Abstract
Chronic inflammation is inextricably linked to cardiovascular disease (CVD). Macrophages themselves play important roles in atherosclerosis, as well as acute and chronic heart failure. Although the role of macrophages in CVD pathophysiology is well-recognized, little is known regarding the precise mechanisms influencing their function in these contexts. Long non-coding RNAs (lncRNAs) have emerged as significant regulators of macrophage function; as such, there is rising interest in understanding how these nucleic acids influence macrophage signaling, cell fate decisions, and activity in health and disease. In this review, we summarize current knowledge regarding lncRNAs in directing various aspects of macrophage function in CVD. These include foam cell formation, Toll-like receptor (TLR) and NF-kβ signaling, and macrophage phenotype switching. This review will provide a comprehensive understanding concerning previous, ongoing, and future studies of lncRNAs in macrophage functions and their importance in CVD.
1. Introduction
Macrophages are a subtype of immune cells that comprise an important fraction of the innate immune system. Initially described by Ilia Metchnikoff in 1882, the macrophage was identified as a cell capable of ingesting particles and/or other cells in a process termed phagocytosis—a function heralded as a protective mechanism against pathogens [1]. Since then, our knowledge regarding the role of macrophages in human physiology and pathology has substantially evolved [2]. Macrophages are actively recruited to various tissues during early development to facilitate organ development and function [3,4]. Tissue-resident macrophages are deposited in two independent waves during embryonic development. First, a transient hematopoietic wave of erythro-myeloid progenitors (EMPs) emerges from the posterior plate mesoderm and forms yolk sac blood islands. These yolk sac-derived macrophages are formed independently of hematopoietic stem/progenitor cells and monocytes. They are called “primitive macrophages” due to their immature immunophenotype (i.e., absence of typical macrophage markers) and lack of detectable phagocytic activity. The second wave starts when EMPs seed fetal liver and differentiate to fetal monocytes and further form macrophages. Thus, tissue-resident macrophages deposited during embryogenesis are composed of yolk sac “primitive macrophages” and fetal liver monocyte-derived macrophages [4,5,6,7].
Many tissue macrophages deposited in organs during development remain in adult tissues. These tissue-resident macrophages have a distinct phenotype and function from monocyte-derived macrophages, which are recruited to tissues and organs during injury and/or infection [4,8]. Tissue-resident macrophages are highly abundant in all organs, including the skin, gastrointestinal tract, lymphoid organs, and lungs. Macrophage populations are aptly named according to the tissue in which they reside, as well as their function; for example, Kupffer cells refer to macrophages in the liver, microglia in the brain, and alveolar macrophages in the lung [4,9]. In general, all macrophages share several important functions: (1) they play a role in tissue development and maintenance; (2) tissue surveillance and sampling; and (3) pathogen clearance, antigen presentation, inflammation resolution, and tissue repair. Depending on the organ or tissue localization, macrophages may fulfill several of these tasks and, as such, comprise highly heterogeneous cell populations with various phenotypes and functions [9]. Macrophages are also known for their plasticity, as they may undergo phenotype switching in response to various stimuli [4,10]. For example, lipopolysaccharide (LPS) exposure stimulates macrophage activation and the production of proinflammatory cytokines, while interleukin 4 (IL-4) stimulation elicits anti-inflammatory cytokine secretion [11]. Although many signaling pathways have been identified that direct macrophage polarization and function, it is not fully understood how such pathways are regulated; however, RNA-mediators, such as long non-coding RNAs (lncRNAs), have emerged as important signaling components dictating various aspects of macrophage biology [12,13,14]. To this end, here, we review the current knowledge regarding the regulation of macrophage function by lncRNAs in the context of cardiovascular disease (CVD). Compared to other similar review articles in recent years [15,16], we summarize current knowledge regarding lncRNAs in directing various aspects of macrophage functions in CVD, including foam cell formation, Toll-like receptor (TLR) and NF-kβ signaling, and macrophage phenotype switching.
2. Role of Macrophage in Cardiovascular Disease
CVD broadly describes a class of diseases that affects the heart and/or blood vessels. A host of risk factors (including high blood pressure/hypertension, smoking, diabetes and obesity, and sedentary lifestyle) have been identified as major contributors to CVD. Such risk factors contribute to the accumulation of fats, cholesterol, and inflammatory cells in coronary vasculature, which can lead to plaque formation and blood flow restriction—a pathology commonly referred to as atherosclerosis. The role of macrophages in the pathogenesis of atherosclerosis has been well-established. Much of what is now known regarding macrophages in this process is derived from preclinical animal models of atherosclerosis and clinical cohorts [17,18,19,20]. Animal models include mice deficient in apolipoprotein E (ApoE) supplemented with a Western-style diet. In these models, elevated fat and sugar levels trigger an emergency-mediated hematopoietic response in the bone marrow and spleen, resulting in the overproduction of monocytes [19,21,22]. The surplus of splenic- and bone marrow-derived monocytes is released into the circulation, where they adhere to the atherosclerotic endothelium and extravasate into lesions, where they differentiate into macrophages. The combinatorial accumulation of monocyte-derived macrophages and lipids in the perivascular space leads to the retention of lipid droplets within the macrophage’s cytoplasm, giving them the characteristic appearance of foam-like structures. The accumulation of foam cells in arterial walls is a hallmark of early atherosclerotic lesion formations [19,20]. An unremitting accumulation of monocytes and their lineage descendant macrophages can contribute to fibrous cap thickening, hematoma, thrombi, calcification, and the degeneration of plaque integrity. The relative abundance of macrophages in atherosclerotic plaques is regulated by their exit and/or death but, also, the sustained recruitment of monocytes. An efficient macrophage exit, or reduced monocyte recruitment, results in a reduction in the lesional macrophage number and regression of the disease. On the other hand, factors contributing to increased monocyte recruitment accelerate atherosclerosis [23,24].
The destabilization and rupture of arterial plaque may lead to partial or complete occlusion of the coronary artery, which can lead to acute cardiac ischemic events, such as myocardial infarction (MI). Ischemic injury to the myocardium triggers a sterile systemic inflammatory response that is required for the activation of the tissue-healing program [25,26,27]. In response to MI, the bone marrow and spleen generate a surplus of myeloid cells, mostly neutrophils and monocytes, which are released to the peripheral blood and, subsequently, home to the damaged myocardium [28,29]. Recruited neutrophils and monocytes are necessary for the removal of necrotic tissue, the initialization of angiogenesis, and the stimulation of myofibroblasts for collagen synthesis and wound healing [27]. This initial myocardial myeloid response is imperative for proper healing. A reduction of neutrophil or monocyte/macrophage infiltration, either through systemic depletion or splenectomy, impairs optimal post-MI healing and results in progressive myocyte death, excessive fibrosis, and possible cardiac rupture [29,30]. On the other hand, excess neutrophils and monocyte/macrophage recruitments lead to an abundant secretion of proinflammatory mediators that negatively affect tissue healing and contribute to myocardial damage [31]. Thus, a balanced immune response due to ischemic injury is necessary for proper infarct healing and recovery after MI.
On the other hand, despite the apparent beneficial effects of myeloid cells on infarct healing, macrophages also contribute to the progression of fibrosis and impaired pump functions in chronic heart failure. Following MI, the heart experiences significant cell death and tissue necrosis. The myocardium is then rapidly overwhelmed by an intense inflammatory response that facilitates both the removal of dead/necrotic tissue, as well as supports tissue replacement with an akinetic, collagen-based scar [25,27]. This remodeling process is an adaptive response that serves to preserve the structural integrity of the myocardium after ischemia-mediated tissue loss. However, the remodeling process, characterized by ongoing collagen deposition (defined as reactive fibrosis) and reduced ventricular compliance, is considered maladaptive in the long term—contributing to sustained myocyte dropout, cardiac hypertrophy, and progressive decrements in ventricular function, which eventually culminate in end-stage heart failure [32]. Systemic inflammation is considered a hallmark feature of chronic heart failure in patients [25,26,27]. Proinflammatory cytokine levels, such as tumor necrosis factor-alpha (TNF-α) and IL-6, are closely associated with the heart failure status, suggesting that inflammatory cytokine signaling contributes to progressive pump failure. Likewise, studies in mice models of heart failure demonstrate that, after MI, there is a sequential (4-8 weeks after MI events) accumulation of macrophages in nonischemic regions of the heart, which may provoke continued matrix deposition, myocardial stiffening, and impaired ventricular performance [32,33]. Compatible with this view, the inhibition of monocyte infiltration via the blockade of adhesion molecules, or splenectomy in mice with established heart failure, dampens and improves adverse cardiac remodeling [32,33]. These findings highlight a potentially detrimental role for macrophages in chronic heart failure. While the precise molecular mechanisms underlying macrophage development, polarization, and biologic activity/function in atherosclerosis and heart failure pathophysiology remain a focus of intense scientific investigation, much remains unknown. Nevertheless, recent studies have brought attention to the importance of lncRNAs in macrophage biology—providing evidence that they actively participate in gene regulatory networks during complex biological processes.
3. Non-Coding RNA Nomenclature
Though lncRNAs are broadly defined as any non-protein-coding RNAs whose lengths are longer than 200 nucleotides, they are ultimately stratified according to their genomic position in relation to nearby protein-coding genes (i.e., messenger RNAs (mRNAs)). If a lncRNA is located on the same genomic DNA strand as a nearby protein-coding gene, it is defined as a sense lncRNA. In many instances, a sense lncRNA may overlap with an adjacent protein-coding gene; these lncRNAs are referred to as sense-overlapping lncRNAs. If a lncRNA is located on an opposite genomic DNA strand relative to a neighboring protein-coding gene, it is defined as an antisense lncRNA. Both sense and antisense lncRNAs are commonly named after their nearest protein-coding genes [34]. A lncRNA can also arise from intronic sequences of protein-coding genes, which are aptly called intronic lncRNA. Furthermore, a lncRNA can be distally located far from, but between, two protein-coding genes. Such types of lncRNAs are defined as long intergenic lncRNAs (lincRNAs). When lincRNAs are located in the enhancer or promoter region of a protein-coding gene, these lncRNAs are defined as enhancer- or promoter-associated lincRNAs (eRNAs and pRNAs, respectively). More recently, a new type of lncRNA was defined, which are called circular RNAs (circRNAs). These derive from splicing events and possess stable circular conformations. Although lncRNAs are subdivided based on their genomic locations and in conjunction with nearby protein-coding genes, the definition of non-protein-coding genes remains contested, as some lncRNAs encode micropeptides, as in the case of Myoregulin (MLN) [35]. Furthermore, pseudogenes, whose genomic sequences are similar to functional protein-coding genes, should be included in the category of lncRNAs, as pseudogenes do not encode for proteins [36].
