Fusion Genes and RNAs in Cancer Development

Abstract
1. Introduction
2. Biological Functions of Fusion Genes and RNAs
3. Biosynthesis Patterns of Fusion Genes and RNAs
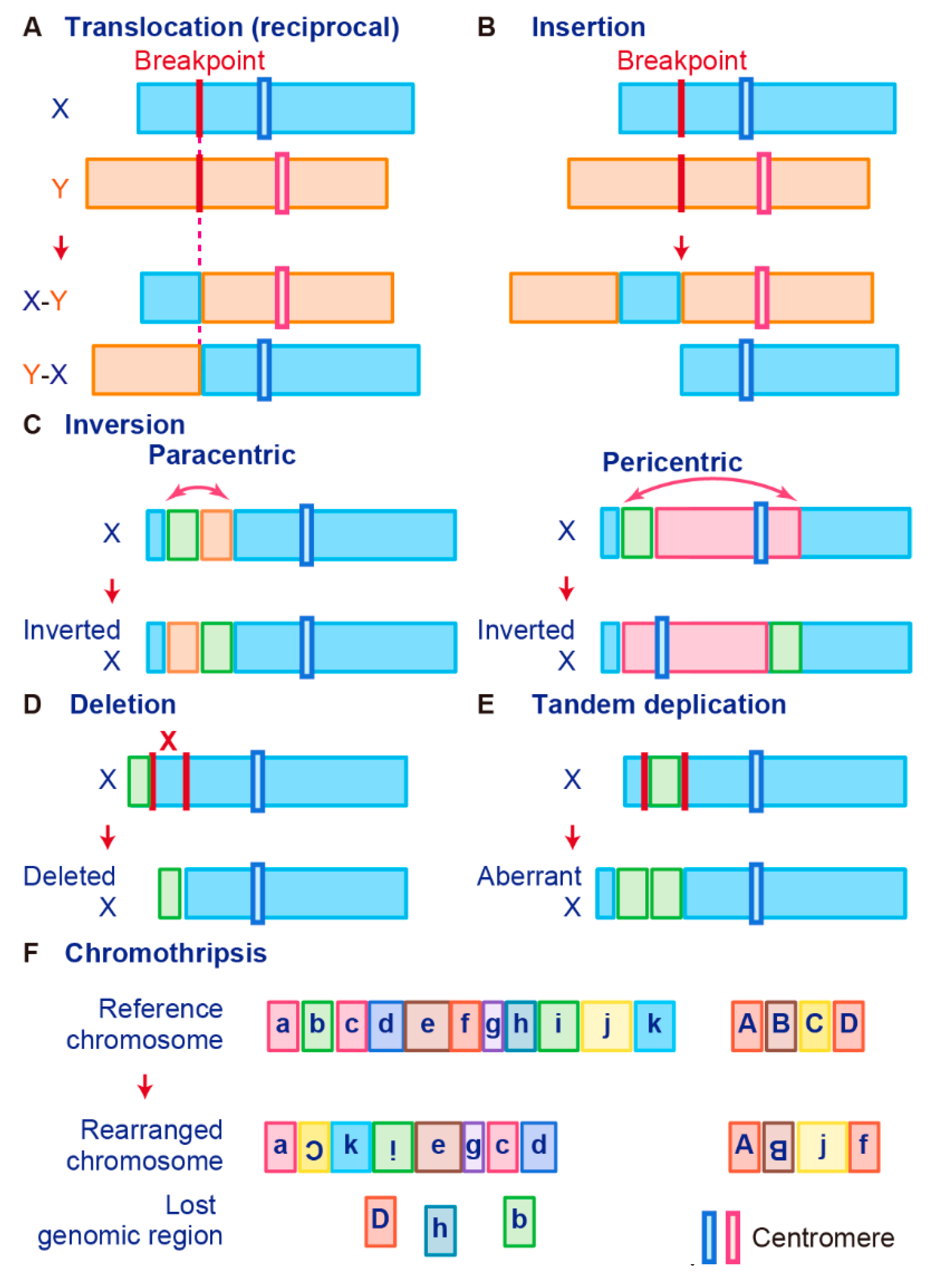
3.1. Chromosomal Rearrangement
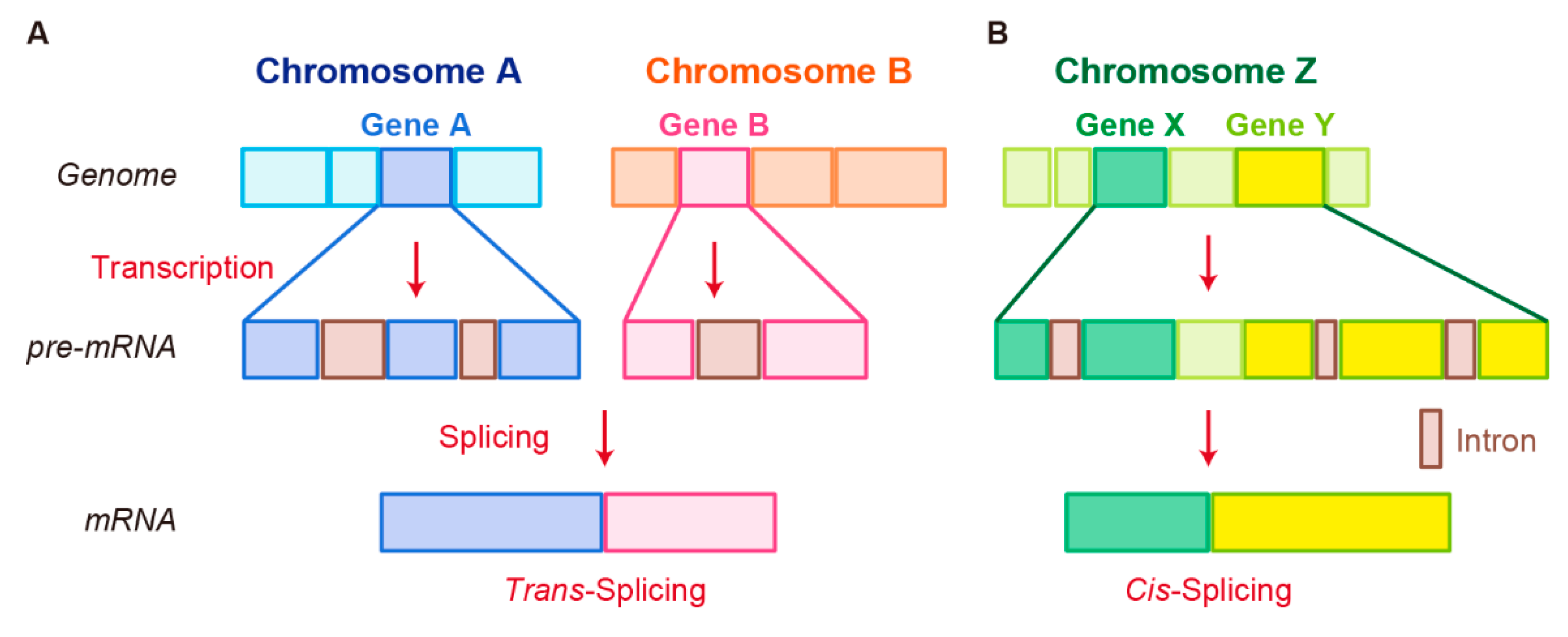
3.2. Trans-Splicing
3.3. Cis-Splicing
4. Detection of Fusion Genes and RNAs
4.1. Guided Approaches to Detect Fusions
4.2. Unbiased Approaches to Detect Fusions
4.3. Databases for Fusion Genes and RNAs
5. Clinical Relevance of Fusion Genes
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Campbell, P.J.; Getz, G.; Korbel, J.O.; Stuart, J.M.; Jennings, J.L.; Stein, L.D.; Perry, M.D.; Nahal-Bose, H.K.; Ouellette, B.F.F.; Li, C.H.; et al. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93. [Google Scholar] [CrossRef]
- Vogelstein, B.; Kinzler, K.W. Cancer genes and the pathways they control. Nat. Med. 2004, 10, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Mitelman, F.; Johansson, B.; Mertens, F. The impact of translocations and gene fusions on cancer causation. Nat. Rev. Cancer 2007, 7, 233–245. [Google Scholar] [CrossRef]
- Mertens, F.; Johansson, B.; Fioretos, T.; Mitelman, F. The emerging complexity of gene fusions in cancer. Nat. Rev. Cancer 2015, 15, 371–381. [Google Scholar] [CrossRef]
- Gao, Q.; Liang, W.W.; Foltz, S.M.; Mutharasu, G.; Jayasinghe, R.G.; Cao, S.; Liao, W.W.; Reynolds, S.M.; Wyczalkowski, M.A.; Yao, L.; et al. Driver Fusions and Their Implications in the Development and Treatment of Human Cancers. Cell Rep. 2018, 23, 227–238. [Google Scholar] [CrossRef]
- Nowell, P.C.; Hungerford, D.A. Chromosome Studies on Normal and Leukemic Human Leukocytes. J. Natl. Cancer Inst. 1960, 25, 85–109. [Google Scholar] [PubMed]
- Dai, X.; Theobard, R.; Cheng, H.; Xing, M.; Zhang, J. Fusion genes: A promising tool combating against cancer. Biochim. Biophys. Acta-Rev. Cancer 2018, 1869, 149–160. [Google Scholar] [CrossRef]
- Shtivelman, E.; Lifshitz, B.; Gale, R.P.; Cananni, E. Fused transcript of abl and bcr genes in chronic myelogenous leukaemia. Nature 1985, 315, 550–554. [Google Scholar] [CrossRef] [PubMed]
- Laemmli, U.K. A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature 1973, 243, 290–293. [Google Scholar]
- Han, C.; Sun, L.Y.; Wang, W.T.; Sun, Y.M.; Chen, Y.Q. Non-coding RNAs in cancers with chromosomal rearrangements: The signatures, causes, functions and implications. J. Mol. Cell Biol. 2019, 11, 886–898. [Google Scholar] [CrossRef]
- Chen, J.; Odenike, O.; Rowley, J.D. Leukaemogenesis: More than mutant genes. Nat. Rev. Cancer 2010, 10, 23–36. [Google Scholar] [CrossRef]
- Schram, A.M.; Chang, M.T.; Jonsson, P.; Drilon, A. Fusions in solid tumours: Diagnostic strategies, targeted therapy, and acquired resistance. Nat. Rev. Clin. Oncol. 2017, 14, 735–748. [Google Scholar] [CrossRef]
- Parker, B.C.; Zhang, W. Fusion genes in solid tumors: An emerging target for cancer diagnosis and treatment. Chin. J. Cancer 2013, 32, 594–603. [Google Scholar] [CrossRef]
- Kas, K.; Voz, M.L.; Roijer, E.; Astrom, A.; Meyen, E.; Stenman, G.; Ven, W.J.M. Van De Promoter swapping between the genes for a novel zinc finger protein and b-catenin in pleiomorphic adenomas with t(3;8)(p21;q12) translocations. Nat. Genet. 1997, 15, 170–174. [Google Scholar] [CrossRef] [PubMed]
- Mark, J.; Dahlenfors, R.; Ekedahl, C.; Stenman, G. The mixed salivary gland tumor—A normally benign human neoplasm frequently showing specific chromosomal abnormalities. Cancer Genet. Cytogenet. 1980, 2, 231–241. [Google Scholar] [CrossRef]
- Kopp, F.; Mendell, J.T. Functional Classification and Experimental Dissection of Long Noncoding RNAs. Cell 2018, 172, 393–407. [Google Scholar] [CrossRef]
- Palazzo, A.F.; Koonin, E.V. Functional Long Non-coding RNAs Evolve from Junk Transcripts. Cell 2020, 183, 1151–1161. [Google Scholar] [CrossRef]
- Rinn, J.L.; Chang, H.Y. Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 2012, 81, 145–166. [Google Scholar] [CrossRef] [PubMed]
- Cech, T.R.; Steitz, J.A. The noncoding RNA revolution-trashing old rules to forge new ones. Cell 2014, 157, 77–94. [Google Scholar] [CrossRef] [PubMed]
