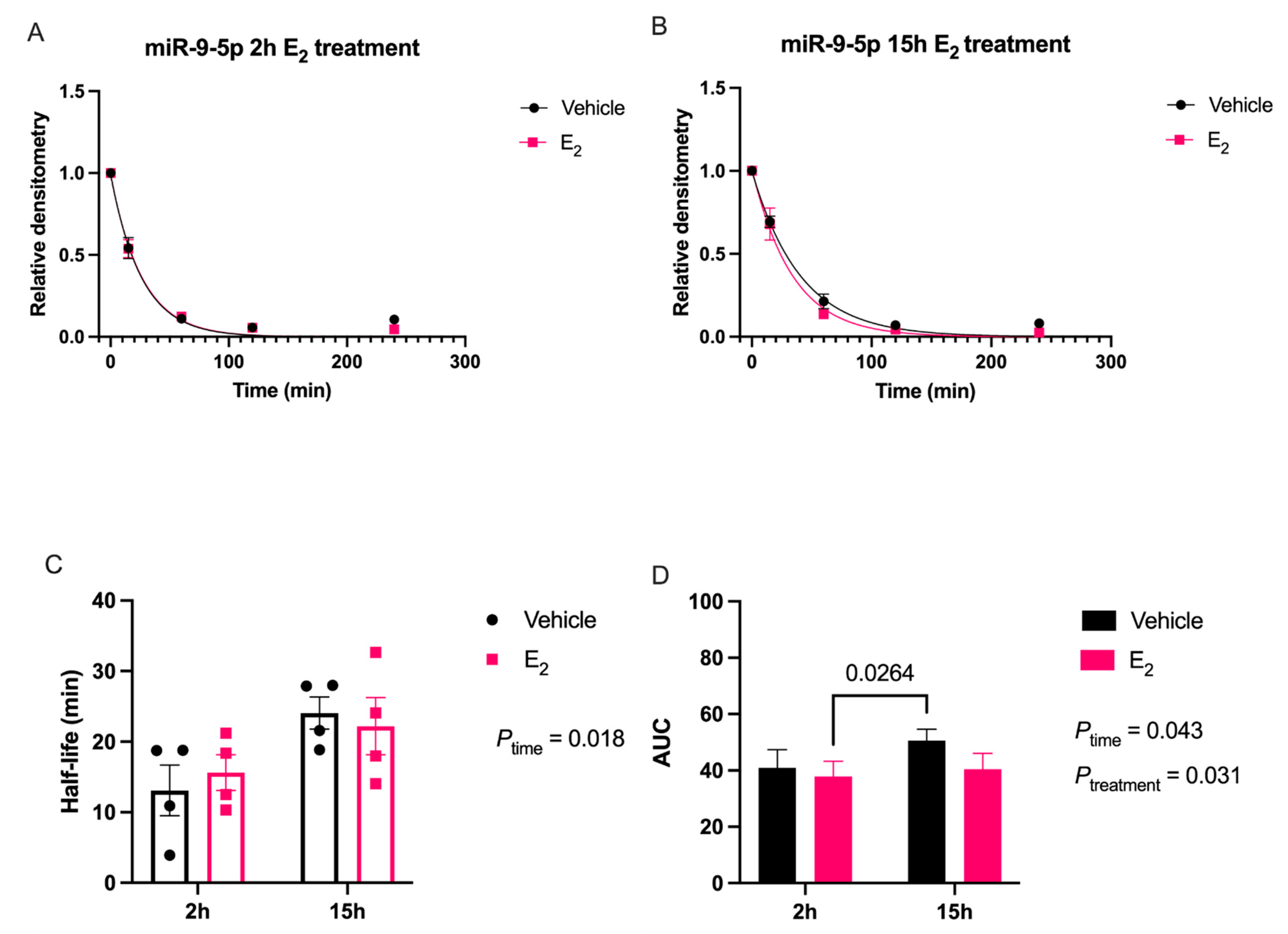
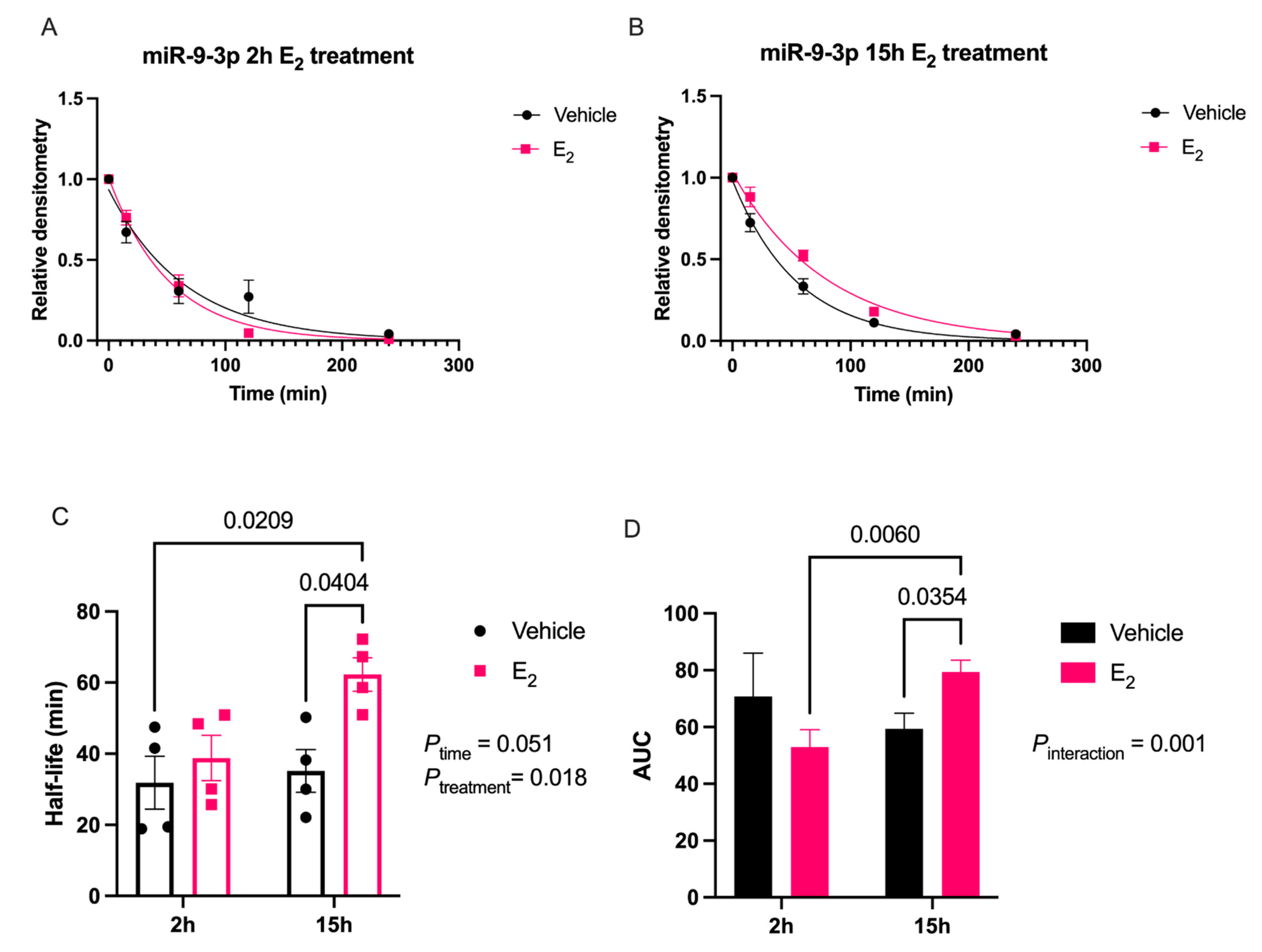
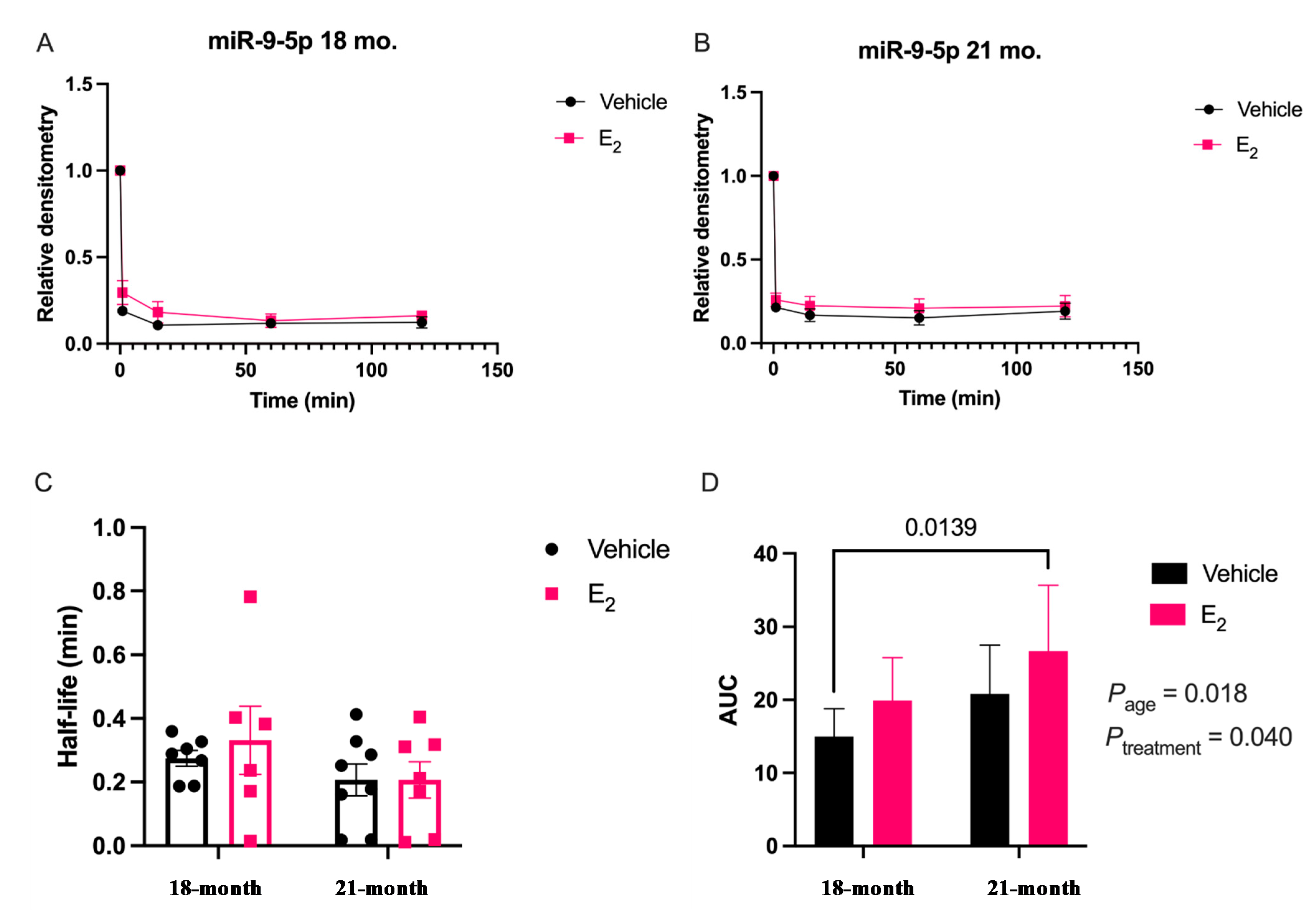
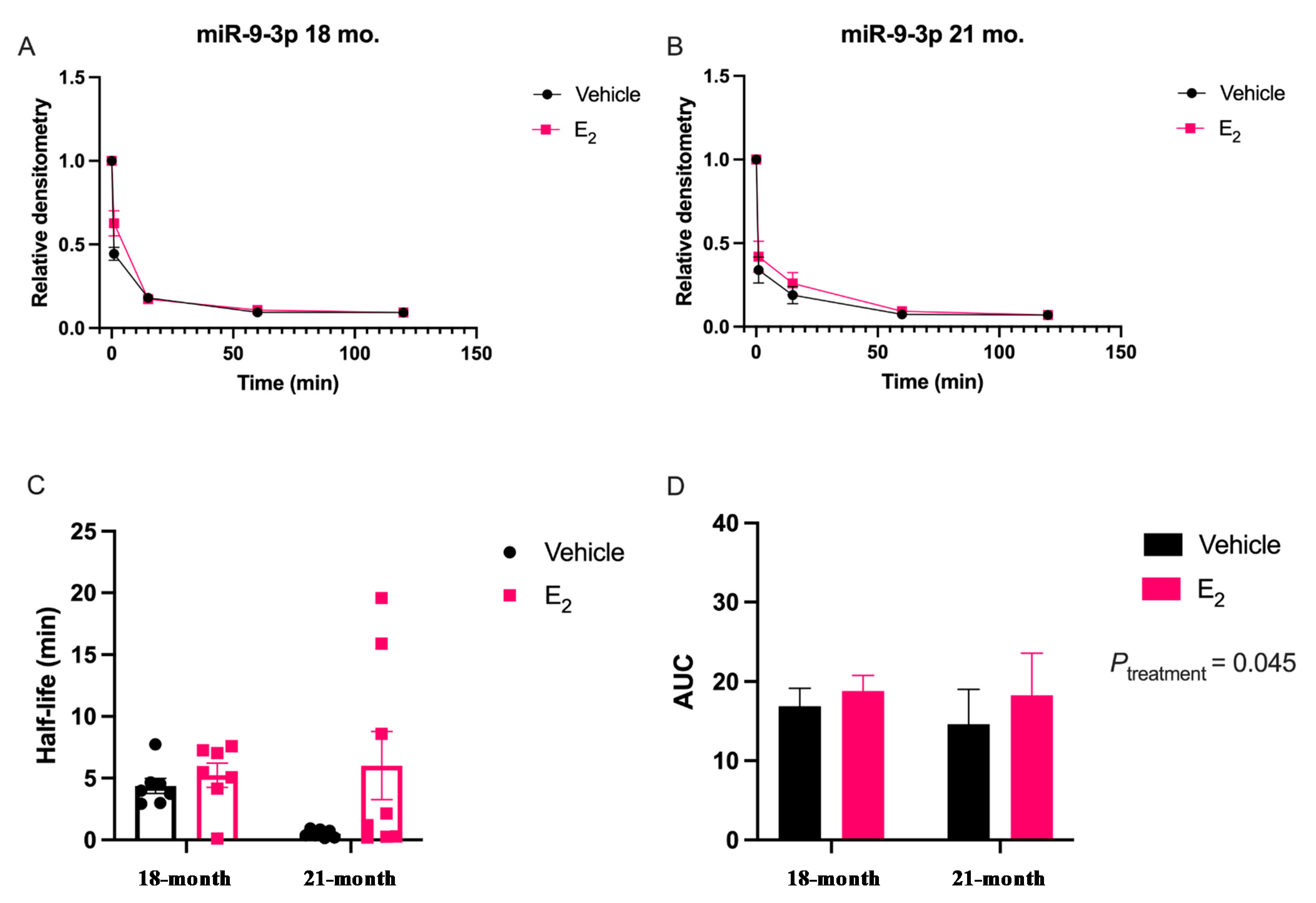
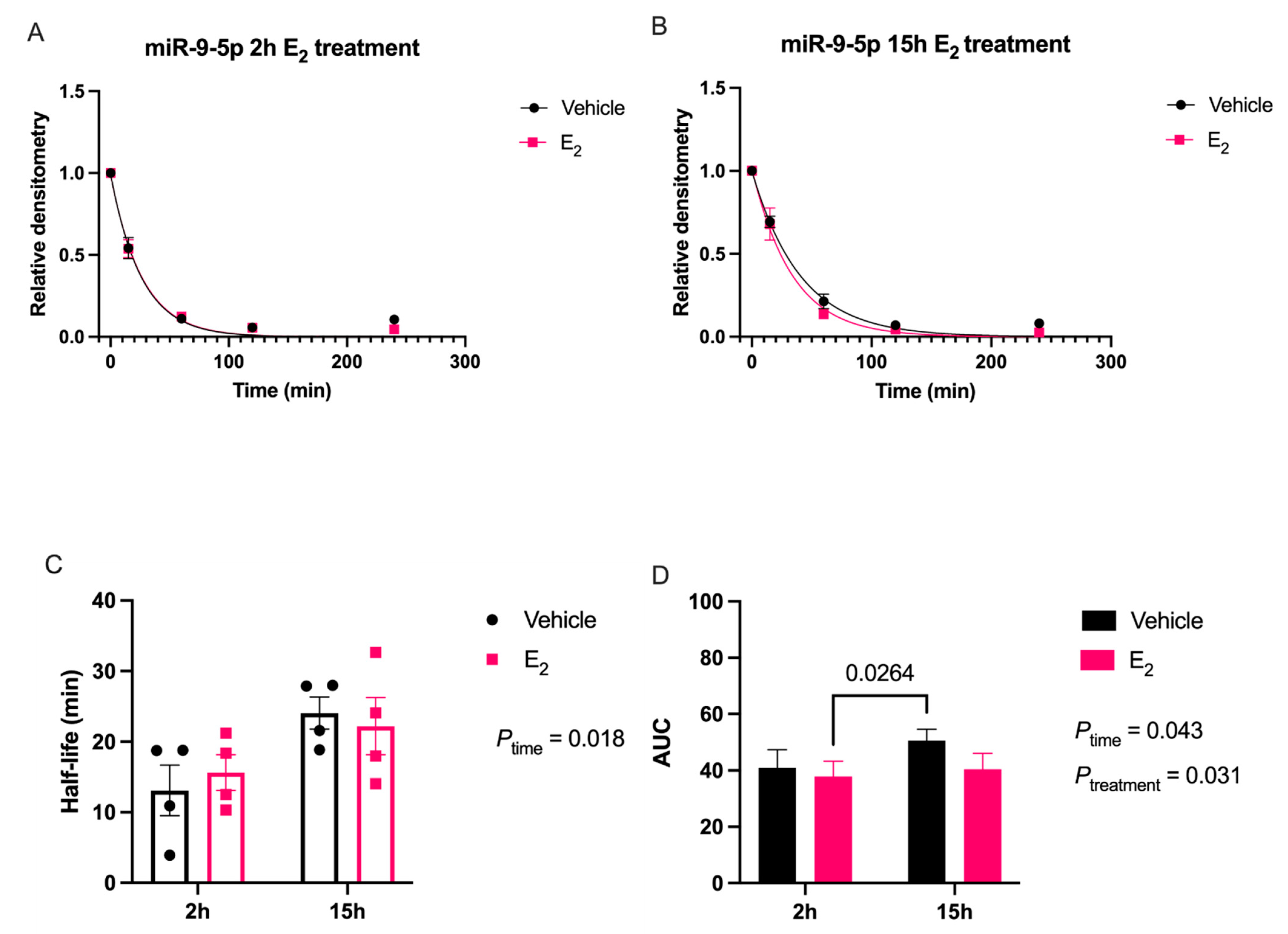
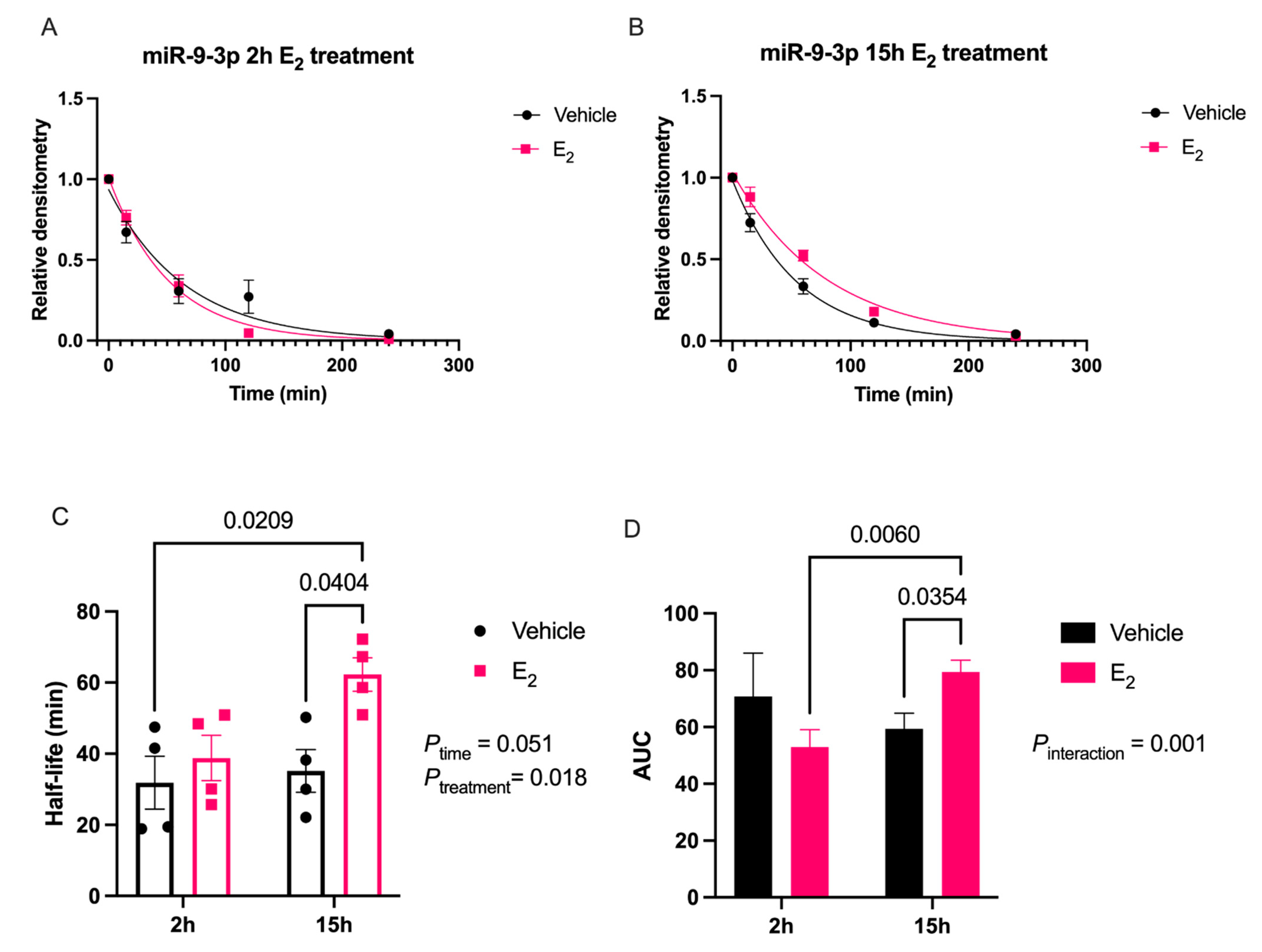
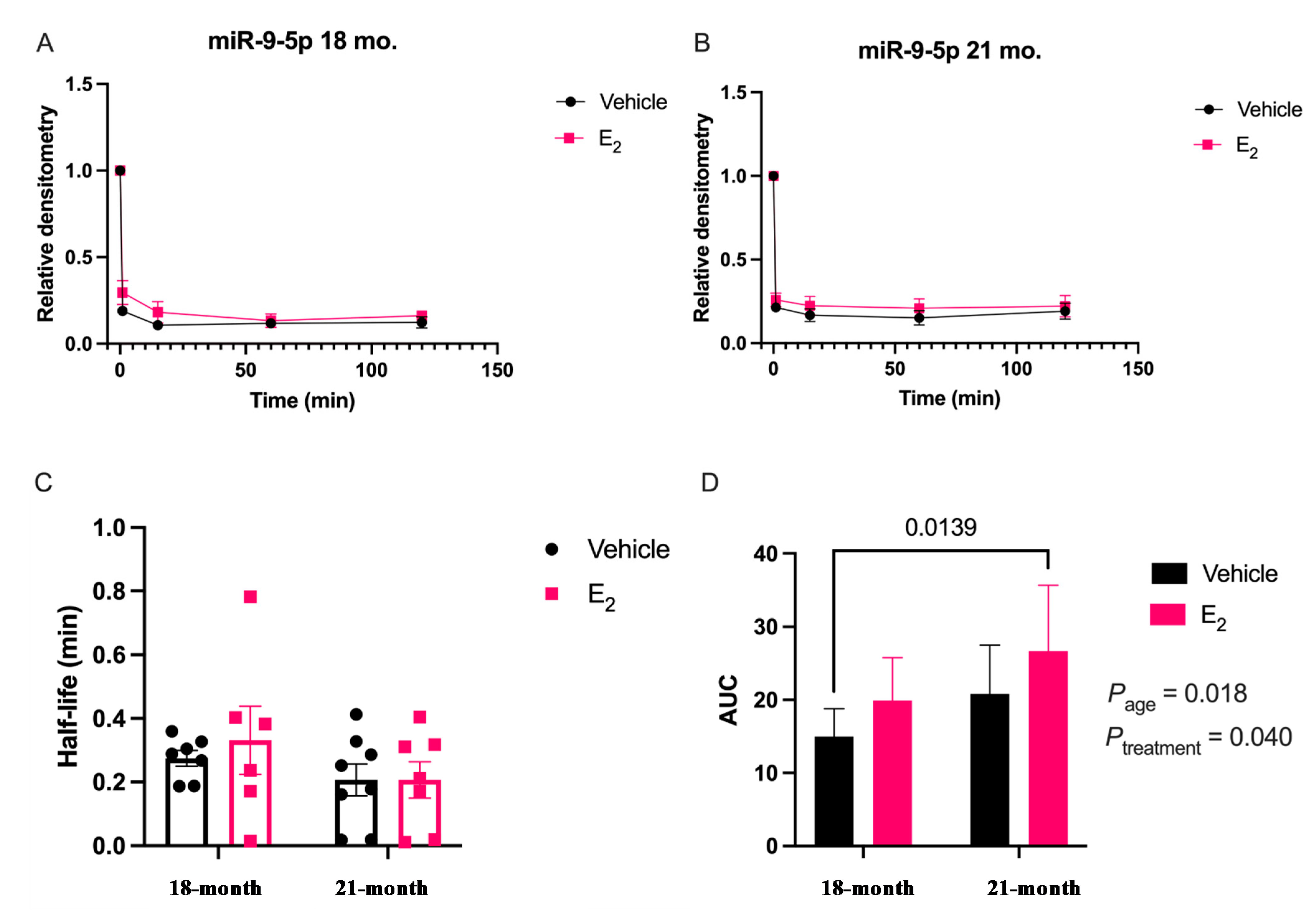
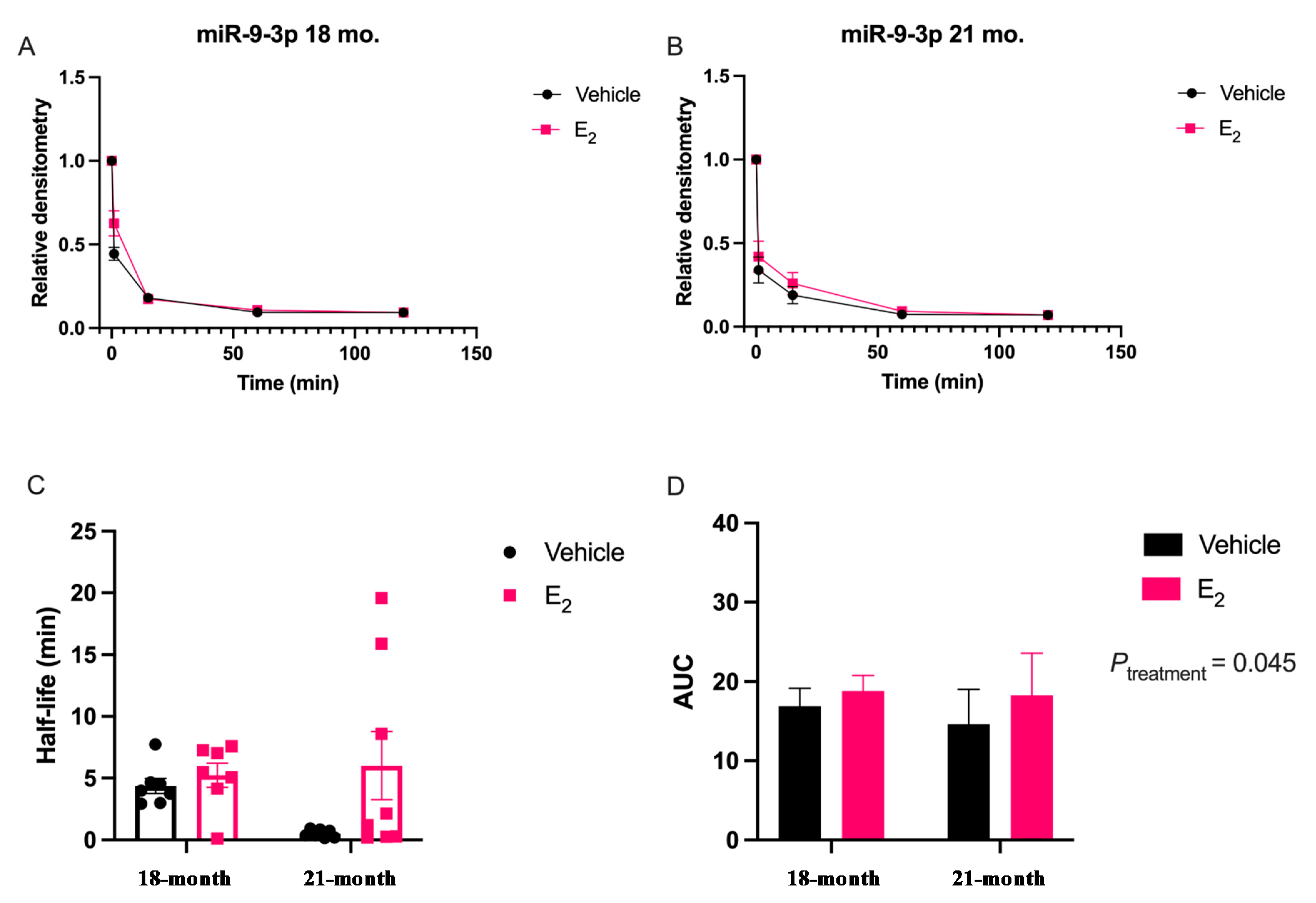