4. Mechanism of lncRNA Function
As many lncRNAs are not known to possess any intrinsic catalytic activity themselves, it is believed that lncRNAs influence biological processes by interacting with macromolecules (e.g., DNA, RNA, and proteins). Since the number of lncRNAs well-surpasses that of protein-coding genes, the categorization of lncRNA functions is challenging. Nevertheless, four broad categories of lncRNA functions were defined. The first category is genomic imprinting, which is an epigenetic phenomenon whereby genes are expressed in a parent-of-origin-dependent manner [37]. Such imprinting lncRNAs include the antisense of IGF2R non-protein coding RNA (Airn) [38], H19-imprinted maternally expressed transcript (H19) [39,40], maternally expressed 3 (MEG3) [41], maternally expressed 8, small nucleolar RNA host gene (MEG8) [42], and X-inactive specific transcript (XIST) [43]. The second category of lncRNA functions is transcription regulation. LncRNAs can directly bind to genomic sequences and form a scaffold for the recruitment of epigenetic and/or transcriptional factors, which influence transcriptional activity [44]. This functional category has been investigated extensively in recent years, as many binding partners (i.e., epigenetic and transcription factors) have been studied in the field of epigenetics. Furthermore, the methods to investigate such mechanisms (i.e., chromatin immunoprecipitation followed by sequencing (ChIP-seq)) are well-established, which makes it easier to elucidate the function of lncRNAs in this context. However, several laboratories have reported the promiscuity of RNA binding of epigenetic factors (especially, enhancer of the zeste homolog 2 (EZH2), a catalytic component of the polycomb repressive complex 2 (PRC2) [45,46,47,48]), suggesting that caution be taken when interpreting lncRNAs as transcriptional regulators. The third category of lncRNA functions is that of post-transcriptional regulation, where their complementation with mRNAs may influence RNA stability. Further, one should note that some lncRNAs encode microRNAs (miRNAs) themselves in their genomic sequences, and others can bind miRNAs and function as miRNA sponges (also known as competing endogenous RNA (CeRNA)) [49]. This is an attractive mechanism to control the availability of miRNAs, as miRNAs bind to the 3’-untranslated regions (3’-UTR) of target mRNAs to induce mRNA degradation and translational repression. However, some lncRNAs (e.g., tumor protein p53 pathway corepressor 1 (TRP53COR1, also known as lincRNA-p21), HOX transcript antisense RNA (HOTAIR), and metastasis-associated lung adenocarcinoma transcript 1 (MALAT1)) are degraded by miRNAs rather than functioning as miRNA sponges [50]; thus, a thorough investigation of the interactions between lncRNAs and miRNAs is necessary. The last category of lncRNA functions is translation control. Through binding to RNA-binding proteins (RBPs), lncRNAs can influence the translational efficiency of mRNAs. As more and more RBPs are being identified from high-throughput screening technologies [51,52,53], there is a growing number of lncRNAs identified as RBP sponges, including those of circRNAs [54]. Thus, just as there exist multifunctional proteins, many lncRNAs, too, exhibit versatility in terms of function—which highlights a likely complex and multidimensional role for lncRNA signaling in immune cell biology.
5. Role of Macrophage lncRNA in Foam Cell Formation
The hallmark feature of atherosclerosis is the formation of atherosclerotic plaques in the intimal layer of the artery [55,56]. At the early stages of atherosclerosis, low-density lipoproteins (LDLs) pass through arterial endothelial junctions and become retained in the subendothelial space via interactions with matrix proteoglycans. Trapped LDLs aggregate and become oxidized to form oxidized low-density lipoproteins (ox-LDLs), which divert their binding from LDL receptors to scavenger receptors (SRs) expressed on monocytes and their descendant macrophages. This interaction ultimately results in the recruitment of monocyte/macrophage populations to the intima of the arterial wall [57]. These SRs include CTD-associated factor 1 (SCAF1, also known as SR-A1), cluster of differentiation 36 (CD36), and lectin-like ox-LDL receptor-1 (LOX-1), which are upregulated after stimulation with ox-LDLs or pro-inflammatory stimuli [58]. After binding to macrophage SRs, ox-LDLs are internalized and transported to lysosomes. There, lipoprotein cholesteryl esters (CE) are hydrolyzed by lysosomal acid lipase (LAL) to generate free cholesterol (FC) [59]. FC are then trafficked from lysosomes through the coordinated action of Niemann-Pick Type C1 and 2 (NPC1 and 2) membrane proteins and, eventually, transported to endoplasmic reticulum (ER), where they become converted back to CE by Acyl-CoA cholesterol acyltransferase (ACAT) through a process of re-esterification. FC can also be transported out of cells by ATP-binding cassette transporters A1 (ABCA1) to form new high-density lipoprotein (HDL) particles [60]. Excess accumulation of CE in lysosomes and the ER leads to the formation of lipid droplets in the macrophage with the appearance of foam-like structures, commonly referred to as macrophage-derived foam cells (Figure 1) [55,56]. Further, CE and FC accumulation leads to the inhibition of LAL and ABCA1 activity, which is believed to contribute to the pathogenesis of atherosclerosis. The main cells involved in the process of foam cell formation include smooth muscle cells, endothelial cells, and macrophages [55,56]. Several methods have been used to identify lncRNAs involved in the pathogenesis of atherosclerosis. Patient-sourced samples (including atherosclerotic plaques and blood samples), as well as animal models of atherosclerosis, have facilitated the identification of numerous candidate lncRNAs (discussed in 5.1-3) [61,62,63]. Additionally, large numbers of lncRNAs involved in atherosclerosis were identified during foam cell formations in vitro, an approach that effectively recapitulates the accumulation of lipid droplets and acquisition of a macrophage foam cell-like phenotype through the incubation of macrophages with ox-LDL [62,64].
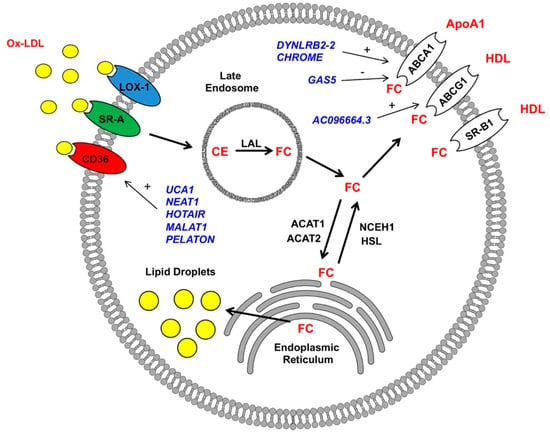
Figure 1.
Long non-coding RNAs (LncRNAs) regulate macrophage-derived foam cell formations. LncRNAs (blue) have positive (+) and negative (-) impacts on the expressions of scavenger receptor CD36 and reverse cholesterol transporters ABCA1 and ABCG1 in macrophages (details in text). Ox-LDL: oxidized low-density lipoproteins, HDL: high-density lipoproteins, FC: free cholesterol, LAL: lysosomal acid lipase, and CE: cholesteryl esters.
5.1. Cholesterol Update—Regulation of Scavenger Receptor Expression
The first step in forming macrophage-derived foam cells involves cholesterol uptake. Three key scavenger receptors were described in macrophages to bind and internalize ox-LDL. As mentioned previously, these are CD36, SR-A1, and LOX-1 [55,56]. CD36 belongs to the scavenger receptor class B family. It consists of an extracellular loop domain with two transmembrane and two cytoplasmic domains. CD36 is a high-affinity ox-LDL receptor that is highly expressed in macrophages [65,66]. The screening utilizing ox-LDL-stimulated macrophages in vitro has resulted in the identification of three candidates lncRNAs that regulate CD36 receptor expression via acting as miRNA sponges. Hu et al. [67] found that the lncRNA urothelial cancer-associated 1 (UCA1) was upregulated in the human monocytic cell line (THP-1 macrophages) after stimulation with ox-LDLs. UCA1 knockdown inhibited the expression of CD36 and limited foam cell formation in response to ox-LDL. The overexpression of miR-206, identified as a predicted target of UCA1, decreased oxidative stress induced by ox-LDL and was reversed by UCA1 upregulation in vitro, suggesting that UCA1 regulates CD36 expression via miR-206 sequestration (“sponging”) [67]. In an analogous study, Wang et al. [68] found that ox-LDL-stimulated THP-1 macrophages yielded an increased expression of the lncRNA nuclear paraspeckle assembly transcript 1 (NEAT1) and a decreased expression of miR342-3p. The inhibition of NEAT1 in THP-1 macrophages stimulated with ox-LDL repressed the expression of CD36 and cholesterol accumulation. Additionally, a decreased expression of NEAT1 led to repression of the inflammatory gene expression program (cytochrome c oxidase subunit II (COX2), interleukin 6 (IL6), interleukin 1 beta (IL1B, also known as IL-1β), and tumor necrosis factor (TNF, also known as TNF-α)) in vitro. Bioinformatics screening identified miR342-3p as a direct target of NEAT1, indicating its potential sponging activity. In further studies, the overexpression of miR-342-3p impaired ox-LDL-induced foam cell formation through the downregulation of CD36 expression [68]. Independently, Chen et al. [69] found that NEAT1 is upregulated in the mouse RAW264 macrophage cell line stimulated with ox-LDLs, which coincided with an upregulation of CD36. Mechanistically, NEAT1 was shown to sponge miR-128, which led to the upregulation of CD36. On the contrary, NEAT1 knockdown restored the expression of miR-128 and reduced the expression of CD36 in response to ox-LDL stimulation [69]. In a similar study, Liu et al. [70] found the lncRNA HOTAIR to regulate CD36 expression, cholesterol uptake, reactive oxygen species (ROS) production, and proinflammatory transcriptional program activation in THP-1 cells stimulated with ox-LDL via the sponging of miR330-5p. These studies show that the expression of CD36 is subject to significant lncRNA-mediated regulation and further provides evidence that this mechanism involves the lncRNA-mediated sequestration of numerous downstream miRNA targets; precisely how those key downstream miRNA targets regulate CD36 expression was not addressed [70].
