Accurate Adapter Information Is Crucial for Reproducibility and Reusability in Small RNA Seq Studies


{kind=link}
Abstract
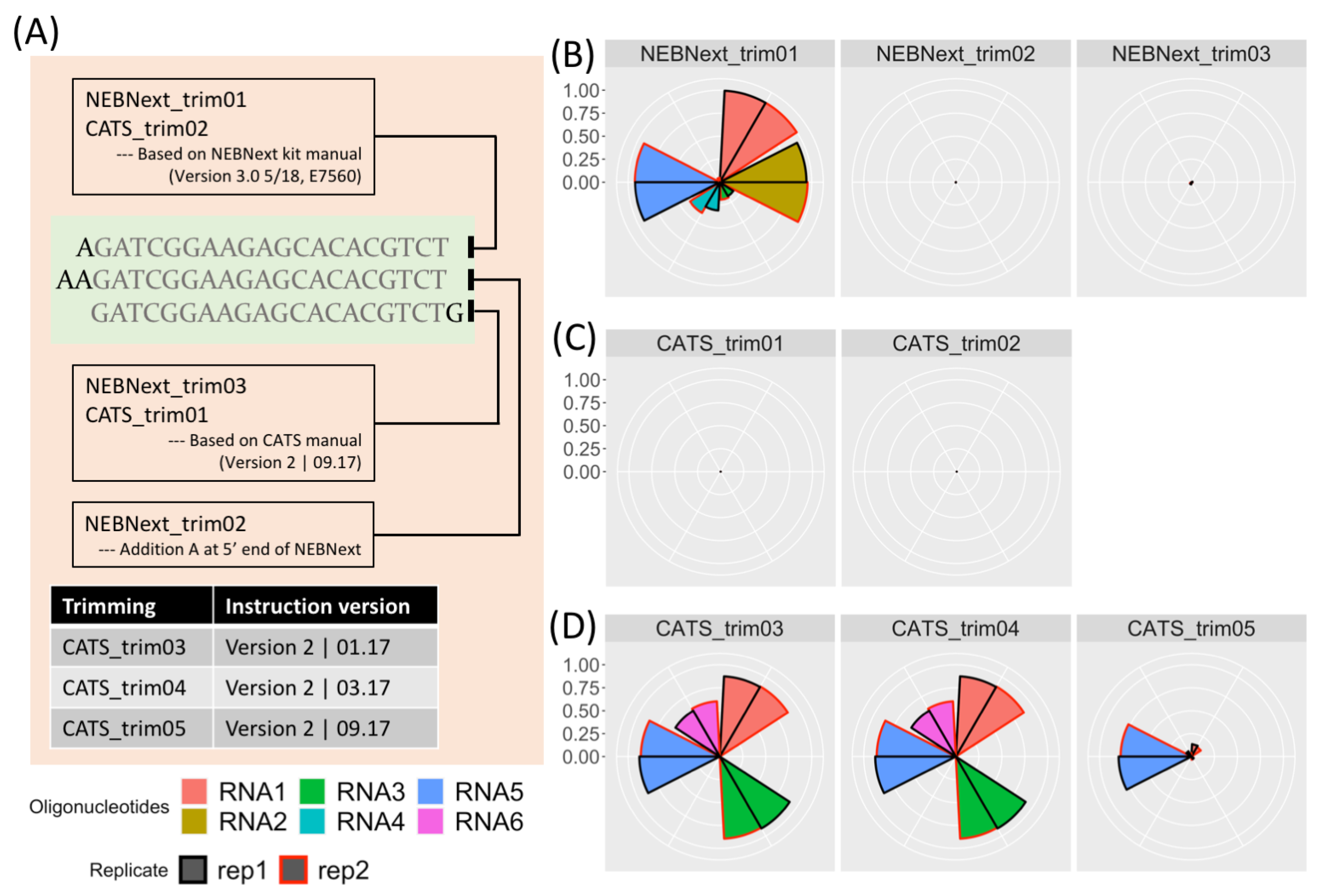
where the rApp, a 5’-adenylated termini, is removed when the 3’ adapter and inserted RNA fragment are ligated by the T4 RNA ligase [3].5’-rAppAGATCGGAAGAGCACACGTCT-NH2-3’
AAGATCGGAAGAGCACACGTCT
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| NGS | Next Generation Sequencing |
| SRA | Sequence Read Archive |
| GEO | Gene Expression Omnibus |
| FAIR | Findable, Accessible, Interoperable and Re-usable |
References
- Hafner, M.; Landgraf, P.; Ludwig, J.; Rice, A.; Ojo, T.; Lin, C.; Holoch, D.; Lim, C.; Tuschl, T. Identification of microRNAs and other small regulatory RNAs using cDNA library sequencing. Methods 2008, 44, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Meyers, B.C.; Green, P.J. Construction of small RNA cDNA libraries for deep sequencing. Methods 2007, 43, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Raabe, C.A.; Tang, T.H.; Brosius, J.; Rozhdestvensky, T.S. Biases in small RNA deep sequencing data. Nucleic Acids Res. 2014, 42, 1414–1426. [Google Scholar] [CrossRef] [PubMed]
- Van Goethem, A.; Yigit, N.; Everaert, C.; Moreno-Smith, M.; Mus, L.M.; Barbieri, E.; Speleman, F.; Mestdagh, P.; Shohet, J.; Van Maerken, T.; et al. Depletion of tRNA-halves enables effective small RNA sequencing of low-input murine serum samples. Sci. Rep. 2016, 6, 37876. [Google Scholar] [CrossRef] [PubMed]
- Zovoilis, A.; Cifuentes-Rojas, C.; Chu, H.P.; Hernandez, A.J.; Lee, J.T. Destabilization of B2 RNA by EZH2 Activates the Stress Response. Cell 2016, 167, 1788–1802.e13. [Google Scholar] [CrossRef] [PubMed]
- Gümürdü, A.; Yildiz, R.; Eren, E.; Karakülah, G.; Ünver, T.; Genç, Ş.; Park, Y. MicroRNA exocytosis by large dense-core vesicle fusion. Sci. Rep. 2017, 7, 45661. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Davis, M.P.A.; van Dongen, S.; Abreu-Goodger, C.; Bartonicek, N.; Enright, A.J. Kraken: A set of tools for quality control and analysis of high-throughput sequence data. Methods 2013, 63, 41–49. [Google Scholar] [CrossRef]