Our data are consistent with previous reports describing rapid miRNA degradation in the central nervous system [
17,
18,
38], which has been linked to neuronal activity. Here, we demonstrated that miR-9-5p and miR-9-3p degrade on a seconds-to-minutes time scale in hypothalamic-derived neuronal cell lines and in the PVN of aged female rats. Of note, miR-9-5p and miR-9-3p degradation in the neuronal cells exhibited a clear one-phase exponential decay that was not observed in the brain tissue, which instead appeared to have a single-phase decay followed by a plateau. These results could be reflective of the homogenous vs. heterogenous cell types in the brain tissue sample, which is a limitation of this technique. For example, it is possible that miRNA degradation factors differ in neurons compared to glial cells, but to our knowledge these specific factors have not yet been discovered. It is also important to consider that the half-lives derived from this study are not necessarily reflective of endogenous miRNA half-lives, as the lysis process required for the degradation assay disrupts the subcellular. organization of its molecular constituents. This could potentially introduce artificial interactions with degradation factors that would not occur in an endogenous setting. Despite these limitations, it is evident from the obtained biochemical data that E
2 treatment is altering the cellular milieu (and perhaps degradation factors) in such a way that it is protective of miR-9-5p and miR-9-3p degradation in a time and age-dependent manner. However, delineating how E
2 treatment impacts the specific kinetics and mechanisms which regulate miRNA stability require further investigation, as previous research has indicated that decay rates influence biological processes and constitute potential drug targets [
39].
