Formation of a Family of Long Intergenic Noncoding RNA Genes with an Embedded Translocation Breakpoint Motif in Human Chromosomal Low Copy Repeats of 22q11.2—Some Surprises and Questions

Abstract
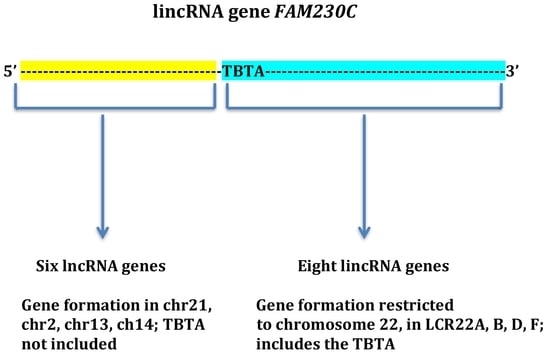
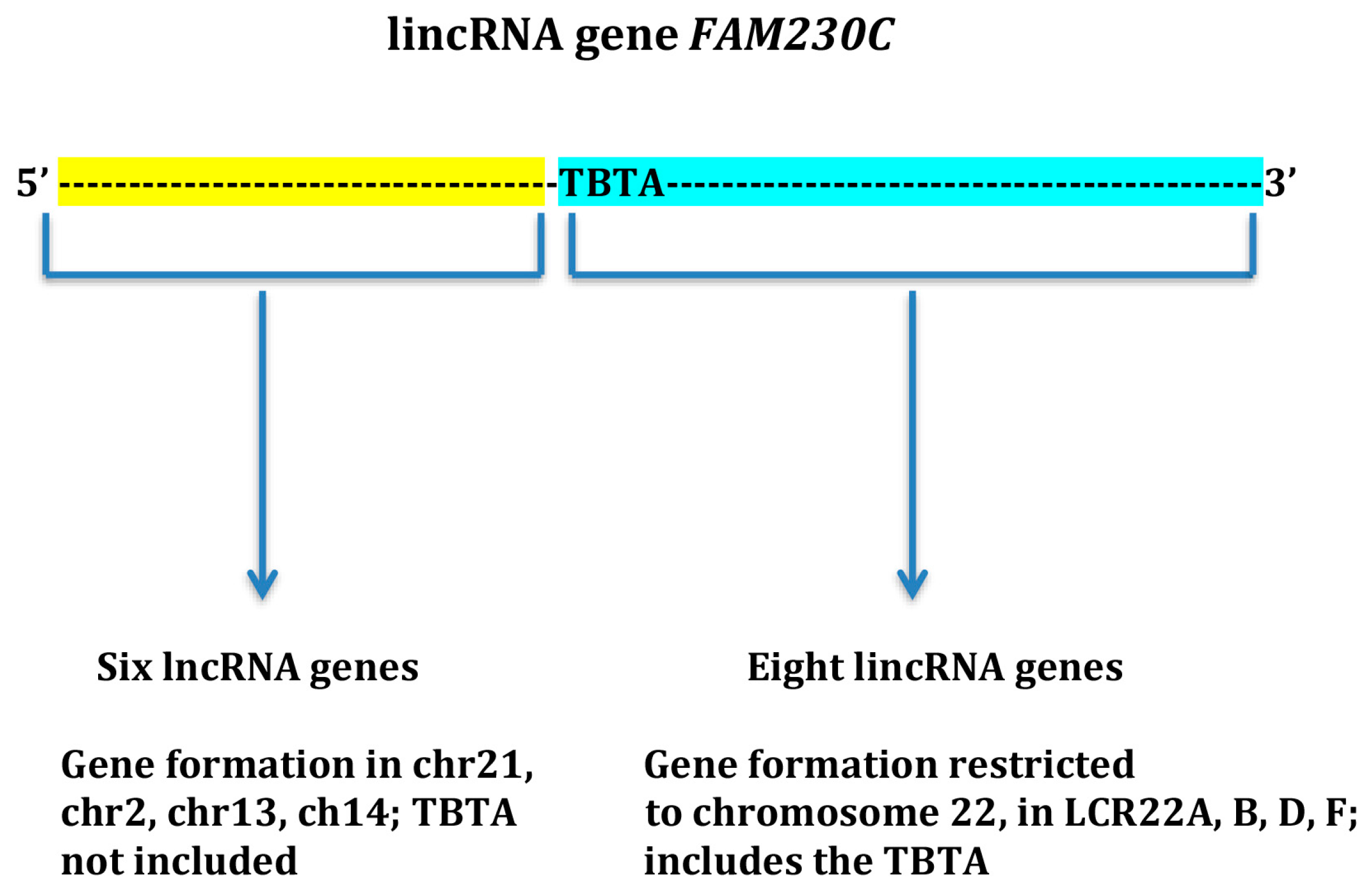
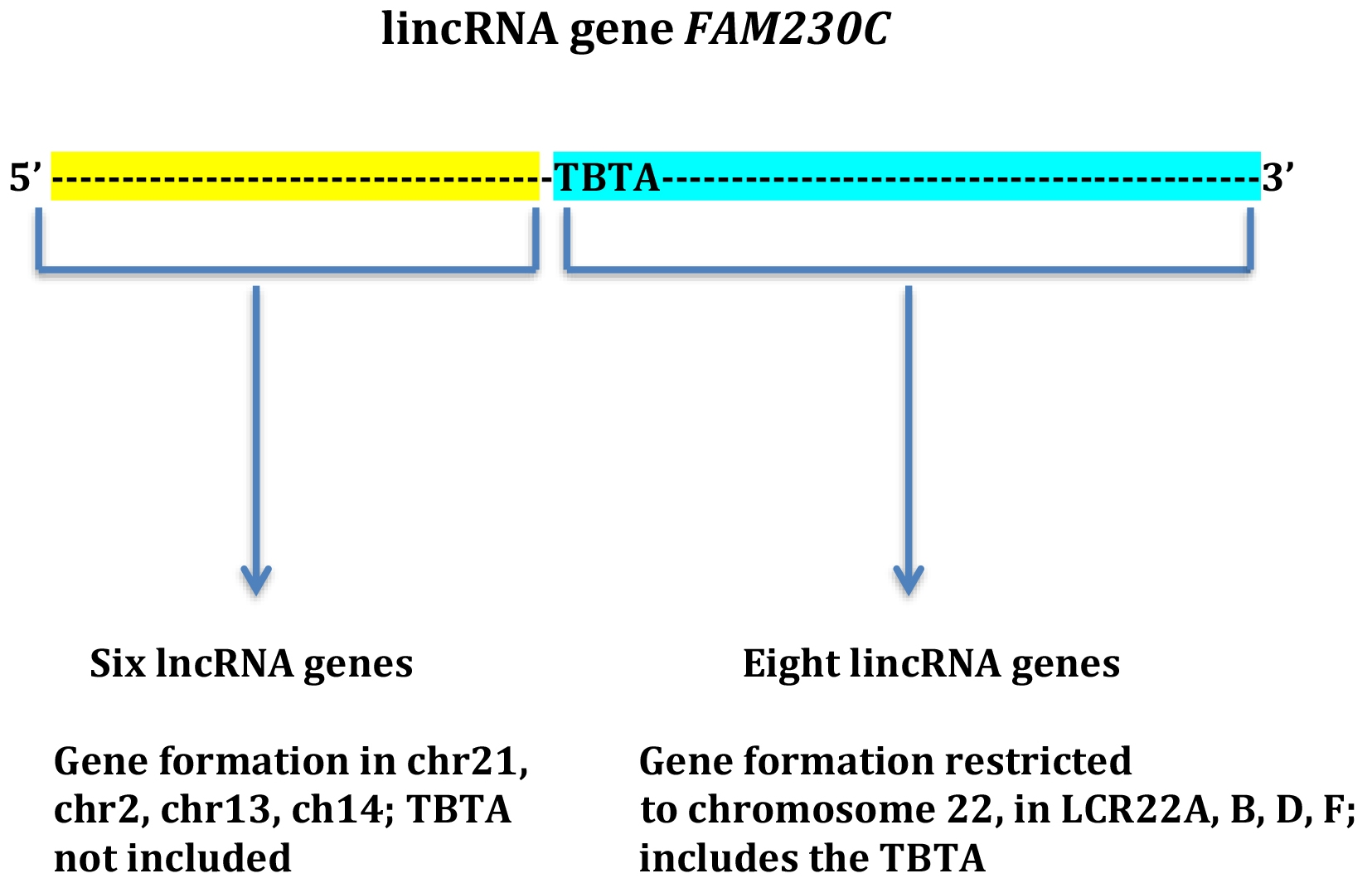{kind=link}
{kind=link}
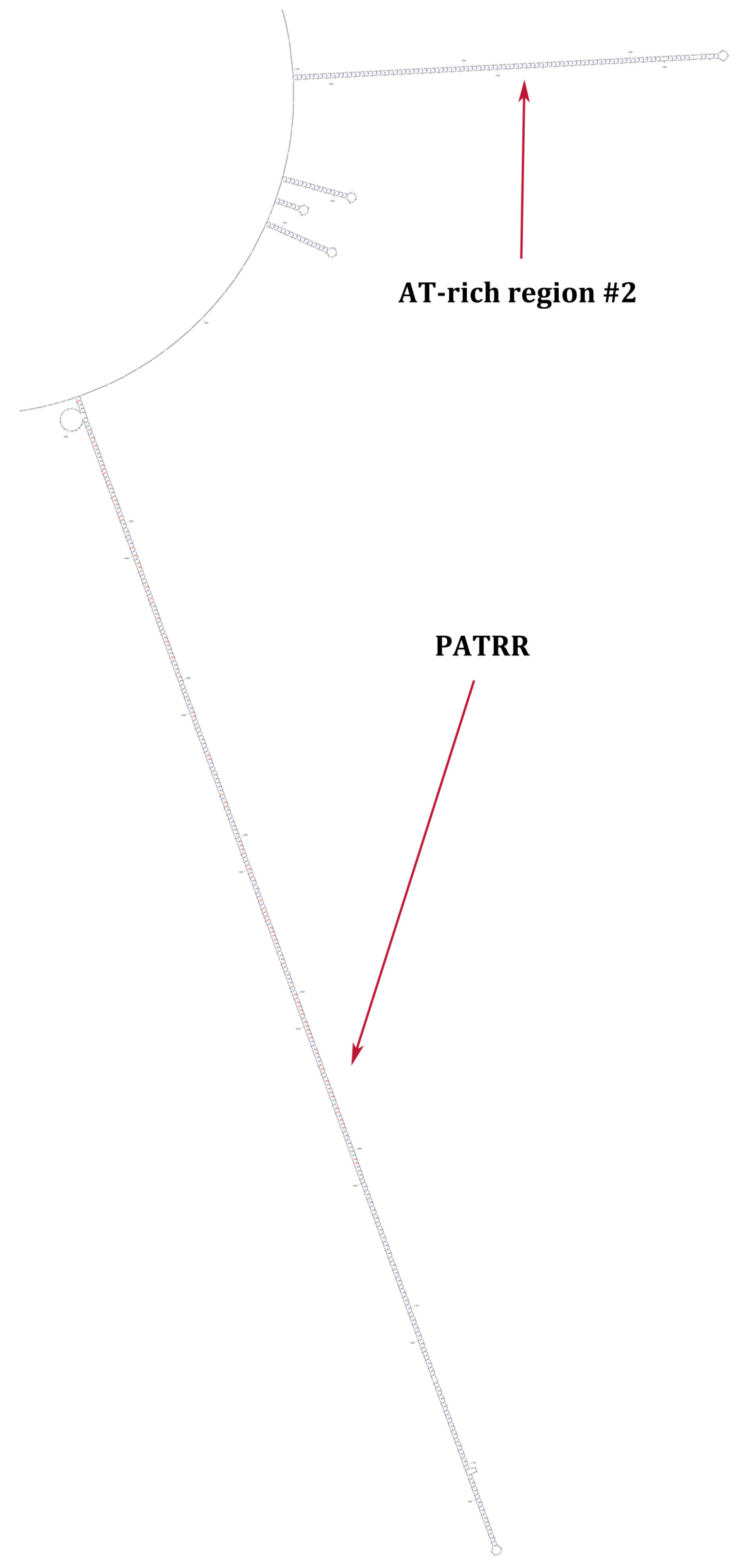{kind=link}
{kind=link}
{kind=link}
Supplementary Materials
Conflicts of Interest
References
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef] [PubMed]
- Iyer, M.K.; Niknafs, Y.S.; Malik, R.; Singhal, U.; Sahu, A.; Hosono, Y.; Barrette, T.R.; Prensner, J.R.; Evans, J.R.; Zhao, S.; et al. The landscape of long noncoding RNAs in the human transcriptome. Nat. Genet. 2015, 47, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Goff, L.A.; Rinn, J.L. Linking RNA biology to lncRNAs. Genome Res. 2015, 25, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Morris, K.V.; Mattick, J.S. The rise of regulatory RNA. Nat. Rev. Genet. 2014, 15, 423–437. [Google Scholar] [CrossRef] [PubMed]
- Kapusta, A.; Feschotte, C. Volatile evolution of long noncoding RNA repertoires: Mechanisms and biological implications. Trends Genet. 2014, 30, 439–452. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Lin, M.; Rockowitz, S.; Lachman, H.M.; Zheng, D. Characterization of human pseudogene-derived non-coding RNAs for functional potential. PLoS ONE 2014, 9, e93972. [Google Scholar] [CrossRef] [PubMed]
- Ulitsky, I. Evolution to the rescue: Using comparative genomics to understand long non-coding RNAs. Nat. Rev. Genet. 2016, 17, 601–614. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.H.; Tsai, Z.T.; Tsai, H.K. Comparative genomic analyses highlight the contribution of pseudogenized protein-coding genes to human lincRNAs. BMC Genom. 2017, 18, 786. [Google Scholar] [CrossRef] [PubMed]