- Anastasiadou, E.; Jacob, L.S.; Slack, F.J. Non-coding RNA networks in cancer. Nat. Rev. Cancer 2017, 18, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.M.; Tsang, F.H.; Ng, I.O.L. Non-coding RNAs in hepatocellular carcinoma: Molecular functions and pathological implications. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 137–151. [Google Scholar] [CrossRef]
- Batista, P.J.; Chang, H.Y. Long noncoding RNAs: Cellular address codes in development and disease. Cell 2013, 152, 1298–1307. [Google Scholar] [CrossRef]
- Huarte, M. The emerging role of lncRNAs in cancer. Nat. Med. 2015, 21, 1253–1261. [Google Scholar] [CrossRef] [PubMed]
- Shirahama, S.; Miki, A.; Kaburaki, T.; Akimitsu, N. Long Non-coding RNAs Involved in Pathogenic Infection. Front. Genet. 2020, 11, 454. [Google Scholar] [CrossRef]
- Imamura, K.; Akimitsu, N. Long non-coding RNAs involved in immune responses. Front. Immunol. 2014, 5, 3–6. [Google Scholar] [CrossRef]
- Tano, K.; Akimitsu, N. Long non-coding RNAs in cancer progression. Front. Genet. 2012, 3, 1–6. [Google Scholar] [CrossRef]
- Ransohoff, J.D.; Wei, Y.; Khavari, P.A. The functions and unique features of long intergenic non-coding RNA. Nat. Rev. Mol. Cell Biol. 2018, 19, 143–157. [Google Scholar] [CrossRef]
- Taniue, K.; Kurimoto, A.; Takeda, Y.; Nagashima, T.; Okada-Hatakeyama, M.; Katou, Y.; Shirahige, K.; Akiyama, T. ASBEL-TCF3 complex is required for the tumorigenicity of colorectal cancer cells. Proc. Natl. Acad. Sci. USA 2016, 113, 12739–12744. [Google Scholar] [CrossRef] [PubMed]
- Taniue, K.; Kurimoto, A.; Sugimasa, H.; Nasu, E.; Takeda, Y.; Iwasaki, K.; Nagashima, T.; Okada-Hatakeyama, M.; Oyama, M.; Kozuka-Hata, H.; et al. Long noncoding RNA UPAT promotes colon tumorigenesis by inhibiting degradation of UHRF1. Proc. Natl. Acad. Sci. USA 2016, 113, 1273–1278. [Google Scholar] [CrossRef] [PubMed]
- Yanagida, S.; Taniue, K.; Sugimasa, H.; Nasu, E.; Takeda, Y.; Kobayashi, M.; Yamamoto, T.; Okamoto, A.; Akiyama, T. ASBEL, an ANA/BTG3 antisense transcript required for tumorigenicity of ovarian carcinoma. Sci. Rep. 2013, 3, 1305. [Google Scholar] [CrossRef] [PubMed]
- Slack, F.J.; Chinnaiyan, A.M. The Role of Non-coding RNAs in Oncology. Cell 2019, 179, 1033–1055. [Google Scholar] [CrossRef] [PubMed]
- Dixon-McIver, A.; East, P.; Mein, C.A.; Cazier, J.B.; Molloy, G.; Chaplin, T.; Lister, T.A.; Young, B.D.; Debernardi, S. Distinctive patterns of microRNA expression associated with karyotype in acute myeloid leukaemia. PLoS ONE 2008, 3, e2141. [Google Scholar] [CrossRef] [PubMed]
- Garzon, R.; Volinia, S.; Liu, C.G.; Fernandez-Cymering, C.; Palumbo, T.; Pichiorri, F.; Fabbri, M.; Coombes, K.; Alder, H.; Nakamura, T.; et al. MicroRNA signatures associated with cytogenetics and prognosis in acute myeloid leukemia. Blood 2008, 111, 3183–3189. [Google Scholar] [CrossRef]
- Li, Z.; Lu, J.; Sun, M.; Mi, S.; Zhang, H.; Luo, R.T.; Chen, P.; Wang, Y.; Yan, M.; Qian, Z.; et al. Distinct microRNA expression profiles in acute myeloid leukemia with common translocations. Proc. Natl. Acad. Sci. USA 2008, 105, 15535–15540. [Google Scholar] [CrossRef]
- Guarnerio, J.; Bezzi, M.; Jeong, J.C.; Paffenholz, S.V.; Berry, K.; Naldini, M.M.; Lo-Coco, F.; Tay, Y.; Beck, A.H.; Pandolfi, P.P. Oncogenic Role of Fusion-circRNAs Derived from Cancer-Associated Chromosomal Translocations. Cell 2016, 165, 289–302. [Google Scholar] [CrossRef]
- Qin, F.; Zhang, Y.; Liu, J.; Li, H. SLC45A3-ELK4 functions as a long non-coding chimeric RNA. Cancer Lett. 2017, 404, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Brien, G.L.; Stegmaier, K.; Armstrong, S.A. Targeting chromatin complexes in fusion protein-driven malignancies. Nat. Rev. Cancer 2019, 19, 255–269. [Google Scholar] [CrossRef]
- Crompton, B.D.; Stewart, C.; Taylor-Weiner, A.; Alexe, G.; Kurek, K.C.; Calicchio, M.L.; Kiezun, A.; Carter, S.L.; Shukla, S.A.; Mehta, S.S.; et al. The genomic landscape of pediatric Ewing sarcoma. Cancer Discov. 2014, 4, 1326–1341. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 340, 1546–1558. [Google Scholar] [CrossRef]
- Yoshihara, K.; Wang, Q.; Torres-Garcia, W.; Zheng, S.; Vegesna, R.; Kim, H.; Verhaak, R.G.W. The landscape and therapeutic relevance of cancer-associated transcript fusions. Oncogene 2015, 34, 4845–4854. [Google Scholar] [CrossRef]