The molecular switch underlying the time-dependent effects of E
2 is likely represented by a convergence of multiple signaling pathways. Here, we identify another potential molecular component to this switch in E
2 action, namely via stabilization of mature miR-9-5p and miR-9-3p, which is observed dependent on age. Due to the importance of these miRNAs in governing normal neuronal function in the adult brain [
7,
11,
40], the results reported herein allow for new avenues of research in understanding how differential miRNA stabilization and activity can determine the efficacy of hormone replacement therapy, particularly in the central nervous system of postmenopausal women.
4. Materials and Methods
4.1. Animals
Animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at Loyola University Chicago (#2009018). All necessary measures were taken to minimize the pain and suffering of animals subject to the Exp. procedures. 18-month-old Fischer 344 rats were obtained from the National Institutes of Aging (NIA) colony at Charles River Laboratories. Rats were pair-housed upon arrival and allowed to acclimate to their environment for one week prior to further experimentation. Rats were supplied with standard rat chow and tap water ad libitum and were kept on a 12/12 h. light/dark cycle with Zeitgeber time (ZT) 0 at 7 A.M. Animals were ovariectomized (OVX) at 18 months of age after the acclimation period and then left undisturbed for 1 or 12 weeks following OVX, therefore animals were 18 months and 21 months, respectively, at the time of euthanasia. These animal procedures were followed as outlined by Rao and colleagues [
5]. After the designated time intervals post-OVX, the animals were given a subcutaneous injection of either safflower oil or 2.5 μg/kg 17β-estradiol E
2 dissolved in safflower oil once/day for 3 consecutive days. This dose has been previously reported to achieve circulating E
2 concentrations within the physiological range for postmenopausal women receiving HRT (17–75 pg/mL) [
6,
41,
42]). Animals were euthanized 24-h after the final injection.
4.2. Ovariectomy
Animals were deeply anesthetized with vaporized isoflurane and bilaterally ovariectomized (OVX), as described previously [
6]. Briefly, the ovary and distal end of the uterine horn were excised from the body cavity after the uterine horn was clamped with a hemostat and ligated proximal to the clamp. Animals were singly housed and provided with acetaminophen analgesic in tap water for 3 days following the procedure. After 3 days of analgesia, the animals were pair-housed with their previous cage mate and were undisturbed for the duration of the experiment.
4.3. Tissue Processing
The animals were deeply anesthetized using vaporized isofluorane and euthanized by rapid decapitation. Brains were rapidly dissected, flash-frozen in 2-methylbutane at −30 °C, and then sectioned coronally at 200 μm on a freezing microtome (Leica Biosystems, Lincolnshire, IL, USA). The paraventricular nucleus (PVN) was microdissected using a 0.75-mm Palkovit’s brain punch tool (Stoelting, Inc., Wood Dale, IL, USA) at −1.49 to −2.12 relative to bregma, as defined by
The Rat Brain in Stereotaxic Coordinates [
43]. Frozen PVN microdissections were transferred to a microcentrifuge tube and stored at −80 °C.
4.4. Cell Culture
For in vitro studies, we used a neuronal cell line derived from the paraventricular nucleus (PVN) of the rat hypothalamus (IVB cells, provided by John Kaskow, University of Cincinnati, Cincinnati, OH, USA). Cells were maintained in normal growth media (DMEM media containing glucose, L-glutamine, sodium pyruvate, and 10% fetal bovine serum (FBS) and grown to 60–70% confluency prior to experiments. For all Exp. conditions, cells were maintained in media containing 10% charcoal/dextran stripped FBS (substituted for regular FBS) for 48 h to eliminate all endogenous sources of hormones from the FBS. After 48 h, cells were treated with 100 nM E2 or an equivalent volume of media containing 0.001% ethanol (vehicle) for 2 or 15 h before lysis.
4.5. RNA Isolation and cDNA Synthesis
Total RNA was isolated from IVB cells and PVN tissue microdissections using the Zymogen DirectZol kit (Zymo Research, Irvine, CA, USA). Total RNA (1.0 μg) was reversed transcribed using the Norgen miRNA cDNA Synthesis kit (Thorold, ON, Canada) or Invitrogen Superscript IV mRNA synthesis kit for mRNA Cdna (Waltham, MA, USA), according to manufacturer instructions.