- Terracciano, D.; Terreri, S.; de Nigris, F.; Costa, V.; Calin, G.A.; Cimmino, A. The role of a new class of long noncoding RNAs transcribed from ultraconserved regions in cancer. Biochim. Biophys. Acta 2017, 1868, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, J.M. On the Origin of lncRNAs: Missing Link Found. Trends Genet. 2017, 33, 660–662. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Yang, L.; Chen, L.L. The Diversity of Long Noncoding RNAs and Their Generation. Trends Genet. 2017, 33, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Awan, H.M.; Shah, A.; Rashid, F.; Shan, G. Primate-specific Long Non-coding RNAs and MicroRNAs. Genom. Proteom. Bioinform. 2017, 15, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Delihas, N. A family of long intergenic non-coding RNA genes in human chromosomal region 22q11.2 carry a DNA translocation breakpoint/AT-rich sequence. PLoS ONE 2018, 13, e0195702. [Google Scholar] [CrossRef] [PubMed]
- Edelmann, L.; Pandita, R.K.; Morrow, B.E. Low-copy repeats mediate the common 3-Mb deletion in patients with velo-cardio-facial syndrome. Am. J. Hum. Genet. 1999, 64, 1076–1086. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, T.H.; Kurahashi, H.; Saitta, S.C.; O’Hare, A.M.; Hu, P.; Roe, B.A.; Driscoll, D.A.; McDonald-McGinn, D.M.; Zackai, E.H.; Budarf, M.L.; et al. Chromosome 22-specific low copy repeats and the 22q11.2 deletion syndrome: Genomic organization and deletion endpoint analysis. Hum. Mol. Genet. 2000, 9, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Eichler, E.E. Masquerading repeats: Paralogous pitfalls of the Human Genome. Genome Res. 1998, 8, 758–762. [Google Scholar] [CrossRef] [PubMed]