Differential mechanisms for the lncRNA-mediated regulation of CD36 expression were noted in a study by Huangfu et al. [71]. There, the lncRNA, MALAT1, was identified as a binding partner of β-catenin. An increased MALAT1 expression in response to ox-LDL led to an enhanced β-catenin binding to the CD36 receptor promoter, which was reversed by the inhibition of MALAT1 [71]. In another study, which employed an RNA sequencing-based approach, identified LINC01272 as a highly expressed lncRNA in patient-derived atherosclerotic plaques [67]. As LINC01272 was markedly upregulated in unstable plaques, it was aptly named the “plaque-enriched lncRNA in atherosclerotic and inflammatory bowel macrophage regulation” (PELATON). PELATON was notably enriched in macrophages and exclusively localized in the nuclear fraction, implicating a likely role in the regulation of gene transcription. As anticipated, in vitro experiments demonstrated that PELATON knockdown effectually impaired macrophage phagocytosis, lipid uptake, and reactive oxygen species production, as well as CD36 expression. Detailed mechanistic studies providing greater insights into the PELATON-mediated regulation of CD36 were not conducted [72]. Regardless of the many advancements in RNA biology to date, no known studies are investigating the impact of lncRNAs on those other receptors (e.g., SR-A1 and LOX-1) in influencing foam cell formation. However, recent studies have begun to focus on the contributions of endothelial and smooth muscle cells to foam cell formation from the perspective of SR-A1 and LOX-1-dependent signaling [73,74,75].
5.2. Reverse Cholesterol Transport – ABCA1 and ABCG1 Regulation
In addition to cholesterol uptake by scavenger receptors, the balance of FC and CE is also a critical factor in the regulation of cholesterol content in macrophage-derived foam cells. In homeostasis, after internalization, lipoproteins are delivered to the late endosome, where CE is hydrolyzed into FC by lysosomal acid lipase (LAL). Subsequently, FC is transported to the ER, where it is re-esterified by acetyl-CoA acetyltransferase 1 (ACAT1) and then stored in the form of lipid droplets [55,56]. When the flux of FC from lysosomes is high, oxysterol levels, including 27-hydroxycholesterol, are increased and bind to the nuclear liver X receptor (LXR). This interaction leads to the activation of a transcriptional program that directs the removal of cholesterol from cells via the initiation of reverse cholesterol transport [76]. One of these includes the ATP-binding cassette transporter ATP-binding cassette subfamily A member 1 (ABCA1). This promotes the cholesterol efflux to Apolipoprotein A1 (APOA1), which is the rate-limiting step in the formation of HDL. In addition to ABCA1, two other transporters are involved in reverse cholesterol transport: ATP-binding cassette subfamily G member 1 (ABCG1) and scavenger receptor class B member 1 (SCARB1, also known as SR-BI) [60,62,64]. During the initial steps of atherosclerosis, an increased uptake of cholesterol and defects in cholesterol efflux ultimately results in the accumulation of lipid droplets and, in turn, foam cell formation [55,56].
ABCA1 is a member of the larger superfamily of ABC transporters. It is well-established that ABCA1 plays a critical role in the prevention of macrophage foam cell formation. ABCA1 mutants yield reduced HDL cholesterol levels and heightened atherosclerotic burdens compared with the controls [77]. The expression of ABCA1 is highly regulated at the transcriptional and post-transcriptional levels. At the transcriptional level, several nuclear receptors, including peroxisome proliferator-activated receptors (PPARs), liver X-receptor (LXR), and farnesoid X receptor (FXR), influence ABCA1 expression. Moreover, at the post-transcriptional level, the 3′-UTR of the ABCA1 gene is directly targeted by multiple miRNAs, including miR-33, miR-758, miR-145, miR-27, miR-144, miR-26, and miR-106, to inhibit the cholesterol efflux [78,79]. Besides the above microRNAs, recent observations below suggest that lncRNAs play an active role in the dysregulation of cholesterol efflux from macrophages and foam cell formation.
Studies from three independent laboratories reported that long intergenic non-protein-coding RNA 1228 (LINC01228, also known as DYNLRB2-2) is upregulated in macrophages in response to ox-LDL. This lncRNA was associated with an enhanced cholesterol efflux and reduced macrophage-derived foam cell formation—findings that were ultimately shown to involve three distinct mechanisms [80,81,82]. In the study by Li Y et al. [80], THP-1 macrophages stimulated with ox-LDL exhibited elevated expression of DYNLRB2-2; the lncRNA promoted cholesterol efflux and inhibited foam cell formation through the activation of autophagy. Mechanistically, DYNLRB2-2 modulated the miR-298/Sirtuin 3 (SIRT3) axis, which subsequently resulted in the LKB1/AMPK/mTOR signaling pathway-mediated initiation of an autophagic program in macrophages [80]. This mechanism supports previous observations indicating that autophagy (in this case, termed “lipophagy”) is a key mechanism of CE reverse transport from lipid droplets to lysosomes and the eventual ABCA1 transporter-mediated efflux [83]. Consistent with this idea, Li Y et al. [81] also provided evidence that elevated DYNLRB2-2 in ox-LDL-stimulated THP-1 and RAW264.7 macrophages is associated with alterations in the ABCA1 expression and cholesterol efflux in vitro. Specifically, DYNLRB2-2 overexpression inhibited the macrophage-derived foam cell formation, a finding that was accompanied by an enhanced cholesterol efflux and ABCA1 expression. Further, this study indicated that DYNLRB2-2 negatively regulated the Toll-like receptor 2 (TLR2) expression. The TLR2 overexpression reversed the effects of DYNLRB2-2 on the cholesterol efflux and ABCA1 expression levels in THP-1 and RAW264.7 macrophages [81]. These data are consistent with reports that macrophage TLR-2 is a protagonist of foam cell formation and atherosclerosis [84]. In another study [82], Hu et al. revealed that the DYNLRB2-2-mediated upregulation of ABCA1 in ox-LDL-stimulated THP-1 macrophages requires G protein-coupled receptor 119 (GPR119). Supporting this, the in vitro overexpression of GRP119 increased the cholesterol efflux, inhibited foam cell formation, and activated a proinflammatory genetic program. Furthermore, the in vivo viral overexpression in high-fat fed ApoE-/- mice (a murine model of atherosclerosis) showed that GRP119 has a protective effect against atherosclerosis via increasing the cholesterol efflux and reducing the proinflammatory cytokine expression. These studies ultimately suggest that DYNLRB2-2 is a promising therapeutically exploitable target to increase cholesterol homeostasis and reduce atherosclerotic plaque formation.
Another lncRNA, termed the “cholesterol-induced regulator of metabolism RNA” (CHROME), was demonstrated to be an imperative component of proper cholesterol homeostasis. Hennessy et al. [85] identified CHROME as elevated in the plasma and atherosclerotic plaques of individuals with coronary artery disease. CHROME expression is influenced by dietary and cellular cholesterol via the sterol-activated liver X receptor transcription factors, which control gene-mediating responses to cholesterol overloads. Using gain- and loss-of-function approaches, the authors show that CHROME promotes cholesterol efflux and foam cell formation, most likely through the regulation of miRNA expression. CHROME knockdown in human macrophages increases the levels of miR-27b, miR-33a, miR-33b, and miR-128, thereby reducing the expressions of their overlapping target gene networks, including ABCA1. Since CHROME is also expressed in hepatocytes, it can play an important role in systemic cholesterol homeostasis in atherosclerosis [85].
Macrophage-expressed lncRNAs can also have detrimental effects on foam cell formation through the inhibition of ABCA1. In the study by Meng et al. [86], the lncRNA growth arrest-specific 5 (GAS5) was highly expressed in the THP-1 macrophage-derived foam cells and localized primarily to the nucleus. The overexpression of GAS5 facilitated lipid accumulation and foam cell formation. Further studies showed that GAS5 inhibits ABCA1 by binding to the histone methyltransferase EZH2. The in vitro overexpression of EZH2 reduced the cholesterol efflux and facilitated lipid accumulation. ApoE-/- mice with overexpression of GAS5 or EZH2 showed increased total cholesterol, free cholesterol, cholesterol ester, low-density lipoprotein levels, aortic plaque, and lipid accumulation, accompanied by reduced HDL levels and cholesterol outflow. These data suggest GAS5 as a promising target to restore cholesterol homeostasis and inhibit atherosclerotic plaque formation in patients [86].
Numerous studies show that ABCG1 and SR-BI are also key components of cholesterol homeostasis in macrophages. In a recent study by Xu et al. [87], authors show that THP-1 macrophages and vascular smooth muscle cells (VSMC) stimulated with ox-LDL express a novel lncRNA, AC096664.3. Further studies in VSMC demonstrated that AC096664.3 regulates the expression of ABCA1 via the inhibition of PPAR-γ expression. However, these findings were not confirmed in macrophages. As of late, no lncRNAs have been found to regulate scavenger receptor class B type 1 (SCARB1, also known as SR-BI).