- Chwalenia, K.; Facemire, L.; Li, H. Chimeric RNAs in cancer and normal physiology. Wiley Interdiscip. Rev. RNA 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kim, N.C.; Wang, Y.D.; Scarborough, E.A.; Moore, J.; Diaz, Z.; MacLea, K.S.; Freibaum, B.; Li, S.; Molliex, A.; et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 2013, 495, 467–473. [Google Scholar] [CrossRef]
- Stransky, N.; Cerami, E.; Schalm, S.; Kim, J.L.; Lengauer, C. The landscape of kinase fusions in cancer. Nat. Commun. 2014, 5, 4846. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Hsu, P.P.; Awad, M.M.; Engelman, J.A. Tyrosine kinase gene rearrangements in epithelial malignancies. Nat. Rev. Cancer 2013, 13, 772–787. [Google Scholar] [CrossRef]
- Rabbitts, T.H. Commonality but Diversity in Cancer Gene Fusions. Cell 2009, 137, 391–395. [Google Scholar] [CrossRef] [PubMed]
- Deplus, R.; Delliaux, C.; Marchand, N.; Flourens, A.; Vanpouille, N.; Leroy, X.; de Launoit, Y.; Duterque-Coquillaud, M. TMPRSS2-ERG fusion promotes prostate cancer metastases in bone. Oncotarget 2017, 8, 11827–11840. [Google Scholar] [CrossRef]
- Delattre, O.; Zucman, J.; Plougastel, B.; Desmaze, C.; Melot, T.; Peter, M.; Kovar, H.; Joubert, I.; De Jong, P.; Rouleau, G.; et al. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature 1992, 359, 162–165. [Google Scholar] [CrossRef] [PubMed]
- May, W.A.; Gishizky, M.L.; Lessnick, S.L.; Lunsford, L.B.; Lewis, B.C.; Delattre, O.; Zucman, J.; Thomas, G.; Denny, C.T. Ewing sarcoma 11;22 translocation produces a chimeric transcription factor that requires the DNA-binding domain encoded by FLI1 for transformation. Proc. Natl. Acad. Sci. USA 1993, 90, 5752–5756. [Google Scholar] [CrossRef]
- Li, Z.; Qin, F.; Li, H. Chimeric RNAs and their implications in cancer. Curr. Opin. Genet. Dev. 2018, 48, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Tuna, M.; Amos, C.I.; Mills, G.B. Molecular mechanisms and pathobiology of oncogenic fusion transcripts in epithelial tumors. Oncotarget 2019, 10, 2095–2111. [Google Scholar] [CrossRef] [PubMed]
- Nilsen, T.W.; Graveley, B.R. Expansion of the eukaryotic proteome by alternative splicing. Nature 2010, 463, 457–463. [Google Scholar] [CrossRef]
- Irimia, M.; Blencowe, B.J. Alternative splicing: Decoding an expansive regulatory layer. Curr. Opin. Cell Biol. 2012, 24, 323–332. [Google Scholar] [CrossRef]
- Jurica, M.S.; Moore, M.J. Pre-mRNA splicing: Awash in a sea of proteins. Mol. Cell 2003, 12, 5–14. [Google Scholar] [CrossRef]
- Ule, J.; Blencowe, B.J. Alternative Splicing Regulatory Networks: Functions, Mechanisms, and Evolution. Mol. Cell 2019, 76, 329–345. [Google Scholar] [CrossRef]
- Shi, Y. Mechanistic insights into precursor messenger RNA splicing by the spliceosome. Nat. Rev. Mol. Cell Biol. 2017, 18, 655–670. [Google Scholar] [CrossRef] [PubMed]
- Neckles, C.; Sundara Rajan, S.; Caplen, N.J. Fusion transcripts: Unexploited vulnerabilities in cancer? Wiley Interdiscip. Rev. RNA 2020, 11, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Lucas, G.S.; Ardern, J.C.; Bartram, C.R. BCR-ABL rearrangements in acute lymphoblastic leukaemia. Lancet 1991, 337, 1548. [Google Scholar] [CrossRef]
- Cuenco, G.M.; Ren, R. Cooperation of BCR-ABL and AML1/MDS1/EVI1 in blocking myeloid differentiation and rapid induction of an acute myelogenous leukemia. Oncogene 2001, 20, 8236–8248. [Google Scholar] [CrossRef] [PubMed]
- Nucifora, G.; Birn, D.J.; Erickson, P.; Gao, J.; LeBeau, M.M.; Drabkin, H.A.; Rowle, J.D. Detection of DNA rearrangements in the AML1 and ETO loci and of an AML1/ETO fusion mRNA in patients with t(8;21) acute myeloid leukemia. Blood 1993, 81, 883–888. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, M.; O’Brien, D.; Kumaravelu, P.; Lenny, N.; Yeoh, E.J.; Downing, J.R. Expression of a conditional AML1-ETO oncogene bypasses embryonic lethality and establishes a murine model of human t(8;21) acute myeloid leukemia. Cancer Cell 2002, 1, 63–74. [Google Scholar] [CrossRef]
