
Rare Splice Variants in Long Non-Coding RNAs



Abstract
:1. Introduction
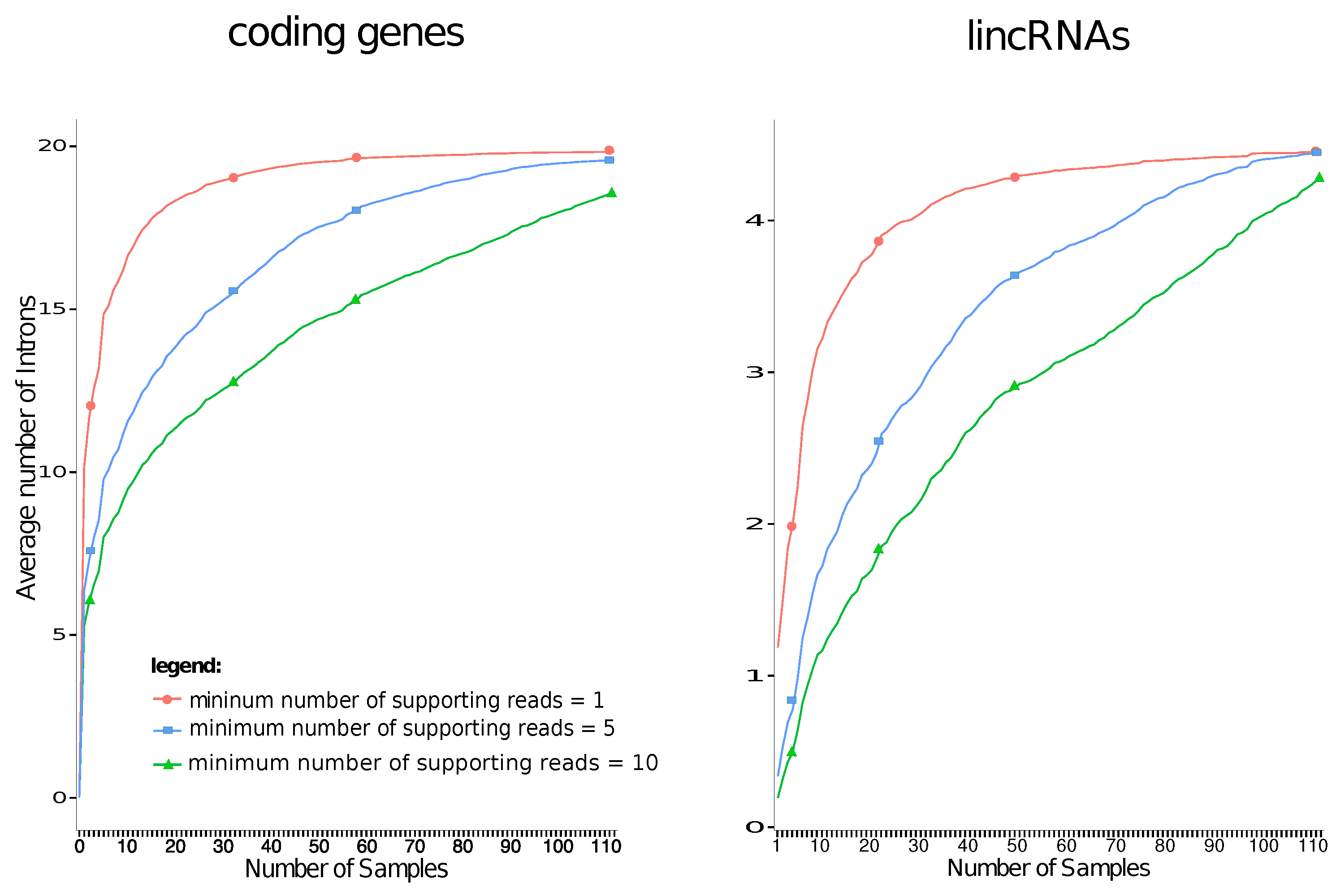