6. Role of lncRNA in the Regulation of Macrophage Polarization
In addition to foam cell formation, macrophages may also contribute to atherosclerosis pathophysiology via the secretion of a broad spectrum of pro- and anti-inflammatory cytokines. Monocytes recruited to the sites of inflammation (e.g., atherosclerotic plaque or infarcted heart) differentiate to macrophages, which engage in shaping the inflammatory response through the secretion of cytokines, chemokines, and reactive oxygen species (ROS) [19,20]. Depending on the inflammatory milieu, tissue-resident macrophages can acquire a pro-inflammatory (M1) or anti-inflammatory/pro-resolving phenotype (M2) [4,10]. Naïve macrophage exposure to danger-associated molecular patterns (DMAPs), oxidized low-density lipoproteins (ox-LDLs), or interferon-gamma (IFN-γ) initiate the activation of an M1 phenotype, which leads to the secretion of proinflammatory cytokines IL-1, IL-6, tumor necrosis factor-alpha (TNF-α), IL-6, and interleukin 12 (IL-12) [57,88]. These cytokines perpetuate non-resolving inflammation and contribute to the pathogenesis of CVD, including atherosclerosis and heart failure. Toll-like receptor (TLR) stimulation leads to the activation of signaling pathways, culminating in proinflammatory gene activation—a chief characteristic of the M1 phenotype (Figure 2) [57,89]. Alternatively, stimulation with interleukin 4 (IL-4) and interleukin 13 (IL-13), as well as exposure to apoptotic cells and the activation of engulfment through TAM receptors (i.e., Tyro3, Axl, and Mer), can activate an anti-inflammatory and pro-resolving M2 phenotype in macrophages [57,89]. This results in the secretion of anti-inflammatory cytokines (IL-10 and TGFβ) but, also, pro-resolving lipid mediators (e.g., resolvins, maresins, and lipoxins), which are associated with the pro-reparative and anti-inflammatory actions of macrophages [90,91,92]. Recent literature suggests that lncRNAs are involved in the modulation of the macrophage phenotype, hence their likely involvement in the pathogenesis of CVD.
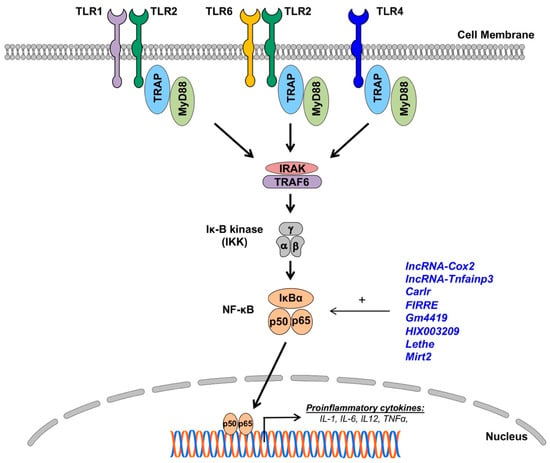
Figure 2.
The impact of lncRNAs on Toll-like receptor (TLR) expressions and nuclear factor (NF)-κB signaling in macrophages. LncRNAs (blue) enhance (+) NF-κB signaling and proinflammatory gene expressions in macrophages (details in text).
6.1. The Role of TLR Signaling in Macrophage Polarization
TLRs are the most extensively studied and characterized pattern-recognition receptors involved in CVD. To date, 13 TLRs have been discovered and characterized. They can be broadly divided into two categories: cell membrane TLRs (TLR1, TLR2, TLR4, TLR5, TLR6, and TLR11) and cytoplasmic TLRs residing in ER, endosomes, and lysosomes, which are nucleic acid-sensing receptors (TLR3, TLR7, TLR8, TLR9, and TLR13). Recognition of the ligands results in TLR dimerization that activates intracellular Toll/interleukin-1 receptor/resistance protein (TIR) domains to enable the recruitment of adapter proteins like myeloid differentiation primary response 88 (MyD88), TIR-domain-containing adapter-inducing interferon-β (TRIF), transforming growth factor-β-activated kinase 1 (TAK1; official gene name, mitogen-activated protein kinase kinase kinase 7 (MAP3K7)), and mal T cell differentiation protein (MAL) [93,94,95,96]. All TLRs, except for TLR3, signal via the adapter protein MyD88, which recruits interleukin-1 receptor-associated kinases (IRAKs) to activate nuclear factor-κB (NF-κB)-dependent proinflammatory cytokines (all TLRs) and IRF7-dependent type I IFN (TLR7-9). TLR4 activates the MyD88-dependent pathway through the activation of the endosomal TIR domain-containing adapter-inducing interferon-β (TRIF)-dependent pathway to activate the expression of type 1 IFNs. TLR3 exclusively uses the TRIF-dependent pathway for downstream signaling. The activation of TLR signaling is associated with M1 macrophage phenotype and pro-inflammatory cytokine profiles (Figure 2) [93,94,95,96]. Thus, it will have a detrimental effect on the progression of CVD.
The participation of the TLR signaling pathways in the pathogenesis of CVD is well-studied in both CVD patients and small animal models [94]. It has been recognized that the pattern of TLR expression changes in CVD patients [97]. Studies in knockout animals have established that TLR signaling plays an active role in the pathogenesis of both atherosclerosis and heart failure [94,97]. Mice with a deficiency of TLR2 and TLR4 or downstream signaling proteins (i.e., IRAK4, TRAF6, TRIF, or MYD88) shows protection from atherosclerosis and various models of heart failure [94,96,98,99]. Therefore, understanding the molecular mechanisms regulating TLR signaling could lead to the discovery of new therapeutic targets. Since TLR signaling is regulated at multiple levels (including TLR expression) and epigenetic and post-translational mechanisms, it is possibly influenced by lncRNAs.
6.2. LncRNA Involved in the Macrophage TLR Activation and Signaling
The growing interest in lncRNAs has prompted the identification of lncRNAs that are involved in TLR signaling. For example, the lncRNA AS-IL1a, encoded within the IL-1α locus, is required for the recruitment of RNA polymerase II to the IL-1α promoter for its transcription in mouse primary bone marrow-derived macrophages [100]. Another example is lnc-MARCKS/ROCKI (Regulator of Cytokines and Inflammation), which interacts with the multifunctional DNA repair enzyme, apurinic/apyrimidinic endodeoxyribonuclease 1 (APEX1), to form a ribonucleoprotein complex and recruit histone deacetylase 1 (HDAC1) to remove the active enhancer mark, histone H3 acetylated at lysine 27 (H3K27ac). The removal of the H3K27ac mark results in the reduced expression of the most prominent cellular substrate for protein kinase C, myristoylated alanine-rich protein kinase C substrate (MARCKS), thereby regulating the expression of proinflammatory cytokines in macrophages [101].
To date, most loss-of-function experiments employed to elucidate the functions of lncRNAs are based on the usage of knockdown methods (e.g., antisense oligos (ASO), shRNAs, and siRNAs). As it is well-known that such acute silencing is associated with technical limitations, including off-target effects [102], the gold standard in developmental biology is to use knockout mice [103,104,105]. To address this point, Atianand et al. genetically deleted the entire 4-kb genomic locus harboring the lncRNA Ttc39a opposite strand RNA 1 (Ttc39aos1, also known as lincRNA-EPS) to generate lincRNA-EPS−/− mice [106]. Using macrophages isolated from lincRNA-EPS-deficient mice, the authors uncovered that lincRNA-EPS interacts with heterogeneous nuclear ribonucleoprotein L (hnRNPL) to suppress the transcription of immune response genes (i.e., interferon-induced protein with tetratricopeptide repeats 2 (Ifit2) and radical S-adenosyl methionine domain containing 2 (Rsad2)).
Besides the transcriptional control above, the post-transcriptional regulation of TLR signaling via lncRNAs are reported. The most prominent ones are those investigating ceRNAs/miRNA sponges to sequester miRNAs to interfere with mRNA degradation. For example, the imprinting lncRNA, Maternally Expressed Gene 3 (Meg3; more specifically, MEG3-4 transcript), binds to miR-138, which targets the proinflammatory cytokine IL-1β. Another example is NEAT1, which binds to miR-15a [107] and miR-17-5p [108] to control the translation of TLR4. Taken together, lncRNAs play active roles in regulating TLR signaling in macrophages.
NF-κB signaling is one of the most studied immune pathways since its discovery in 1986 [109]. It consists of five subunits (NF-κB1 p50, NF-κB2 p52, RelA/p65, RelB, and cREel) that can dimerize to form unique transcription factors that can either interact with other gene promoters via κB consensus motifs or with five inhibitory proteins that comprise the IκB family [110]. IκB binding to NF-κB dimers inhibits translocation to the nucleus by occluding DNA-binding Rel proteins and masking the nuclear localization signal region of the NF-κB subunit. The diversity of NF-κB activation is vast, and canonical and noncanonical signaling pathways have been reviewed extensively elsewhere [110,111,112]. NF-κB signaling in macrophages largely follows the same pattern of regulation, albeit mostly on the activation of pattern recognition receptors like TLRs and a heavier reliance on c-Rel as a vital component for the activity of NF-κB dimers [110,112]. NF-κB dimers are henceforth uninhibited by their release from the inhibitory IκB subunit and can be translocated into the nucleus, where they function as a transcription factor for a plethora of inflammatory genes, such as those that make up the well-known NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3) inflammasome, a hallmark of the M1 phenotype [113].
LncRNAs have been found to both upregulate and suppress immune-responsive genes. Several of these lncRNAs have now substantiated roles in the regulation of NF-κB signaling in many human disease models in mice, including CVD [111,114,115,116,117,118,119,120,121,122,123,124]. The crosstalk between lncRNAs and NF-κB signaling is complex, involving both direct and indirect mechanisms and spanning from epigenetic control and even to post-translational modifications. The expression of long intergenic non-coding RNA (lincRNA) prostaglandin-endoperoxide synthase 2, opposite strand 2 (Ptgs2os2, also known as lncRNA-Cox2), is known to be highly induced in macrophages upon TLR activation. Hu et al. [125] found that lncRNA-Cox2 is directly involved in modulating the assembly of NF-κB subunits to the SWI/SNF complex, thereby acting as an epigenetic coactivator of the NF-κB-mediated late-primary response gene transcription in murine macrophage (microglia and RAW264.7) cells [125]. Further evidence for chromatin remodeling via lncRNAs was shown in a study where lincRNA-Tnfaip3 was found in the LPS-stimulated macrophages to regulate high-mobility group box 1 (Hmgb1)-associated histone modification and the subsequent NF-κB-mediated transactivation of pro-inflammatory genes [126]. Other studies in murine bone marrow-derived macrophages demonstrated that the expression of the lncRNA cardiac and apoptosis-related lncRNA (Carlr) is not only directly increased by NF-κB activation but, more importantly, its knockdown results in significantly reduced expression levels of several NF-κB-regulated genes, such as the nuclear factor of the kappa light polypeptide gene enhancer in B cells 2, p49/p100 (Nfkb2), prostaglandin-endoperoxide synthase 2 (Ptgs2), interleukin 1 alpha (Il1a), and interleukin 1 beta (Il1b) [127].