- Jurlander, J.; Caligiuri, M.A.; Ruutu, T.; Baer, M.R.; Strout, M.P.; Oberkircher, A.R.; Hoffmann, L.; Ball, E.D.; Frei-Lahr, D.A.; Christiansen, N.P.; et al. Persistence of the AML1/ETO fusion transcript in patients treated with allogeneic bone marrow transplantation for t(8;21) leukemia. Blood 1996, 88, 2183–2191. [Google Scholar] [CrossRef]
- Nucifora, G.; Rowley, J.D. AML1 and the 8;21 and 3;21 translocations in acute and chronic myeloid leukemia. Blood 1995, 86, 1–14. [Google Scholar] [CrossRef]
- Yuan, Y.; Zhou, L.; Miyamoto, T.; Iwasaki, H.; Harakawa, N.; Hetherington, C.J.; Burel, S.A.; Lagasse, E.; Weissman, I.L.; Akashi, K.; et al. AML1-ETO expression is directly involved in the development of acute myeloid leukemia in the presence of additional mutations. Proc. Natl. Acad. Sci. USA 2001, 98, 10398–10403. [Google Scholar] [CrossRef]
- de Thé, H.; Lavau, C.; Marchio, A.; Chomienne, C.; Degos, L.; Dejean, A. The PML-RARα fusion mRNA generated by the t(15;17) translocation in acute promyelocytic leukemia encodes a functionally altered RAR. Cell 1991, 66, 675–684. [Google Scholar] [CrossRef]
- Goddard, A.D.; Borrow, J.; Freemont, P.S.; Solomon, E. Characterization of a zinc finger gene disrupted by the t(15;17) in acute promyelocytic leukemia. Science 1991, 254, 1371–1374. [Google Scholar] [CrossRef]
- Kakizuka, A.; Miller, W.H.; Umesono, K.; Warrell, R.P.; Frankel, S.R.; Murty, V.V.V.S.; Dmitrovsky, E.; Evans, R.M. Chromosomal translocation t(15;17) in human acute promyelocytic leukemia fuses RARα with a novel putative transcription factor, PML. Cell 1991, 66, 663–674. [Google Scholar] [CrossRef]
- Meroni, G.; Diez-Roux, G. TRIM/RBCC, a novel class of “single protein RING finger” E3 ubiquitin ligases. BioEssays 2005, 27, 1147–1157. [Google Scholar] [CrossRef]
- Reymond, A.; Meroni, G.; Fantozzi, A.; Merla, G.; Cairo, S.; Luzi, L.; Riganelli, D.; Zanaria, E.; Messali, S.; Cainarca, S.; et al. The tripartite motif family identifies cell compartments. EMBO J. 2001, 20, 2140–2151. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, E.; Laukens, K.; Dang, T.H.; Van Ostade, X. A manually curated network of the PML nuclear body interactome reveals an important role for PML-NBs in SUMOylation dynamics. Int. J. Biol. Sci. 2010, 6, 51–67. [Google Scholar] [CrossRef] [PubMed]
- Shima, Y.; Shima, T.; Chiba, T.; Irimura, T.; Pandolfi, P.P.; Kitabayashi, I. PML Activates Transcription by Protecting HIPK2 and p300 from SCFFbx3-Mediated Degradation. Mol. Cell. Biol. 2008, 28, 7126–7138. [Google Scholar] [CrossRef] [PubMed]
- Di Masi, A.; Cilli, D.; Berardinelli, F.; Talarico, A.; Pallavicini, I.; Pennisi, R.; Leone, S.; Antoccia, A.; Noguera, N.I.; Lo-Coco, F.; et al. PML nuclear body disruption impairs DNA double-strand break sensing and repair in APL. Cell Death Dis. 2016, 7, e2308. [Google Scholar] [CrossRef]
- Sorensen, P.H.B.; Lessnick, S.L.; Lopez-terrada, D.; Liu, X.F.; Triche, T.J.; Denny, C.T.; Cherry, H. A second Ewing’s sarcoma translocation, t(21;22), fuses the EWS gene to another ETS–family transcription factor, ERG. Nat. Genet. 1994, 6, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Zinszner, H.; Immanuel, D.; Yin, Y.; Liang, F.X.; Ron, D. A topogenic role for the oncogenic N-terminus of TLS: Nucleolar localization when transcription is inhibited. Oncogene 1997, 14, 451–461. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yang, L.; Chansky, H.A.; Hickstein, D.D. EWS·Fli-1 fusion protein interacts with hyperphosphorylated RNA polymerase II and interferes with serine-arginine protein-mediated RNA splicing. J. Biol. Chem. 2000, 275, 37612–37618. [Google Scholar] [CrossRef] [PubMed]
- Bailly, R.; Bosselut, R.; Zucman, J.; Delattre, O.; Roussel, M.; Thomas, G.; Ghysdaell, J. DNA-binding and transcriptional activation properties of the EWS-FLI-1 fusion protein resulting from the t (11; 22) translocation in Ewing sarcoma. Mol. Cell. Biol. 1994, 14, 3230–3241. [Google Scholar] [CrossRef]
- Braun, B.S.; Frieden, R.; Lessnick, S.L.; May, W.A.; Denny, C.T. Identification of target genes for the Ewing’s sarcoma EWS/FLI fusion protein by representational difference analysis. Mol. Cell. Biol. 1995, 15, 4623–4630. [Google Scholar] [CrossRef]