- Stankiewicz, P.; Lupski, J.R. Molecular-evolutionary mechanisms for genomic disorders. Curr. Opin. Genet. Dev. 2002, 12, 312–319. [Google Scholar] [CrossRef]
- Edelmann, L.; Spiteri, E.; Koren, K.; Pulijaal, V.; Bialer, M.G.; Shanske, A.; Goldberg, R.; Morrow, B.E. AT-rich palindromes mediate the constitutional t(11;22) translocation. Am. J. Hum. Genet. 2001, 68, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Kurahashi, H.; Emanuel, B.S. Chromosomal translocations and palindromic AT-rich repeats. Curr. Opin. Genet. Dev. 2012, 22, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Kurahashi, H.; Inagaki, H.; Hosoba, E.; Kato, T.; Ohye, T.; Kogo, H.; Emanuel, B.S. Molecular cloning of a translocation breakpoint hotspot in 22q11. Genome Res. 2007, 17, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Babcock, M.; Yatsenko, S.; Stankiewicz, P.; Lupski, J.R.; Morrow, B.E. AT-rich repeats associated with chromosome 22q11.2 rearrangement disorders shape human genome architecture on Yq12. Genome Res. 2007, 17, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Fagerberg, L.; Hallström, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteom. 2014, 13, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Zerbino, D.R.; Achuthan, P.; Akanni, W.; Amode, M.R.; Barrell, D.; Bhai, J.; Billis, K.; Cummins, C.; Gall, A.; Girón, C.G.; et al. Ensembl 2018. Nucleic Acids Res. 2017, 46, D754–D761. [Google Scholar] [CrossRef] [PubMed]
- Samonte, R.V.; Eichler, E.E. Segmental duplications and the evolution of the primate genome. Nat. Rev. Genet. 2002, 3, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Stankiewicz, P.; Shaw, C.J.; Withers, M.; Inoue, K.; Lupski, J.R. Serial segmental duplications during primate evolution result in complex human genome architecture. Genome Res. 2004, 14, 2209–2220. [Google Scholar] [CrossRef] [PubMed]