The lncRNA Firre intergenic repeating RNA element (FIRRE) is a species-conserved lncRNA (between humans and mice) that is upregulated by NF-κB signaling. Lu et al. [128] showed an interesting post-transcriptional mechanism whereby FIRRE directly interacts with heterogeneous nuclear ribonucleoproteins (hnRNPs) to stabilize the mRNAs of inflammatory genes (TNF-a, IL1b, and IL6) following LPS stimulation. Similarly, the post-translational modulation of inflammatory cytokines was shown in microglial cells whereby the lncRNA-predicted gene 4419 (Gm4419) was found to be upregulated and promoted the phosphorylation of the inhibitor of nuclear factor kappa B (IκBα) by direct physical association, ultimately leading to the increased transcriptional activation of TNF-α, Il1b, and Il6 [129]. The aforementioned FIRRE has also been shown to form a positive feedback loop to promote the NLR family pyrin domain containing 3 (NLRP3) inflammasome formation in microglial cells [130]. Adding to the complexity of lncRNAs’ roles in inflammatory diseases, the lncRNA HIX003209 was shown to promote macrophage activation in THP-1 cells by acting as a ceRNA for miR-6089 through a TLR4/IκB/NF-κB signaling pathway [131]. The lncRNA Lethe also appears to be important in metabolic diseases, such as diabetes, by regulating the ROS production in RAW264.7 macrophages via NF-κB signaling [132].
The indirect regulation of macrophage functions by lncRNAs and NF-κB crosstalk has also been shown in mice peritoneal macrophages whereby the lncRNA myocardial infarction-associated transcript 2 (Mirt2) acts as a molecular checkpoint to prevent the aberrant activation of a proinflammatory program that potently regulates the macrophage phenotype. Mirt2 is induced in the LPS-stimulated macrophages and inhibits the K-63 ubiquitination of TNF receptor-associated factor 6 (TRAF6) and, thus, attenuates NF-κB signaling via negative feedback. Mirt2 knockdown suppressed the proinflammatory program in LPS-stimulated macrophages [133]. Together, lncRNAs are involved in the regulation of NF-kB functions in macrophages. Increasing our understanding of how the complex lncRNA network interplays with NF-kB signaling would provide new potential molecular targets to limit the proinflammatory activation of macrophages in the pathophysiology of CVD.
7. Conclusions
Recent studies suggest that lncRNAs play a crucial role in the regulation of myeloid recruitment and lineage determination (e.g., the polarization of macrophages to proinflammatory M1 and reparative M2 macrophages), as well as innate and adaptive immune functions. Although significant progress has been made in macrophage biology, little is known regarding lncRNAs in the relationship between macrophages and failing hearts. Furthermore, due to the lack of species conservation of lncRNAs (e.g., between humans and mice), the studies of lncRNAs in model organisms are not well-received in human diseases, including CVD. Adding to this lack of species conservation, most of the recent lncRNA studies fail to provide the detailed characteristics of lncRNAs, including the presence of isoforms and other non-coding RNAs (ncRNAs). For example, one of the most well-studied lncRNA, MEG3, owns 50 human isoforms (Ensemble database accession ENSG00000214548). Within its locus, several other ncRNAs are encoded (i.e., AL117190.1-201, AL117190.3-201, AL117190.7-201, and MIR770-201), which raises a significant caution in interpreting the loss-of-function experimental results. In the case of MEG3, gain-of-function experiments will be difficult to conduct if all 50 MEG3 isoforms are to be cloned and overexpressed in experimental settings. However, it is imperative to conduct more careful studies of the lncRNA locus itself, as well as the validation of isoforms via the rapid amplification of cDNA ends (RACE) and Northern blotting assays to move the lncRNA field forward to avoid the misinterpretation of experimental results due to ambiguity surrounding the functionalities of lncRNAs.
Author Contributions
M.W., J.K., J.B.M.I., and S.U. wrote the manuscript. M.W. and S.U. revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded in part by the NIH Grants P01-HL078825 (M.W.), R01-141191 (M.W.), and R01-HL141081 (J.M.B.); the V.V. Cooke Foundation (S.U.); the Mansbach Foundation (S.U); and the Gheens Foundation (S.U.).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Metschnikoff, E. Ueber den Kampf der Zellen gegen Erysipel-kokken. Arch. Patholog. Anatom. Physiol. Klin. Med. 1887, 107, 209–249. [Google Scholar] [CrossRef]
- Frodermann, V.; Nahrendorf, M. Macrophages and cardiovascular health. Physiol. Rev. 2018, 98, 2523–2569. [Google Scholar] [CrossRef]
- Wysoczynski, M.; Moore, J.B.T.; Uchida, S. A novel macrophage subtype directs hematopoietic stem cell homing and retention. Ann. Transl. Med. 2019, 7, S79. [Google Scholar] [CrossRef]
- Epelman, S.; Lavine, K.J.; Randolph, G.J. Origin and functions of tissue macrophages. Immunity 2014, 41, 21–35. [Google Scholar] [CrossRef]
- Hoeffel, G.; Ginhoux, F. Fetal monocytes and the origins of tissue-resident macrophages. Cell. Immunol. 2018, 330, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Leid, J.; Carrelha, J.; Boukarabila, H.; Epelman, S.; Jacobsen, S.E.; Lavine, K.J. Primitive embryonic macrophages are required for coronary development and maturation. Circ. Res. 2016, 118, 1498–1511. [Google Scholar] [CrossRef] [PubMed]
- Perdiguero, E.G.; Geissmann, F. The development and maintenance of resident macrophages. Nat. Immunol. 2016, 17, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Epelman, S.; Lavine, K.J.; Beaudin, A.E.; Sojka, D.K.; Carrero, J.A.; Calderon, B.; Brija, T.; Gautier, E.L.; Ivanov, S.; Satpathy, A.T.; et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 2014, 40, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Ovchinnikov, D.A. Macrophages in the embryo and beyond: Much more than just giant phagocytes. Genesis 2008, 46, 447–462. [Google Scholar] [CrossRef]
- Wysoczynski, M.; Khan, A.; Bolli, R. New paradigms in cell therapy: Repeated dosing, intravenous delivery, immunomodulatory actions, and new cell types. Circ. Res. 2018, 123, 138–158. [Google Scholar] [CrossRef]
- Mills, C.D. M1 and M2 Macrophages: Oracles of health and disease. Crit. Rev. Immunol. 2012, 32, 463–488. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, I.; Valverde, A.; Ahmad, F.; Naqvi, A.R. Long noncoding RNA in myeloid and lymphoid cell differentiation, polarization and function. Cells 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Robinson, E.K.; Covarrubias, S.; Carpenter, S. The how and why of lncRNA function: An innate immune perspective. Biochim. Biophys. Acta Gene Regul. Mech. 2020, 1863, 194419. [Google Scholar] [CrossRef]
- Scacalossi, K.R.; van Solingen, C.; Moore, K.J. Long non-coding RNAs regulating macrophage functions in homeostasis and disease. Vascul. Pharmacol. 2019, 114, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Gurha, P. Noncoding RNAs in cardiovascular diseases. Curr. Opin. Cardiol. 2019, 34, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Kok, F.O.; Baker, A.H. The function of long non-coding RNAs in vascular biology and disease. Vascul. Pharmacol. 2019, 114, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Nahrendorf, M.; Swirski, F.K. Immunology. Neutrophil-macrophage communication in inflammation and atherosclerosis. Science 2015, 349, 237–238. [Google Scholar] [CrossRef]
- Nahrendorf, M.; Swirski, F.K. Cholesterol, CCR2, and monocyte phenotypes in atherosclerosis. Eur. Heart J. 2017, 38, 1594–1596. [Google Scholar] [CrossRef][Green Version]
- Swirski, F.K.; Nahrendorf, M. Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science 2013, 339, 161–166. [Google Scholar] [CrossRef]
- Swirski, F.K.; Nahrendorf, M.; Libby, P. Mechanisms of myeloid cell modulation of atherosclerosis. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef]
- van der Valk, F.M.; Kuijk, C.; Verweij, S.L.; Stiekema, L.C.A.; Kaiser, Y.; Zeerleder, S.; Nahrendorf, M.; Voermans, C.; Stroes, E.S.G. Increased haematopoietic activity in patients with atherosclerosis. Eur. Heart J. 2017, 38, 425–432. [Google Scholar] [CrossRef]