- Boulay, G.; Sandoval, G.J.; Riggi, N.; Iyer, S.; Buisson, R.; Naigles, B.; Awad, M.E.; Rengarajan, S.; Volorio, A.; McBride, M.J.; et al. Cancer-Specific Retargeting of BAF Complexes by a Prion-like Domain. Cell 2017, 171, 163–178. [Google Scholar] [CrossRef]
- Pierotti, M.A.; Santoro, M.; Jenkins, R.B.; Sozzi, G.; Bongarzone, I.; Grieco, M.; Monzini, N.; Miozzo, M.; Herrmann, M.A.; Fusco, A.; et al. Characterization of an inversion on the long arm of chromosome 10 juxtaposing D10S170 and RET and creating the oncogenic sequence RET/PTC. Proc. Natl. Acad. Sci. USA 1992, 89, 1616–1620. [Google Scholar] [CrossRef]
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.I.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007, 448, 561–566. [Google Scholar] [CrossRef]
- Rikova, K.; Guo, A.; Zeng, Q.; Possemato, A.; Yu, J.; Haack, H.; Nardone, J.; Lee, K.; Reeves, C.; Li, Y.; et al. Global Survey of Phosphotyrosine Signaling Identifies Oncogenic Kinases in Lung Cancer. Cell 2007, 131, 1190–1203. [Google Scholar] [CrossRef]
- Pederzoli, F.; Bandini, M.; Marandino, L.; Ali, S.M.; Madison, R.; Chung, J.; Ross, J.S.; Necchi, A. Targetable gene fusions and aberrations in genitourinary oncology. Nat. Rev. Urol. 2020, 17, 613–625. [Google Scholar] [CrossRef]
- Tomlins, S.A.; Laxman, B.; Dhanasekaran, S.M.; Helgeson, B.E.; Cao, X.; Morris, D.S.; Menon, A.; Jing, X.; Cao, Q.; Han, B.; et al. Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature 2007, 448, 595–599. [Google Scholar] [CrossRef]
- Hollenhorst, P.C.; McIntosh, L.P.; Graves, B.J. Genomic and biochemical insights into the specificity of ETS transcription factors. Annu. Rev. Biochem. 2011, 80, 437–471. [Google Scholar] [CrossRef] [PubMed]
- Perner, S.; Demichelis, F.; Beroukhim, R.; Schmidt, F.H.; Mosquera, J.M.; Setlur, S.; Tchinda, J.; Tomlins, S.A.; Hofer, M.D.; Pienta, K.G.; et al. TMPRSS2:ERG fusion-associated deletions provide insight into the heterogeneity of prostate cancer. Cancer Res. 2006, 66, 8337–8341. [Google Scholar] [CrossRef]
- Singh, D.; Chan, J.M.; Zoppoli, P.; Niola, F.; Sullivan, R.; Castano, A.; Liu, E.M.; Reichel, J.; Porrati, P.; Pellegatta, S.; et al. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science 2012, 337, 1231–1235. [Google Scholar] [CrossRef] [PubMed]
- Lipson, D.; Capelletti, M.; Yelensky, R.; Otto, G.; Parker, A.; Jarosz, M.; Curran, J.A.; Balasubramanian, S.; Bloom, T.; Brennan, K.W.; et al. Identification of new ALK and RET gene fusions from colorectal and lung cancer biopsies. Nat. Med. 2012, 18, 382–384. [Google Scholar] [CrossRef] [PubMed]
- Holland, A.J.; Cleveland, D.W. Chromoanagenesis and cancer: Mechanisms and consequences of localized, complex chromosomal rearrangements. Nat. Med. 2012, 18, 1630–1638. [Google Scholar] [CrossRef]
- Northcott, P.A.; Shih, D.J.H.; Peacock, J.; Garzia, L.; Sorana Morrissy, A.; Zichner, T.; Stútz, A.M.; Korshunov, A.; Reimand, J.; Schumacher, S.E.; et al. Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature 2012, 487, 49–56. [Google Scholar] [CrossRef]
- Li, H.; Wang, J.; Mor, G.; Sklar, J. A Neoplastic Gene Fusion Mimics Trans-Splicing of RNAs in Normal Human Cells. Science 2008, 321, 1357–1361. [Google Scholar] [CrossRef]
- Li, B.L.; Li, X.L.; Duan, Z.J.; Lee, O.; Lin, S.; Ma, Z.M.; Chang, C.C.Y.; Yang, X.Y.; Park, J.P.; Mohandas, T.K.; et al. Human acyl-CoA:cholesterol acyltransferase-1 (ACAT-1) gene organization and evidence that the 4.3-kilobase ACAT-1 mRNA is produced from two different chromosomes. J. Biol. Chem. 1999, 274, 11060–11071. [Google Scholar] [CrossRef]
- Breen, M.A.; Ashcroft, S.J.H. A truncated isoform of Ca2+/calmodulin-dependent protein kinase II expressed in human islets of Langerhans may result from trans-splicing. FEBS Lett. 1997, 409, 375–379. [Google Scholar] [CrossRef]
- Horiuchi, T.; Giniger, E.; Aigaki, T. Alternative trans-splicing of constant and variable exons of a Drosophila axon guidance gene, lola. Genes Dev. 2003, 17, 2496–2501. [Google Scholar] [CrossRef] [PubMed]
- Dorn, R.; Reuter, G.; Loewendorf, A. Transgene analysis proves mRNA trans-splicing at the complex mod(mdg4) locus in Drosophila. Proc. Natl. Acad. Sci. USA 2001, 98, 9724–9729. [Google Scholar] [CrossRef] [PubMed]