4.6. RT-qPCR
All mRNA transcripts (i.e., miRNA biogenesis components and targets) were quantified by RT-qPCR using the procedures and primers previously described [
6]. Each individual biological sample was assayed as triplicate replicates within an assay. We used 18S rRNA as a housekeeping gene that has previously been verified as unaffected by E
2 treatment [
44]. Transcripts were quantified relative to vehicle-treated control using the ΔΔCt method [
45]. The following conditions were used for the thermocycler: (1) 95 °C for 10 min, (2) 95 °C for 15 s, (3) 59 °C for 20 s, and (4) 72 °C for 12s in addition to melting curve analysis.
4.7. miRNA Degradation Assay
The miRNA degradation assay was adapted for tissue and whole cell lysates according to methods originally described by [
46], and then modified by Kim and colleagues [
18]. Briefly, oligonucleotide constructs were synthesized with the exact nucleotide sequence of the mature transcript for miR-9-5p and miR-9-3p, [UCUUUGGUUAUCUAGCUGUAUGA] and [AUAAAGCUAGAUAACCGAAAGU], respectively (Integrated DNA Technologies, Coralville, IA, USA). These single stranded oligonucleotide sequences were then radiolabeled on the 5′ end using [γ32P] ATP (3000 Ci/mmol; PerkinElmer, Waltham, MA, USA); at a ratio of 10 fmols: 20 μg for the radiolabeled RNA and protein from either the IVB cell lysate or brain tissue lysate, respectively. Incubation of the radiolabeled miRNA oligonucleotides with the lysate was terminated at five different time points by boiling at 95 °C for 2 min following the addition of 2 × RNA Loading Dye (New England Biolabs, Ipswich, MA, USA). The resulting mixture was then resolved on an 8% urea gel by electrophoresis. Finally, the gel was visualized by phosphoimaging (Typhoon GE Healthcare, Chicago, IL, USA) to detect levels of the radiolabeled miRNA at the various time points. Gel bands were quantified using densitometry analyses with ImageJ (RRID:SCR_003070) v5.2 software and averaged densitometry values from multiple replicates were plotted on a scatterplot using OriginLab software (v8.5.1, Northampton, MA, USA). Prism software (v9.1.0, San Diego, CA, USA) was used to determine degradation kinetics using a non-linear regression, one-phase decay, half-life calculated from the best fit exponential decay function; specifically, T
1/2 = ln2*tau and area under the curve (AUC). Data were analyzed by two-factor ANOVA, followed by Tukey post-hoc for multiple pair-wise comparisons.4.8. Western Blot
Protein lysate from IVB cells and tissue was prepared using a 0.5% NP40 buffer with protease and phosphatase inhibitors (Thermo Fisher Scientific, Skokie, IL, USA). Following lysis procedures, protein concentration was determined using a bicinchoninic acid (BCA) assay according to manufacturer instructions (Thermo Fisher Scientific, Skokie, IL, USA). Total protein (30 μg) was boiled with 4X Laemmli buffer (BioRad, Hercules, CA, USA) at 95 °C for 5 min before electrophoresis on a 10% polyacrylamide gel. Following gel electrophoresis at 120 V for 1 h, proteins were transferred to Immobilon™ PVDF membranes (Millipore, Burlington, MA, USA) at 100 V for 1 h at 4 °C. Membranes were blocked with a 1:1 solution of TBS and Odyssey blocking solution (Li-cor Biosciences, Lincoln, NE, USA) for 1 h at room temperature. Following the blocking procedure, membranes were incubated with the indicated primary antibody overnight with constant agitation at 4 °C. Membranes were then incubated with 1:1000 secondary antibodies, as appropriate (Li-cor Biosciences, Lincoln, NE, USA), for 1 h at room temperature. Protein bands were visualized using the Azure Biosystems imaging system (Dublin, CA, USA). Densitometry values were calculated after subtracting background and relative fold changes were made compared to vehicle controls.
4.8. Polysome Profiling
Hypothalamic-derived neuronal IVB cells were grown in media containing 10% charcoal-stripped FBS for 48 h, then treated with E
2 (100 nM) or vehicle (0.001% EtOH) for 15 h, followed by 0.1 mg/mL cycloheximide (Sigma-Aldrich, St. Louis, MO, USA) for 10 min prior to lysis. Cells were lysed in polysome extraction buffer [
47] and protein was quantified by BCA to ensure equal loading between each sample. 1.5 mg lysate was loaded onto a 15–45% sucrose gradient. Gradients were centrifuged for 90 min at 19,0000×
g at 4 °C. Fractions were collected on ice at 1.5 mL/min into 10 tubes per gradient. Absorbance was measured at 254 nm using a Brandel UA-6 Type 11 absorbance detector and data was recorded with Brandel Peak Chart data capture software to record polysome profiles (Gaithersburg, MD, USA). Trizol was immediately added to fractions and stored at −80 °C until RNA isolation. All RNA samples were collected using column purification (Zymo, Irvine, CA, USA). 200 ng of total RNA per fraction was used for miRNA or mRNA cDNA synthesis, as described above. RT-qPCR analysis of miRNAs in each fraction was normalized to 18S mRNA, which did not show shifts in distribution patterns with treatment. Each assay was repeated in 3–4 independent experiments, with 3 technical replicates per experiment.
4.9. Statistics
Statistical analyses were performed using Prism software (v9.1.0, San Diego, CA, USA). Data were analyzed by two-factor ANOVA followed by Tukey post-hoc test for multiple pair-wise comparisons unless otherwise noted in figure captions. Data are displayed as mean ± SEM and statistical significance was determined at p < 0.05.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}