- Robbins, C.S.; Chudnovskiy, A.; Rauch, P.J.; Figueiredo, J.L.; Iwamoto, Y.; Gorbatov, R.; Etzrodt, M.; Weber, G.F.; Ueno, T.; van Rooijen, N.; et al. Extramedullary hematopoiesis generates Ly-6C (high) monocytes that infiltrate atherosclerotic lesions. Circulation 2012, 125, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Feig, J.E.; Pineda-Torra, I.; Sanson, M.; Bradley, M.N.; Vengrenyuk, Y.; Bogunovic, D.; Gautier, E.L.; Rubinstein, D.; Hong, C.; Liu, J.; et al. LXR promotes the maximal egress of monocyte-derived cells from mouse aortic plaques during atherosclerosis regression. J. Clin. Invest. 2010, 120, 4415–4424. [Google Scholar] [CrossRef] [PubMed]
- Potteaux, S.; Gautier, E.L.; Hutchison, S.B.; van Rooijen, N.; Rader, D.J.; Thomas, M.J.; Sorci-Thomas, M.G.; Randolph, G.J. Suppressed monocyte recruitment drives macrophage removal from atherosclerotic plaques of Apoe₋/₋ mice during disease regression. J. Clin. Invest. 2011, 121, 2025–2036. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Pathophysiology of myocardial infarction. Compr. Physiol. 2015, 5, 1841–1875. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Inflammation in cardiac injury, repair and regeneration. Curr. Opin. Cardiol. 2015, 30, 240–245. [Google Scholar] [CrossRef]
- Prabhu, S.D.; Frangogiannis, N.G. The biological basis for cardiac repair after myocardial infarction: From Inflammation to fibrosis. Circ. Res. 2016, 119, 91–112. [Google Scholar] [CrossRef]
- Heidt, T.; Sager, H.B.; Courties, G.; Dutta, P.; Iwamoto, Y.; Zaltsman, A.; von Zur Muhlen, C.; Bode, C.; Fricchione, G.L.; Denninger, J.; et al. Chronic variable stress activates hematopoietic stem cells. Nat. Med. 2014, 20, 754–758. [Google Scholar] [CrossRef]
- Heidt, T.; Courties, G.; Dutta, P.; Sager, H.B.; Sebas, M.; Iwamoto, Y.; Sun, Y.; Da Silva, N.; Panizzi, P.; van der Laan, A.M.; et al. Differential contribution of monocytes to heart macrophages in steady-state and after myocardial infarction. Circ. Res. 2014, 115, 284–295. [Google Scholar] [CrossRef]
- Horckmans, M.; Ring, L.; Duchene, J.; Santovito, D.; Schloss, M.J.; Drechsler, M.; Weber, C.; Soehnlein, O.; Steffens, S. Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur. Heart J. 2017, 38, 187–197. [Google Scholar] [CrossRef]
- Nahrendorf, M.; Pittet, M.J.; Swirski, F.K. Monocytes: Protagonists of infarct inflammation and repair after myocardial infarction. Circulation 2010, 121, 2437–2445. [Google Scholar] [CrossRef] [PubMed]
- Sager, H.B.; Hulsmans, M.; Lavine, K.J.; Moreira, M.B.; Heidt, T.; Courties, G.; Sun, Y.; Iwamoto, Y.; Tricot, B.; Khan, O.F.; et al. Proliferation and recruitment contribute to myocardial macrophage expansion in chronic heart failure. Circ. Res. 2016, 119, 853–864. [Google Scholar] [CrossRef] [PubMed]
- Ismahil, M.A.; Hamid, T.; Bansal, S.S.; Patel, B.; Kingery, J.R.; Prabhu, S.D. Remodeling of the mononuclear phagocyte network underlies chronic inflammation and disease progression in heart failure: Critical importance of the cardiosplenic axis. Circ. Res. 2014, 114, 266–282. [Google Scholar] [CrossRef]
- Mattick, J.S.; Rinn, J.L. Discovery and annotation of long noncoding RNAs. Nat. Struct. Mol. Biol. 2015, 22, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.M.; Anderson, K.M.; Chang, C.L.; Makarewich, C.A.; Nelson, B.R.; McAnally, J.R.; Kasaragod, P.; Shelton, J.M.; Liou, J.; Bassel-Duby, R.; et al. A micropeptide encoded by a putative long noncoding RNA regulates muscle performance. Cell 2015, 160, 595–606. [Google Scholar] [CrossRef]
- Cheetham, S.W.; Faulkner, G.J.; Dinger, M.E. Overcoming challenges and dogmas to understand the functions of pseudogenes. Nat. Rev. Genet. 2020, 21, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Peters, J. The role of genomic imprinting in biology and disease: An expanding view. Nat. Rev. Genet. 2014, 15, 517–530. [Google Scholar] [CrossRef]
- Latos, P.A.; Pauler, F.M.; Koerner, M.V.; Senergin, H.B.; Hudson, Q.J.; Stocsits, R.R.; Allhoff, W.; Stricker, S.H.; Klement, R.M.; Warczok, K.E.; et al. Airn transcriptional overlap, but not its lncRNA products, induces imprinted Igf2r silencing. Science 2012, 338, 1469–1472. Available online: https://pubmed.ncbi.nlm.nih.gov/23239737/ (accessed on 10 July 2020). [CrossRef]
- Bartolomei, M.S.; Zemel, S.; Tilghman, S.M. Parental imprinting of the mouse H19 gene. Nature 1991, 351, 153–155. [Google Scholar] [CrossRef]
- Rachmilewitz, J.; Goshen, R.; Ariel, I.; Schneider, T.; de Groot, N.; Hochberg, A. Parental imprinting of the human H19 gene. FEBS Lett. 1992, 309, 25–28. [Google Scholar] [CrossRef]
- Miyoshi, N.; Wagatsuma, H.; Wakana, S.; Shiroishi, T.; Nomura, M.; Aisaka, K.; Kohda, T.; Surani, M.A.; Kaneko-Ishino, T.; Ishino, F. Identification of an imprinted gene, Meg3/Gtl2 and its human homologue MEG3, first mapped on mouse distal chromosome 12 and human chromosome 14q. Genes Cells Devot. Mol. Cell. Mech. 2000, 5, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Charlier, C.; Segers, K.; Karim, L.; Shay, T.; Gyapay, G.; Cockett, N.; Georges, M. The callipyge mutation enhances the expression of coregulated imprinted genes in cis without affecting their imprinting status. Nat. Genet. 2001, 27, 367–369. [Google Scholar] [CrossRef]
- Norris, D.P.; Patel, D.; Kay, G.F.; Penny, G.D.; Brockdorff, N.; Sheardown, S.A.; Rastan, S. Evidence that random and imprinted Xist expression is controlled by preemptive methylation. Cell 1994, 77, 41–51. [Google Scholar] [CrossRef]
- Tsai, M.C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long noncoding RNA as modular scaffold of histone modification complexes. Science 2010, 329, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Goodrich, K.J.; Gooding, A.R.; Naeem, H.; Archer, S.; Paucek, R.D.; Youmans, D.T.; Cech, T.R.; Davidovich, C. Targeting of polycomb repressive complex 2 to RNA by short repeats of consecutive guanines. Mol. Cell 2017, 65, 1056–1067.e1055. [Google Scholar] [CrossRef]
- Davidovich, C.; Cech, T.R. The recruitment of chromatin modifiers by long noncoding RNAs: Lessons from PRC2. RNA 2015, 21, 2007–2022. [Google Scholar] [CrossRef]
- Cifuentes-Rojas, C.; Hernandez, A.J.; Sarma, K.; Lee, J.T. Regulatory interactions between RNA and polycomb repressive complex 2. Mol. Cell 2014, 55, 171–185. [Google Scholar] [CrossRef]
- Zhao, J.; Ohsumi, T.K.; Kung, J.T.; Ogawa, Y.; Grau, D.J.; Sarma, K.; Song, J.J.; Kingston, R.E.; Borowsky, M.; Lee, J.T. Genome-wide identification of polycomb-associated RNAs by RIP-seq. Mol. Cell 2010, 40, 939–953. [Google Scholar] [CrossRef]
- Militello, G.; Weirick, T.; John, D.; Doring, C.; Dimmeler, S.; Uchida, S. Screening and validation of lncRNAs and circRNAs as miRNA sponges. Brief Bioinform. 2017, 18, 780–788. [Google Scholar] [CrossRef]
- Yoon, J.H.; Abdelmohsen, K.; Gorospe, M. Functional interactions among microRNAs and long noncoding RNAs. Semin. Cell. Dev. Biol. 2014, 34, 9–14. [Google Scholar] [CrossRef]
- Perez-Perri, J.I.; Rogell, B.; Schwarzl, T.; Stein, F.; Zhou, Y.; Rettel, M.; Brosig, A.; Hentze, M.W. Discovery of RNA-binding proteins and characterization of their dynamic responses by enhanced RNA interactome capture. Nat. Commun. 2018, 9, 4408. [Google Scholar] [CrossRef] [PubMed]
- Baltz, A.G.; Munschauer, M.; Schwanhausser, B.; Vasile, A.; Murakawa, Y.; Schueler, M.; Youngs, N.; Penfold-Brown, D.; Drew, K.; Milek, M.; et al. The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol. Cell 2012, 46, 674–690. [Google Scholar] [CrossRef] [PubMed]
- Castello, A.; Fischer, B.; Eichelbaum, K.; Horos, R.; Beckmann, B.M.; Strein, C.; Davey, N.E.; Humphreys, D.T.; Preiss, T.; Steinmetz, L.M.; et al. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell 2012, 149, 1393–1406. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wang, T.; Xiao, G.; Xie, Y. Large-scale profiling of RBP-circRNA interactions from public CLIP-seq datasets. Genes 2020, 11. [Google Scholar] [CrossRef]
- Tabas, I.; Williams, K.J.; Boren, J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: Update and therapeutic implications. Circulation 2007, 116, 1832–1844. [Google Scholar] [CrossRef] [PubMed]
- Jerome, W.G. Advanced atherosclerotic foam cell formation has features of an acquired lysosomal storage disorder. Rejuvenation Res. 2006, 9, 245–255. [Google Scholar] [CrossRef]
- Tabas, I.; Bornfeldt, K.E. Macrophage phenotype and function in different stages of atherosclerosis. Circ. Res. 2016, 118, 653–667. [Google Scholar] [CrossRef]
- Pirillo, A.; Norata, G.D.; Catapano, A.L. LOX-1, OxLDL, and atherosclerosis. Mediators Inflamm. 2013, 2013, 152786. [Google Scholar] [CrossRef] [PubMed]