- Lei, Q.; Li, C.; Zuo, Z.; Huang, C.; Cheng, H.; Zhou, R. Evolutionary Insights into RNA trans-Splicing in Vertebrates. Genome Biol. Evol. 2016, 8, 562–577. [Google Scholar] [CrossRef] [PubMed]
- Solnick, D. Trans splicing of mRNA precursors. Cell 1985, 42, 157–164. [Google Scholar] [CrossRef]
- Konarska, M.M.; Padgett, R.A.; Sharp, P.A. Trans splicing of mRNA precursors in vitro. Cell 1985, 42, 165–171. [Google Scholar] [CrossRef]
- Eul, J.; Graessmann, M.; Graessmann, A. Experimental evidence for RNA trans-splicing in mammalian cells. EMBO J. 1995, 14, 3226–3235. [Google Scholar] [CrossRef]
- Eul, J.; Graessmann, M.; Graessmann, A. Trans-splicing and alternative-tandem-cis-splicing: Two ways by which mammalian cells generate a truncated SV40 T-antigen. Nucleic Acids Res. 1996, 24, 1653–1661. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Qin, F.; Movassagh, M.; Park, H.; Golden, W.; Xie, Z.; Zhang, P.; Sklar, J.; Li, H. A chimeric RNA characteristic of rhabdomyosarcoma in normal myogenesis process. Cancer Discov. 2013, 3, 1394–1403. [Google Scholar] [CrossRef]
- Wu, H.; Li, X.; Li, H. Gene fusions and chimeric RNAs, and their implications in cancer. Genes Dis. 2019, 6, 385–390. [Google Scholar] [CrossRef]
- Maher, C.A.; Kumar-Sinha, C.; Cao, X.; Kalyana-Sundaram, S.; Han, B.; Jing, X.; Sam, L.; Barrette, T.; Palanisamy, N.; Chinnaiyan, A.M. Transcriptome sequencing to detect gene fusions in cancer. Nature 2009, 458, 97–101. [Google Scholar] [CrossRef]
- Rickman, D.S.; Pflueger, D.; Moss, B.; VanDoren, V.E.; Chen, C.X.; De la Taille, A.; Kuefer, R.; Tewari, A.K.; Setlur, S.R.; Demichelis, F.; et al. SLC45A3-ELK4 is a novel and frequent erythroblast transformation-specific fusion transcript in prostate cancer. Cancer Res. 2009, 69, 2737–2738. [Google Scholar] [CrossRef]
- Wang, K.; Ubriaco, G.; Sutherland, L.C. RBM6-RBM5 transcription-induced chimeras are differentially expressed in tumours. BMC Genom. 2007, 8, 1–12. [Google Scholar] [CrossRef]
- Nacu, S.; Yuan, W.; Kan, Z.; Bhatt, D.; Rivers, C.S.; Stinson, J.; Peters, B.A.; Modrusan, Z.; Jung, K.; Seshagiri, S.; et al. Deep RNA sequencing analysis of readthrough gene fusions in human prostate adenocarcinoma and reference samples. BMC Med. Genom. 2011, 4, 11. [Google Scholar] [CrossRef]
- Kim, H.P.; Cho, G.A.; Han, S.W.; Shin, J.Y.; Jeong, E.G.; Song, S.H.; Lee, W.C.; Lee, K.H.; Bang, D.; Seo, J.S.; et al. Novel fusion transcripts in human gastric cancer revealed by transcriptome analysis. Oncogene 2014, 33, 5434–5441. [Google Scholar] [CrossRef]
- Tang, Y.; Qin, F.; Liu, A.; Li, H. Recurrent fusion RNA DUS4L-BCAP29 in non-cancer human tissues and cells. Oncotarget 2017, 8, 31415–31423. [Google Scholar] [CrossRef]
- Speicher, M.R.; Carter, N.P. The new cytogenetics: Blurring the boundaries with molecular biology. Nat. Rev. Genet. 2005, 6, 782–792. [Google Scholar] [CrossRef]
- Saiki, R.K.; Scharf, S.; Faloona, F.; Mullis, K.B.; Horn, G.T.; Erlich, H.A.; Arnheim, N. Enzymatic amplification of beta-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. 1985. Biotechnology 1992, 24, 476–480. [Google Scholar] [CrossRef]
- Vandenberg, O.; Martiny, D.; Rochas, O.; van Belkum, A.; Kozlakidis, Z. Considerations for diagnostic COVID-19 tests. Nat. Rev. Microbiol. 2020, 14, 1–13. [Google Scholar] [CrossRef]
- Pinkel, D.; Albertson, D.G. Array comparative genomic hybridization and its applications in cancer. Nat. Genet. 2005, 37, S11–S17. [Google Scholar] [CrossRef]
- Schuster, S.C. Next-generation sequencing transforms today’s biology. Nat. Methods 2008, 5, 16–18. [Google Scholar] [CrossRef]
- Wold, B.; Myers, R.M. Sequence census methods for functional genomics. Nat. Methods 2008, 5, 19–21. [Google Scholar] [CrossRef]
- Shendure, J.; Ji, H. Next-generation DNA sequencing. Nat. Biotechnol. 2008, 26, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- Lou, D.I.; Hussmann, J.A.; McBee, R.M.; Acevedo, A.; Andino, R.; Press, W.H.; Sawyer, S.L. High-throughput DNA sequencing errors are reduced by orders of magnitude using circle sequencing. Proc. Natl. Acad. Sci. USA 2013, 110, 19872–19877. [Google Scholar] [CrossRef] [PubMed]
- Sleep, J.A.; Schreiber, A.W.; Baumann, U. Sequencing error correction without a reference genome. BMC Bioinformatics 2013, 14, 367. [Google Scholar] [CrossRef]