- Dennehey, B.K.; Gutches, D.G.; McConkey, E.H.; Krauter, K.S. Inversion, duplication, and changes in gene context are associated with human chromosome 18 evolution. Genomics 2004, 83, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Magadum, S.; Banerjee, U.; Murugan, P.; Gangapur, D.; Ravikesavan, R. Gene duplication as a major force in evolution. J. Genet. 2013, 92, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Dennis, M.Y.; Eichler, E.E. Human adaptation and evolution by segmental duplication. Curr. Opin. Genet. Dev. 2016, 41, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Freyer, L.; Morrow, B.; Zheng, D. Characterization of the past and current duplication activities in the human 22q11.2 region. BMC Genom. 2011, 12, 71. [Google Scholar] [CrossRef] [PubMed]
- Harel T Pehlivan, D.; Caskey, C.T.; Lupski, J.R. Mendelian, Non-Mendelian, Multigenic Inheritance, and Epigenetics. In Rosenberg’s Molecular and Genetic Basis of Neurological and Psychiatric Disease, 5th ed.; Academic Press: Waltham, MA, USA, 2015; Chapter 1; pp. 3–27. [Google Scholar]
- Shaikh, T.H.; Kurahashi, H.; Emanuel, B.S. Evolutionarily conserved low copy repeats (LCRs) in 22q11 mediate deletions, duplications, translocations, and genomic instability: An update and literature review. Genet. Med. 2001, 3, 6–13. [Google Scholar] [CrossRef] [PubMed]
- McDonald-McGinn, D.M.; Sullivan, K.E.; Marino, B.; Philip, N.; Swillen, A.; Vorstman, J.A.; Zackai, E.H.; Emanuel, B.S.; Vermeesch, J.R.; Morrow, B.E.; et al. 22q11.2 deletion syndrome. Nat. Rev. Dis. Primers 2015, 1, 15071. [Google Scholar] [CrossRef] [PubMed]