- Dubland, J.A.; Francis, G.A. Lysosomal acid lipase: At the crossroads of normal and atherogenic cholesterol metabolism. Front. Cell Dev. Biol. 2015, 3, 3. [Google Scholar] [CrossRef]
- Ouimet, M.; Barrett, T.J.; Fisher, E.A. HDL and reverse cholesterol transport. Circ. Res. 2019, 124, 1505–1518. [Google Scholar] [CrossRef]
- Liu, Y.; Zheng, L.; Wang, Q.; Hu, Y.W. Emerging roles and mechanisms of long noncoding RNAs in atherosclerosis. Int. J. Cardiol. 2017, 228, 570–582. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Ding, J.W.; Wang, X.A.; Zheng, X.X. Long noncoding RNAs and atherosclerosis. Atherosclerosis 2016, 248, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Uchida, S.; Dimmeler, S. Long noncoding RNAs in cardiovascular diseases. Circ. Res. 2015, 116, 737–750. [Google Scholar] [CrossRef] [PubMed]
- Jian, L.; Jian, D.; Chen, Q.; Zhang, L. Long Noncoding RNAs in Atherosclerosis. J. Atheroscler. Thromb. 2016, 23, 376–384. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Bobryshev, Y.V.; Orekhov, A.N. Macrophage-mediated cholesterol handling in atherosclerosis. J. Cell. Mol. Med. 2016, 20, 17–28. [Google Scholar] [CrossRef]
- Collot-Teixeira, S.; Martin, J.; McDermott-Roe, C.; Poston, R.; McGregor, J.L. CD36 and macrophages in atherosclerosis. Cardiovasc. Res. 2007, 75, 468–477. [Google Scholar] [CrossRef]
- Hu, X.; Ma, R.; Fu, W.; Zhang, C.; Du, X. LncRNA UCA1 sponges miR-206 to exacerbate oxidative stress and apoptosis induced by ox-LDL in human macrophages. J. Cell. Physiol. 2019, 234, 14154–14160. [Google Scholar] [CrossRef]
- Wang, L.; Xia, J.W.; Ke, Z.P.; Zhang, B.H. Blockade of NEAT1 represses inflammation response and lipid uptake via modulating miR-342-3p in human macrophages THP-1 cells. J. Cell. Physiol. 2019, 234, 5319–5326. [Google Scholar] [CrossRef]
- Chen, D.D.; Hui, L.L.; Zhang, X.C.; Chang, Q. NEAT1 contributes to ox-LDL-induced inflammation and oxidative stress in macrophages through inhibiting miR-128. J. Cell. Biochem. 2018. [Google Scholar] [CrossRef]
- Liu, J.; Huang, G.Q.; Ke, Z.P. Silence of long intergenic noncoding RNA HOTAIR ameliorates oxidative stress and inflammation response in ox-LDL-treated human macrophages by upregulating miR-330-5p. J. Cell. Physiol. 2019, 234, 5134–5142. [Google Scholar] [CrossRef]
- Huangfu, N.; Xu, Z.; Zheng, W.; Wang, Y.; Cheng, J.; Chen, X. LncRNA MALAT1 regulates oxLDL-induced CD36 expression via activating beta-catenin. Biochem. Biophys. Res. Commun. 2018, 495, 2111–2117. [Google Scholar] [CrossRef] [PubMed]
- Hung, J.; Scanlon, J.P.; Mahmoud, A.D.; Rodor, J.; Ballantyne, M.; Fontaine, M.A.C.; Temmerman, L.; Kaczynski, J.; Connor, K.L.; Bhushan, R.; et al. Novel plaque enriched long noncoding RNA in atherosclerotic macrophage regulation (PELATON). Arterioscler. Thromb. Vasc. Biol. 2020, 40, 697–713. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yang, H.; Li, Y.; Huo, P.; Ma, P. LNCRNA OIP5-AS1 regulates oxidative low-density lipoprotein-mediated endothelial cell injury via miR-320a/LOX1 axis. Mol. Cell. Biochem. 2020, 467, 15–25. [Google Scholar] [CrossRef]
- Zhou, T.; Chen, X. Long intergenic noncoding RNA p21 mediates oxidized LDLinduced apoptosis and expression of LOX1 in human coronary artery endothelial cells. Mol. Med. Rep. 2017, 16, 8513–8519. [Google Scholar] [CrossRef]
- Dai, Y.; Condorelli, G.; Mehta, J.L. Scavenger receptors and non-coding RNAs: Relevance in atherogenesis. Cardiovasc. Res. 2016, 109, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Venkateswaran, A.; Laffitte, B.A.; Joseph, S.B.; Mak, P.A.; Wilpitz, D.C.; Edwards, P.A.; Tontonoz, P. Control of cellular cholesterol efflux by the nuclear oxysterol receptor LXR alpha. Proc. Natl. Acad. Sci. USA 2000, 97, 12097–12102. [Google Scholar] [CrossRef]
- Bochem, A.E.; van Wijk, D.F.; Holleboom, A.G.; Duivenvoorden, R.; Motazacker, M.M.; Dallinga-Thie, G.M.; de Groot, E.; Kastelein, J.J.; Nederveen, A.J.; Hovingh, G.K.; et al. ABCA1 mutation carriers with low high-density lipoprotein cholesterol are characterized by a larger atherosclerotic burden. Eur. Heart J. 2013, 34, 286–291. [Google Scholar] [CrossRef]
- Zaiou, M.; Rihn, B.H.; Bakillah, A. Epigenetic regulation of genes involved in the reverse cholesterol transport through interaction with miRNAs. Front. Biosci. 2018, 23, 2090–2105. [Google Scholar] [CrossRef]
- Zaiou, M.; Bakillah, A. Epigenetic regulation of ATP-binding cassette protein A1 (ABCA1) gene expression: A new era to alleviate atherosclerotic cardiovascular disease. Diseases 2018, 6. [Google Scholar] [CrossRef]
- Li, Y.; Sun, T.; Shen, S.; Wang, L.; Yan, J. LncRNA DYNLRB2-2 inhibits THP-1 macrophage foam cell formation by enhancing autophagy. Biol. Chem. 2019, 400. Available online: https://www.degruyter.com/view/journals/bchm/400/8/article-p1047.xml (accessed on 10 July 2020). [CrossRef]
- Li, Y.; Shen, S.; Ding, S.; Wang, L. LincRNA DYN-LRB2-2 upregulates cholesterol efflux by decreasing TLR2 expression in macrophages. J. Cell. Biochem. 2018, 119, 1911–1921. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.W.; Yang, J.Y.; Ma, X.; Chen, Z.P.; Hu, Y.R.; Zhao, J.Y.; Li, S.F.; Qiu, Y.R.; Lu, J.B.; Wang, Y.C.; et al. A lincRNA-DYNLRB2-2/GPR119/GLP-1R/ABCA1-dependent signal transduction pathway is essential for the regulation of cholesterol homeostasis. J. Lipid Res. 2014, 55, 681–697. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [PubMed]
- Blich, M.; Golan, A.; Arvatz, G.; Sebbag, A.; Shafat, I.; Sabo, E.; Cohen-Kaplan, V.; Petcherski, S.; Avniel-Polak, S.; Eitan, A.; et al. Macrophage activation by heparanase is mediated by TLR-2 and TLR-4 and associates with plaque progression. Arterioscler. Thromb. Vasc. Biol. 2013, 33, e56–e65. [Google Scholar] [CrossRef] [PubMed]
- Hennessy, E.J.; van Solingen, C.; Scacalossi, K.R.; Ouimet, M.; Afonso, M.S.; Prins, J.; Koelwyn, G.J.; Sharma, M.; Ramkhelawon, B.; Carpenter, S.; et al. The long noncoding RNA CHROME regulates cholesterol homeostasis in primate. Nat. Metab. 2019, 1, 98–110. [Google Scholar] [CrossRef]
- Meng, X.D.; Yao, H.H.; Wang, L.M.; Yu, M.; Shi, S.; Yuan, Z.X.; Liu, J. Knockdown of GAS5 inhibits atherosclerosis progression via reducing EZH2-mediated ABCA1 transcription in ApoE-/- Mice. Mol. Ther. Nucleic Acids 2020, 19, 84–96. [Google Scholar] [CrossRef]
- Xu, B.M.; Xiao, L.; Kang, C.M.; Ding, L.; Guo, F.X.; Li, P.; Lu, Z.F.; Wu, Q.; Xu, Y.J.; Bai, H.L.; et al. LncRNA AC096664.3/PPAR-gamma/ABCG1-dependent signal transduction pathway contributes to the regulation of cholesterol homeostasis. J. Cell. Biochem. 2019, 120, 13775–13782. [Google Scholar] [CrossRef] [PubMed]
- Koelwyn, G.J.; Corr, E.M.; Erbay, E.; Moore, K.J. Regulation of macrophage immunometabolism in atherosclerosis. Nat. Immunol. 2018, 19, 526–537. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Huang, C.; Lin, Z.; Zhan, S.; Kong, L.; Fang, C.; Li, J. Macrophage polarization and function with emphasis on the evolving roles of coordinated regulation of cellular signaling pathways. Cell. Signal. 2014, 26, 192–197. [Google Scholar] [CrossRef]
- Back, M.; Yurdagul, A., Jr.; Tabas, I.; Oorni, K.; Kovanen, P.T. Inflammation and its resolution in atherosclerosis: Mediators and therapeutic opportunities. Nat. Rev. Cardiol. 2019, 16, 389–406. [Google Scholar] [CrossRef]
- Kasikara, C.; Doran, A.C.; Cai, B.; Tabas, I. The role of non-resolving inflammation in atherosclerosis. J. Clin. Invest. 2018, 128, 2713–2723. [Google Scholar] [CrossRef]
- Dalli, J.; Serhan, C. Macrophage proresolving mediators—The when and where. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef]
- Leifer, C.A.; Medvedev, A.E. Molecular mechanisms of regulation of Toll-like receptor signaling. J. Leukoc. Biol. 2016, 100, 927–941. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Kakkar, V.; Lu, X. Essential roles of toll-like receptors in atherosclerosis. Curr. Med. Chem. 2016, 23, 431–454. [Google Scholar] [CrossRef]
- Blasius, A.L.; Beutler, B. Intracellular toll-like receptors. Immunity 2010, 32, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K.; Robertson, A.K.; Soderberg-Naucler, C. Inflammation and atherosclerosis. Annu. Rev. Pathol. 2006, 1, 297–329. [Google Scholar] [CrossRef] [PubMed]
- Tall, A.R.; Yvan-Charvet, L. Cholesterol, inflammation and innate immunity. Nat. Rev. Immunol. 2015, 15, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Den Dekker, W.K.; Cheng, C.; Pasterkamp, G.; Duckers, H.J. Toll like receptor 4 in atherosclerosis and plaque destabilization. Atherosclerosis 2010, 209, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Michelsen, K.S.; Arditi, M. Toll-like receptor signaling and atherosclerosis. Curr. Opin. Hematol. 2006, 13, 163–168. [Google Scholar] [CrossRef]