- Kumar, S.; Razzaq, S.K.; Vo, A.D.; Gautam, M.; Li, H. Identifying fusion transcripts using next generation sequencing. Wiley Interdiscip. Rev. RNA 2016, 7, 811–823. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Vo, A.D.; Qin, F.; Li, H. Comparative assessment of methods for the fusion transcripts detection from RNA-Seq data. Sci. Rep. 2016, 6, 21597. [Google Scholar] [CrossRef]
- Liu, S.; Tsai, W.H.; Ding, Y.; Chen, R.; Fang, Z.; Huo, Z.; Kim, S.; Ma, T.; Chang, T.Y.; Priedigkeit, N.M.; et al. Comprehensive evaluation of fusion transcript detection algorithms and a meta-caller to combine top performing methods in paired-end RNA-seq data. Nucleic Acids Res. 2015, 44, e47. [Google Scholar] [CrossRef]
- Carrara, M.; Beccuti, M.; Lazzarato, F.; Cavallo, F.; Cordero, F.; Donatelli, S.; Calogero, R.A. State-of-the-Art Fusion-Finder Algorithms Sensitivity and Specificity. BioMed Res. Int. 2013, 2013, 340620. [Google Scholar] [CrossRef]
- Lu, Z.; Matera, A.G. Vicinal: A method for the determination of ncRNA ends using chimeric reads from RNA-seq experiments. Nucleic Acids Res. 2014, 42, 1–9. [Google Scholar] [CrossRef]
- Houseley, J.; Tollervey, D. Apparent non-canonical trans-splicing is generated by reverse transcriptase in vitro. PLoS ONE 2010, 5, e12271. [Google Scholar] [CrossRef]
- Yu, C.Y.; Liu, H.J.; Hung, L.Y.; Kuo, H.C.; Chuang, T.J. Is an observed non-co-linear RNA product spliced in trans, in cis or just in vitro? Nucleic Acids Res. 2014, 42, 9410–9423. [Google Scholar] [CrossRef]
- Stephens, P.J.; McBride, D.J.; Lin, M.L.; Varela, I.; Pleasance, E.D.; Simpson, J.T.; Stebbings, L.A.; Leroy, C.; Edkins, S.; Mudie, L.J.; et al. Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature 2009, 462, 1005–1010. [Google Scholar] [CrossRef]
- Hammerman, P.S.; Voet, D.; Lawrence, M.S.; Voet, D.; Jing, R.; Cibulskis, K.; Sivachenko, A.; Stojanov, P.; McKenna, A.; Lander, E.S.; et al. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012, 489, 519–525. [Google Scholar] [CrossRef]
- Maher, C.A.; Palanisamy, N.; Brenner, J.C.; Cao, X.; Kalyana-Sundaram, S.; Luo, S.; Khrebtukova, I.; Barrette, T.R.; Grasso, C.; Yu, J.; et al. Chimeric transcript discovery by paired-end transcriptome sequencing. Proc. Natl. Acad. Sci. USA 2009, 106, 12353–12358. [Google Scholar] [CrossRef] [PubMed]
- Muzny, D.M.; Bainbridge, M.N.; Chang, K.; Dinh, H.H.; Drummond, J.A.; Fowler, G.; Kovar, C.L.; Lewis, L.R.; Morgan, M.B.; Newsham, I.F.; et al. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef]
- Getz, G.; Gabriel, S.B.; Cibulskis, K.; Lander, E.; Sivachenko, A.; Sougnez, C.; Lawrence, M.; Kandoth, C.; Dooling, D.; Fulton, R.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [CrossRef]
- Welch, J.S.; Westervelt, P.; Ding, L.; Larson, D.E.; Klco, J.M.; Kulkarni, S.; Wallis, J.; Chen, K.; Payton, J.E.; Fulton, R.S.; et al. Use of whole-genome sequencing to diagnose a cryptic fusion oncogene. JAMA. 2011, 305, 1577–1584. [Google Scholar] [CrossRef]
- Roberts, K.G.; Morin, R.D.; Zhang, J.; Hirst, M.; Zhao, Y.; Su, X.; Chen, S.C.; Payne-Turner, D.; Churchman, M.L.; Harvey, R.C.; et al. Genetic Alterations Activating Kinase and Cytokine Receptor Signaling in High-Risk Acute Lymphoblastic Leukemia. Cancer Cell 2012, 22, 153–166. [Google Scholar] [CrossRef]
- Li, Y.; Roberts, N.D.; Wala, J.A.; Shapira, O.; Schumacher, S.E.; Kumar, K.; Khurana, E.; Waszak, S.; Korbel, J.O.; Haber, J.E.; et al. Patterns of somatic structural variation in human cancer genomes. Nature 2020, 578, 112–121. [Google Scholar] [CrossRef]
- Calabrese, C.; Davidson, N.R.; Demircioğlu, D.; Fonseca, N.A.; He, Y.; Kahles, A.; Van Lehmann, K.; Liu, F.; Shiraishi, Y.; Soulette, C.M.; et al. Genomic basis for RNA alterations in cancer. Nature 2020, 578, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Priestley, P.; Baber, J.; Lolkema, M.P.; Steeghs, N.; de Bruijn, E.; Shale, C.; Duyvesteyn, K.; Haidari, S.; van Hoeck, A.; Onstenk, W.; et al. Pan-cancer whole-genome analyses of metastatic solid tumours. Nature 2019, 575, 210–216. [Google Scholar] [CrossRef]