- Kobrynski, L.J.; Sullivan, K.E. Velocardiofacial syndrome, DiGeorge syndrome: The chromosome 22q11.2 deletion syndromes. Lancet 2007, 370, 1443–1452. [Google Scholar] [CrossRef]
- Kurahashi, H.; Inagaki, H.; Ohye, T.; Kogo, H.; Kato, T.; Emanuel, B.S. Chromosomal translocations mediated by palindromic DNA. Cell Cycle 2006, 5, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, H.; Kato, T.; Tsutsumi, M.; Ouchi, Y.; Ohye, T.; Kurahashi, H. Palindrome-Mediated Translocations in Humans: A New Mechanistic Model for Gross Chromosomal Rearrangements. Front. Genet. 2016, 7, 125. [Google Scholar] [CrossRef] [PubMed]
- Gotter, A.L.; Shaikh, T.H.; Budarf, M.L.; Rhodes, C.H.; Emanuel, B.S. A palindrome-mediated mechanism distinguishes translocations involving LCR-B of chromosome 22q11.2. Hum. Mol. Genet. 2004, 13, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef] [PubMed]
- Needleman, S.B.; Wunsch, C.D. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 1970, 48, 443–453. [Google Scholar] [CrossRef]
- Tong, M.; Kato, T.; Yamada, K.; Inagaki, H.; Kogo, H.; Ohye, T.; Tsutsumi, M.; Wang, J.; Emanuel, B.S.; Kurahashi, H. Polymorphisms of the 22q11.2 breakpoint region influence the frequency of de novo constitutional t(11;22)s in sperm. Hum. Mol. Genet. 2010, 19, 2630–2637. [Google Scholar] [CrossRef] [PubMed]
- Frommer, M.; Prosser, J.; Vincent, P.C. Human satellite I sequences include a male specific 2.47 kb tandemly repeated unit containing one Alu family member per repeat. Nucleic Acids Res. 1984, 12, 2887–2900. [Google Scholar] [CrossRef] [PubMed]
- Delihas, N. Complexity of a small non-protein coding sequence in chromosomal region 22q11.2: Presence of specialized DNA secondary structures and RNA exon/intron motifs. BMC Genom 2015, 16, 785. [Google Scholar] [CrossRef] [PubMed]