- Tsuji, J.; Weng, Z. DNApi: A De Novo Adapter Prediction Algorithm for Small RNA Sequencing Data. PLoS ONE 2016, 11, e0164228. [Google Scholar] [CrossRef]
- Schubert, M.; Lindgreen, S.; Orlando, L. AdapterRemoval v2: Rapid adapter trimming, identification, and read merging. BMC Res. Notes 2016, 9, 88. [Google Scholar] [CrossRef] [PubMed]
- Shore, S.; Henderson, J.M.; Lebedev, A.; Salcedo, M.P.; Zon, G.; McCaffrey, A.P.; Paul, N.; Hogrefe, R.I. Small RNA Library Preparation Method for Next-Generation Sequencing Using Chemical Modifications to Prevent Adapter Dimer Formation. PLoS ONE 2016, 11, e0167009. [Google Scholar] [CrossRef] [PubMed]
- Niu, J.; Smagghe, G.; De Coninck, D.I.M.; Van Nieuwerburgh, F.; Deforce, D.; Meeus, I. In vivo study of Dicer-2-mediated immune response of the small interfering RNA pathway upon systemic infections of virulent and avirulent viruses in Bombus terrestris. Insect Biochem. Mol. Biol. 2016, 70, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Niu, J.; Meeus, I.; De Coninck, D.I.; Deforce, D.; Etebari, K.; Asgari, S.; Smagghe, G. Infections of virulent and avirulent viruses differentially influenced the expression of dicer-1, ago-1, and microRNAs in Bombus terrestris. Sci. Rep. 2017, 7, 45620. [Google Scholar] [CrossRef]
- Dard-Dascot, C.; Naquin, D.; d’Aubenton Carafa, Y.; Alix, K.; Thermes, C.; van Dijk, E. Systematic comparison of small RNA library preparation protocols for next-generation sequencing. BMC Genom. 2018, 19, 118. [Google Scholar] [CrossRef]
- Corpas, M.; Kovalevskaya, N.V.; McMurray, A.; Nielsen, F.G.G. A FAIR guide for data providers to maximise sharing of human genomic data. PLoS Comput. Biol. 2018, 14, 1–10. [Google Scholar] [CrossRef]
- Seguin-Orlando, A.; Schubert, M.; Clary, J.; Stagegaard, J.; Alberdi, M.T.; Prado, J.L.; Prieto, A.; Willerslev, E.; Orlando, L. Ligation bias in illumina next-generation DNA libraries: Implications for sequencing ancient genomes. PLoS ONE 2013, 8, e78575. [Google Scholar] [CrossRef]