- Chan, J.; Atianand, M.; Jiang, Z.; Carpenter, S.; Aiello, D.; Elling, R.; Fitzgerald, K.A.; Caffrey, D.R. Cutting edge: A natural antisense transcript, AS-IL1alpha, controls inducible transcription of the proinflammatory cytokine IL-1alpha. J. Immunol. 2015, 195, 1359–1363. [Google Scholar] [CrossRef]
- Zhang, Q.; Chao, T.C.; Patil, V.S.; Qin, Y.; Tiwari, S.K.; Chiou, J.; Dobin, A.; Tsai, C.M.; Li, Z.; Dang, J.; et al. The long noncoding RNA ROCKI regulates inflammatory gene expression. EMBO J. 2019, 38. [Google Scholar] [CrossRef] [PubMed]
- Watts, J.K.; Corey, D.R. Silencing disease genes in the laboratory and the clinic. J. Pathol. 2012, 226, 365–379. [Google Scholar] [CrossRef]
- Nakagawa, S. Lessons from reverse-genetic studies of lncRNAs. Biochim. Biophys. Acta 2016, 1859, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Chang, H.Y. Physiological roles of long noncoding RNAs: Insight from knockout mice. Trends Cell. Biol. 2014, 24, 594–602. [Google Scholar] [CrossRef] [PubMed]
- Sauvageau, M.; Goff, L.A.; Lodato, S.; Bonev, B.; Groff, A.F.; Gerhardinger, C.; Sanchez-Gomez, D.B.; Hacisuleyman, E.; Li, E.; Spence, M.; et al. Multiple knockout mouse models reveal lincRNAs are required for life and brain development. eLife 2013, 2, e01749. [Google Scholar] [CrossRef]
- Atianand, M.K.; Hu, W.; Satpathy, A.T.; Shen, Y.; Ricci, E.P.; Alvarez-Dominguez, J.R.; Bhatta, A.; Schattgen, S.A.; McGowan, J.D.; Blin, J.; et al. A long noncoding RNA lincRNA-EPS acts as a transcriptional brake to restrain inflammation. Cell 2016, 165, 1672–1685. [Google Scholar] [CrossRef]
- Li, Y.; Guo, W.; Cai, Y. NEAT1 Promotes LPS-induced inflammatory injury in macrophages by regulating MiR-17-5p/TLR4. Open Med. 2020, 15, 38–49. [Google Scholar] [CrossRef]
- Zhang, C.C.; Niu, F. LncRNA NEAT1 promotes inflammatory response in sepsis-induced liver injury via the Let-7a/TLR4 axis. Int. Immunopharmacol. 2019, 75, 105731. [Google Scholar] [CrossRef]
- Sen, R.; Baltimore, D. Inducibility of κ immunoglobulin enhancer-binding protein Nf-κ B by a posttranslational mechanism. Cell 1986, 47, 921–928. [Google Scholar] [CrossRef]
- Dorrington, M.G.; Fraser, I.D.C. NF-κB Signaling in macrophages: Dynamics, crosstalk, and signal integration. Front. Immunol. 2019, 10, 705. [Google Scholar] [CrossRef]
- Gupta, S.C.; Awasthee, N.; Rai, V.; Chava, S.; Gunda, V.; Challagundla, K.B. Long non-coding RNAs and nuclear factor-κB crosstalk in cancer and other human diseases. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188316. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Lenardo, M.J.; Baltimore, D. 30 Years of NF-κB: A blossoming of relevance to human pathobiology. Cell 2017, 168, 37–57. [Google Scholar] [CrossRef] [PubMed]
- Boaru, S.G.; Borkham-Kamphorst, E.; Van de Leur, E.; Lehnen, E.; Liedtke, C.; Weiskirchen, R. NLRP3 inflammasome expression is driven by NF-κB in cultured hepatocytes. Biochem. Biophys. Res. Commun. 2015, 458, 700–706. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cao, X.; Li, P.; Fan, Y.; Zhang, L.; Ma, X.; Sun, R.; Liu, Y.; Li, W. LncRNA NKILA integrates RXFP1/AKT and NF-κB signalling to regulate osteogenesis of mesenchymal stem cells. J. Cell. Mol. Med. 2020, 24, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Guo, Q.; Liu, G.; Zheng, F.; Chen, J.; Huang, D.; Ding, L.; Yang, X.; Song, E.; Xiang, Y.; et al. NKILA represses nasopharyngeal carcinoma carcinogenesis and metastasis by NF-κB pathway inhibition. PLoS Genet. 2019, 15, e1008325. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.D.; Li, Y.; Liao, G.J.; Qiu, H.W. LncRNA NKILA inhibits invasion and migration of osteosarcoma cells via NF-κB/Snail signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 4118–4125. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Chen, F.; Cui, X.; Yang, L.; Chen, J.; Zhao, J.; Huang, D.; Liu, J.; Yang, L.; Zeng, J.; et al. LncRNA NKILA suppresses TGF-β-induced epithelial-mesenchymal transition by blocking NF-κB signaling in breast cancer. Int. J. Cancer 2018, 143, 2213–2224. [Google Scholar] [CrossRef]
- Yang, T.; Li, S.; Liu, J.; Yin, D.; Yang, X.; Tang, Q. lncRNA-NKILA/NF-κB feedback loop modulates laryngeal cancer cell proliferation, invasion, and radioresistance. Cancer Med. 2018, 7, 2048–2063. [Google Scholar] [CrossRef]
- Yu, X.; Tang, W.; Yang, Y.; Tang, L.; Dai, R.; Pu, B.; Feng, C.; Xia, J. Long noncoding RNA NKILA enhances the anti-cancer effects of baicalein in hepatocellular carcinoma via the regulation of NF-κB signaling. Chem. Biol. Interact. 2018, 285, 48–58. [Google Scholar] [CrossRef]
- Zhang, B.; Wang, D.; Ji, T.F.; Shi, L.; Yu, J.L. Overexpression of lncRNA ANRIL up-regulates VEGF expression and promotes angiogenesis of diabetes mellitus combined with cerebral infarction by activating NF-κB signaling pathway in a rat model. Oncotarget 2017, 8, 17347–17359. [Google Scholar] [CrossRef]
- Zhao, G.; Su, Z.; Song, D.; Mao, Y.; Mao, X. The long noncoding RNA MALAT1 regulates the lipopolysaccharide-induced inflammatory response through its interaction with NF-κB. FEBS Lett. 2016, 590, 2884–2895. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Guan, H. Noncoding RNAs Regulating NF-κB Signaling. Adv. Exp. Med. Biol. 2016, 927, 317–336. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Han, X.; Wittfeldt, A.; Sun, J.; Liu, C.; Wang, X.; Gan, L.M.; Cao, H.; Liang, Z. Long non-coding RNA ANRIL regulates inflammatory responses as a novel component of NF-κB pathway. RNA Biol. 2016, 13, 98–108. [Google Scholar] [CrossRef]
- Zhou, X.; Liu, S.; Cai, G.; Kong, L.; Zhang, T.; Ren, Y.; Wu, Y.; Mei, M.; Zhang, L.; Wang, X. Long non coding RNA MALAT1 promotes tumor growth and metastasis by inducing epithelial-mesenchymal transition in oral squamous cell carcinoma. Sci. Rep. 2015, 5, 15972. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Gong, A.Y.; Wang, Y.; Ma, S.; Chen, X.; Chen, J.; Su, C.J.; Shibata, A.; Strauss-Soukup, J.K.; Drescher, K.M.; et al. LincRNA-Cox2 promotes late inflammatory gene transcription in macrophages through modulating SWI/SNF-Mediated chromatin remodeling. J. Immunol. 2016, 196, 2799–2808. [Google Scholar] [CrossRef]
- Ma, S.; Ming, Z.; Gong, A.Y.; Wang, Y.; Chen, X.; Hu, G.; Zhou, R.; Shibata, A.; Swanson, P.C.; Chen, X.M. A long noncoding RNA, lincRNA-Tnfaip3, acts as a coregulator of NF-κB to modulate inflammatory gene transcription in mouse macrophages. FASEB J. 2017, 31, 1215–1225. [Google Scholar] [CrossRef]
- Castellanos-Rubio, A.; Kratchmarov, R.; Sebastian, M.; Garcia-Etxebarria, K.; Garcia, L.; Irastorza, I.; Ghosh, S. Cytoplasmic form of Carlr lncRNA facilitates inflammatory gene expression upon NF-κB Activation. J. Immunol. 2017, 199, 581–588. [Google Scholar] [CrossRef]
- Lu, Y.; Liu, X.; Xie, M.; Liu, M.; Ye, M.; Li, M.; Chen, X.M.; Li, X.; Zhou, R. The NF-κB-Responsive long noncoding RNA FIRRE regulates posttranscriptional regulation of inflammatory gene expression through interacting with hnRNPU. J. Immunol. 2017, 199, 3571–3582. [Google Scholar] [CrossRef]
- Wen, Y.; Yu, Y.; Fu, X. LncRNA Gm4419 contributes to OGD/R injury of cerebral microglial cells via IκB phosphorylation and NF-κB activation. Biochem. Biophys. Res. Commun. 2017, 487, 923–929. [Google Scholar] [CrossRef]
- Zang, Y.; Zhou, X.; Wang, Q.; Li, X.; Huang, H. LncRNA FIRRE/NF-kB feedback loop contributes to OGD/R injury of cerebral microglial cells. Biochem. Biophys. Res. Commun. 2018, 501, 131–138. [Google Scholar] [CrossRef]
- Yan, S.; Wang, P.; Wang, J.; Yang, J.; Lu, H.; Jin, C.; Cheng, M.; Xu, D. Long non-coding RNA HIX003209 promotes inflammation by sponging miR-6089 via TLR4/NF-κB signaling pathway in rheumatoid arthritis. Front. Immunol. 2019, 10, 2218. [Google Scholar] [CrossRef] [PubMed]
- Zgheib, C.; Hodges, M.M.; Hu, J.; Liechty, K.W.; Xu, J. Long non-coding RNA lethe regulates hyperglycemia-induced reactive oxygen species production in macrophages. PLoS ONE 2017, 12, e0177453. [Google Scholar] [CrossRef] [PubMed]
- Du, M.; Yuan, L.; Tan, X.; Huang, D.; Wang, X.; Zheng, Z.; Mao, X.; Li, X.; Yang, L.; Huang, K.; et al. The LPS-inducible lncRNA Mirt2 is a negative regulator of inflammation. Nat. Commun. 2017, 8, 2049. [Google Scholar] [CrossRef] [PubMed]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).