- Bacolla, A.; Collins, J.R.; Gold, B.; Chuzhanova, N.; Yi, M.; Stephens, R.M.; Stefanov, S.; Olsh, A.; Jakupciak, J.P.; Dean, M.; et al. Long homopurine·homopyrimidine sequences are characteristic of genes expressed in brain and the pseudoautosomal region. Nucleic Acids Res. 2006, 34, 2663–2675. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, H.; Ohye, T.; Kogo, H.; Yamada, K.; Kowa, H.; Shaikh, T.H.; Emanuel, B.S. Kurahashi Palindromic AT-rich repeat in the NF1 gene is hypervariable in humans and evolutionarily conserved in primates. Hum. Mutat. 2005, 26, 332–342. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Delihas, N. Formation of a Family of Long Intergenic Noncoding RNA Genes with an Embedded Translocation Breakpoint Motif in Human Chromosomal Low Copy Repeats of 22q11.2—Some Surprises and Questions. Non-Coding RNA 2018, 4, 16. https://doi.org/10.3390/ncrna4030016
Delihas N. Formation of a Family of Long Intergenic Noncoding RNA Genes with an Embedded Translocation Breakpoint Motif in Human Chromosomal Low Copy Repeats of 22q11.2—Some Surprises and Questions. Non-Coding RNA. 2018; 4(3):16. https://doi.org/10.3390/ncrna4030016
Chicago/Turabian StyleDelihas, Nicholas. 2018. "Formation of a Family of Long Intergenic Noncoding RNA Genes with an Embedded Translocation Breakpoint Motif in Human Chromosomal Low Copy Repeats of 22q11.2—Some Surprises and Questions" Non-Coding RNA 4, no. 3: 16. https://doi.org/10.3390/ncrna4030016
APA StyleDelihas, N. (2018). Formation of a Family of Long Intergenic Noncoding RNA Genes with an Embedded Translocation Breakpoint Motif in Human Chromosomal Low Copy Repeats of 22q11.2—Some Surprises and Questions. Non-Coding RNA, 4(3), 16. https://doi.org/10.3390/ncrna4030016