- Tian, G.; Yin, X.; Luo, H.; Xu, X.; Bolund, L.; Zhang, X.; Gan, S.Q.; Li, N. Sequencing bias: Comparison of different protocols of microRNA library construction. BMC Biotechnol. 2010, 10, 64. [Google Scholar] [CrossRef]
- Zhuang, F.; Fuchs, R.T.; Sun, Z.; Zheng, Y.; Robb, G.B. Structural bias in T4 RNA ligase-mediated 3’-adapter ligation. Nucleic Acids Res. 2012, 40, e54. [Google Scholar] [CrossRef]
- Sorefan, K.; Pais, H.; Hall, A.E.; Kozomara, A.; Griffiths-Jones, S.; Moulton, V.; Dalmay, T. Reducing ligation bias of small RNAs in libraries for next generation sequencing. Silence 2012, 3, 4. [Google Scholar] [CrossRef]
- Alon, S.; Vigneault, F.; Eminaga, S.; Christodoulou, D.C.; Seidman, J.G.; Church, G.M.; Eisenberg, E. Barcoding bias in high-throughput multiplex sequencing of miRNA. Genome Res. 2011, 21, 1506–1511. [Google Scholar] [CrossRef] [PubMed]
- Sansone, S.A.; McQuilton, P.; Rocca-Serra, P.; Gonzalez-Beltran, A.; Izzo, M.; Lister, A.L.; Thurston, M.; FAIRsharing Community. FAIRsharing as a community approach to standards, repositories and policies. Nat. Biotechnol. 2019, 37, 358–367. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhong, X.; Heinicke, F.; Lie, B.A.; Rayner, S. Accurate Adapter Information Is Crucial for Reproducibility and Reusability in Small RNA Seq Studies. Non-Coding RNA 2019, 5, 49. https://doi.org/10.3390/ncrna5040049
Zhong X, Heinicke F, Lie BA, Rayner S. Accurate Adapter Information Is Crucial for Reproducibility and Reusability in Small RNA Seq Studies. Non-Coding RNA. 2019; 5(4):49. https://doi.org/10.3390/ncrna5040049
Chicago/Turabian StyleZhong, Xiangfu, Fatima Heinicke, Benedicte A. Lie, and Simon Rayner. 2019. "Accurate Adapter Information Is Crucial for Reproducibility and Reusability in Small RNA Seq Studies" Non-Coding RNA 5, no. 4: 49. https://doi.org/10.3390/ncrna5040049
APA StyleZhong, X., Heinicke, F., Lie, B. A., & Rayner, S. (2019). Accurate Adapter Information Is Crucial for Reproducibility and Reusability in Small RNA Seq Studies. Non-Coding RNA, 5(4), 49. https://doi.org/10.3390/ncrna5040049