- Barresi, V.; Cosentini, I.; Scuderi, C.; Napoli, S.; Di Bella, V.; Spampinato, G.; Condorelli, D.F. Fusion transcripts of adjacent genes: New insights into the world of human complex transcripts in cancer. Int. J. Mol. Sci. 2019, 20, 5252. [Google Scholar] [CrossRef]
- Nashed, A.L.; Rao, K.W.; Gulley, M.L. Clinical applications of BCR-ABL molecular testing in acute leukemia. J. Mol. Diagnostics 2003, 5, 63–72. [Google Scholar] [CrossRef]
- Goldman, J.M.; Druker, B.J. Chronic myeloid leukemia: Current treatment options. Blood 2001, 98, 2039–2042. [Google Scholar] [CrossRef]
- Shaw, A.T.; Yeap, B.Y.; Solomon, B.J.; Riely, G.J.; Gainor, J.; Engelman, J.A.; Shapiro, G.I.; Costa, D.B.; Ou, S.H.I.; Butaney, M.; et al. Effect of crizotinib on overall survival in patients with advanced non-small-cell lung cancer harbouring ALK gene rearrangement: A retrospective analysis. Lancet Oncol. 2011, 12, 1004–1012. [Google Scholar] [CrossRef]
- Adamo, P.; Ladomery, M.R. The oncogene ERG: A key factor in prostate cancer. Oncogene 2016, 35, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Tognon, C.; Knezevich, S.R.; Huntsman, D.; Roskelley, C.D.; Melnyk, N.; Mathers, J.A.; Becker, L.; Carneiro, F.; MacPherson, N.; Horsman, D.; et al. Expression of the ETV6-NTRK3 gene fusion as a primary event in human secretory breast carcinoma. Cancer Cell 2002, 2, 367–376. [Google Scholar] [CrossRef]
- Babiceanu, M.; Qin, F.; Xie, Z.; Jia, Y.; Lopez, K.; Janus, N.; Facemire, L.; Kumar, S.; Pang, Y.; Qi, Y.; et al. Recurrent chimeric fusion RNAs in non-cancer tissues and cells. Nucleic Acids Res. 2016, 44, 2859–2872. [Google Scholar] [CrossRef]
- Wu, C.S.; Yu, C.Y.; Chuang, C.Y.; Hsiao, M.; Kao, C.F.; Kuo, H.C.; Chuang, T.J. Integrative transcriptome sequencing identifies trans-splicing events with important roles in human embryonic stem cell pluripotency. Genome Res. 2014, 24, 25–36. [Google Scholar] [CrossRef]
- Chase, A.; Ernst, T.; Fiebig, A.; Collins, A.; Grand, F.; Erben, P.; Reiter, A.; Schreiber, S.; Cross, N.C.P. TFG, a target of chromosome translocations in lymphoma and soft tissue tumors, fuses to GPR128 in healthy individuals. Haematologica 2010, 95, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, Y.; Sereewattanawoot, S.; Suzuki, A. A new era of long-read sequencing for cancer genomics. J. Hum. Genet. 2020, 65, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Ozaki, H.; Sasagawa, Y.; Umeda, M.; Danno, H.; Nikaido, I. Single-cell full-length total RNA sequencing uncovers dynamics of recursive splicing and enhancer RNAs. Nat. Commun. 2018, 9, 619. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Database Name | Tumor | Non- Tumor | First Release | Last Update | Current Version | Total Fusions | URL |
|---|---|---|---|---|---|---|---|
| Mitleman | Yes | No | 1983 | 2020 | 32,578 | https://mitelmandatabase.isb-cgc.org | |
| ChimerDB | Yes | Yes | 2006 | 2020 | 4.0 | 67,610 | https://www.kobic.re.kr/chimerdb_mirror/ |
| FusionGDB | Yes | No | 2019 | 2020 | 43,895 | https://ccsm.uth.edu/FusionGDB/ | |
| COSMIC | Yes | No | 2004 | 2020 | 92 | 19,369 | https://cancer.sanger.ac.uk/cosmic |
| ChiTaRs | Yes | Yes | 2012 | 2019 | 5.0 | 23,167 | http://chitars.md.biu.ac.il/ |
| Tumor Fusion Gene Data Portal | Yes | Yes | 2015 | 2018 | 20,731 | https://www.tumorfusions.org/ | |
| FusionHub | Yes | Yes | 2018 | 2018 | 150,699 | https://fusionhub.persistent.co.in | |
| TICdb | Yes | No | 2007 | 2013 | 3.3 | 1374 | https://genetica.unav.edu/TICdb/ |
| dbCRID | Yes | Yes | 2010 | 2010 | 0.9 | 2643 | http://c1.accurascience.com/dbCRID/ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taniue, K.; Akimitsu, N. Fusion Genes and RNAs in Cancer Development. Non-Coding RNA 2021, 7, 10. https://doi.org/10.3390/ncrna7010010
Taniue K, Akimitsu N. Fusion Genes and RNAs in Cancer Development. Non-Coding RNA. 2021; 7(1):10. https://doi.org/10.3390/ncrna7010010
Chicago/Turabian StyleTaniue, Kenzui, and Nobuyoshi Akimitsu. 2021. "Fusion Genes and RNAs in Cancer Development" Non-Coding RNA 7, no. 1: 10. https://doi.org/10.3390/ncrna7010010
APA StyleTaniue, K., & Akimitsu, N. (2021). Fusion Genes and RNAs in Cancer Development. Non-Coding RNA, 7(1), 10. https://doi.org/10.3390/ncrna7010010