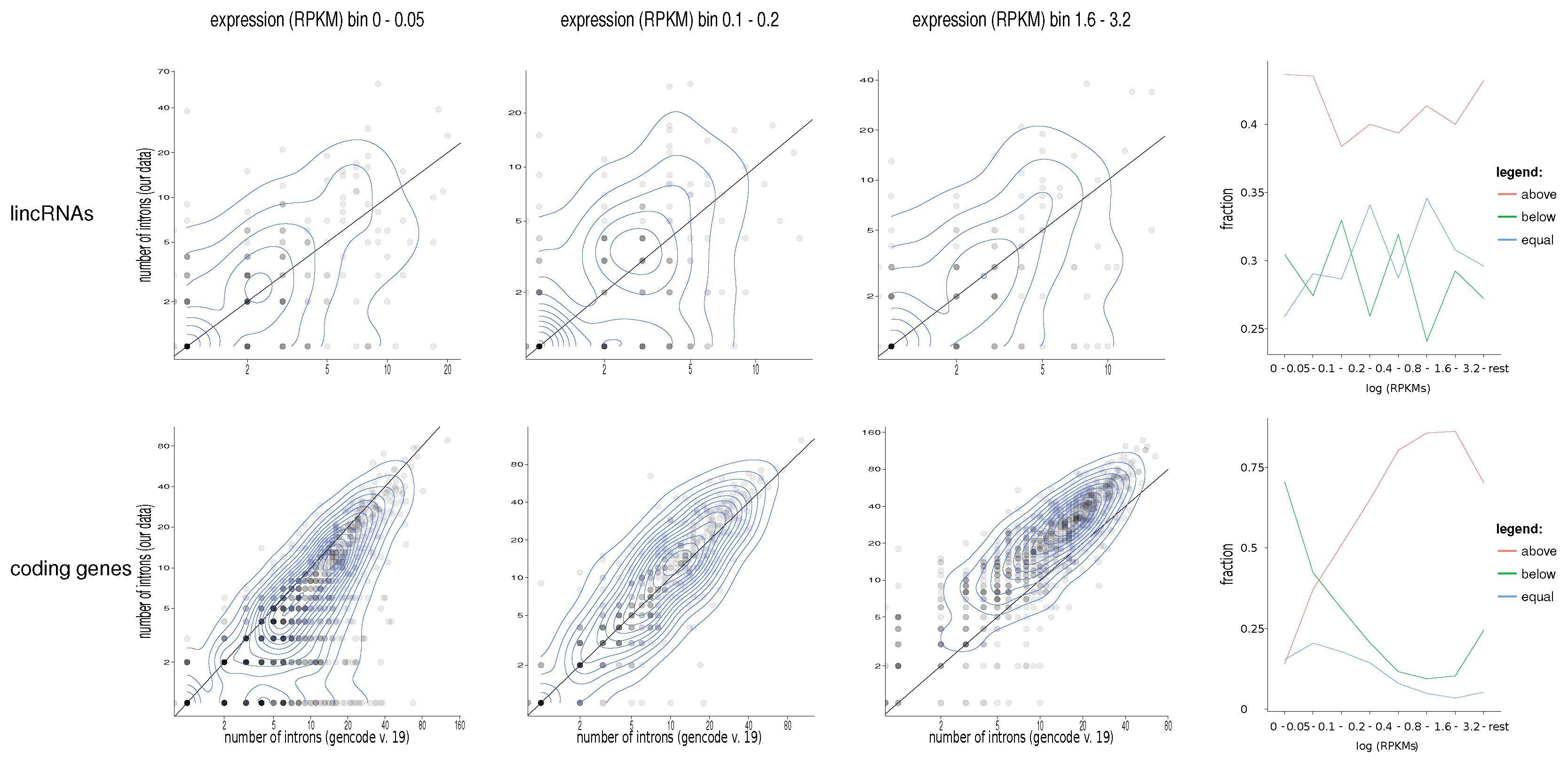
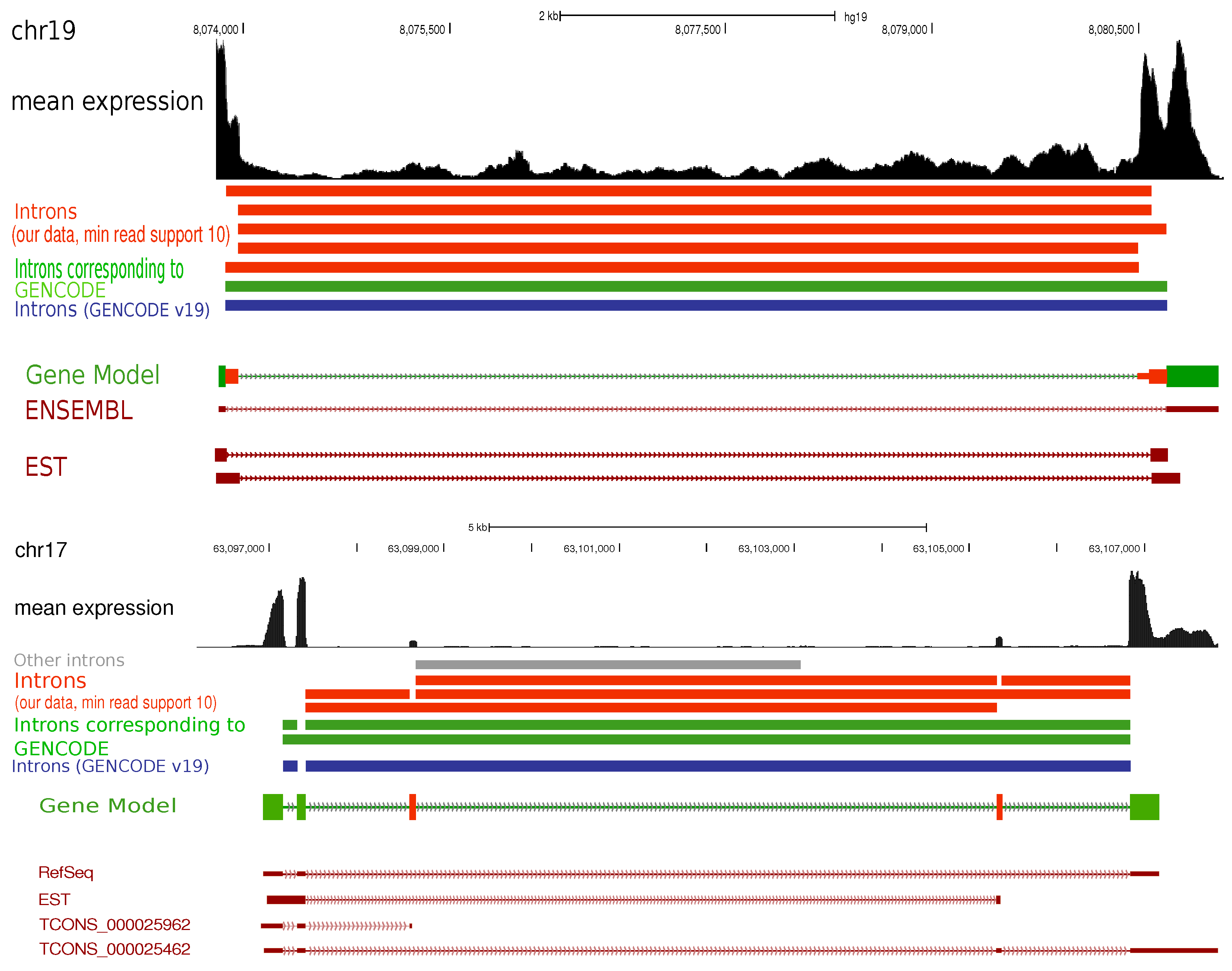
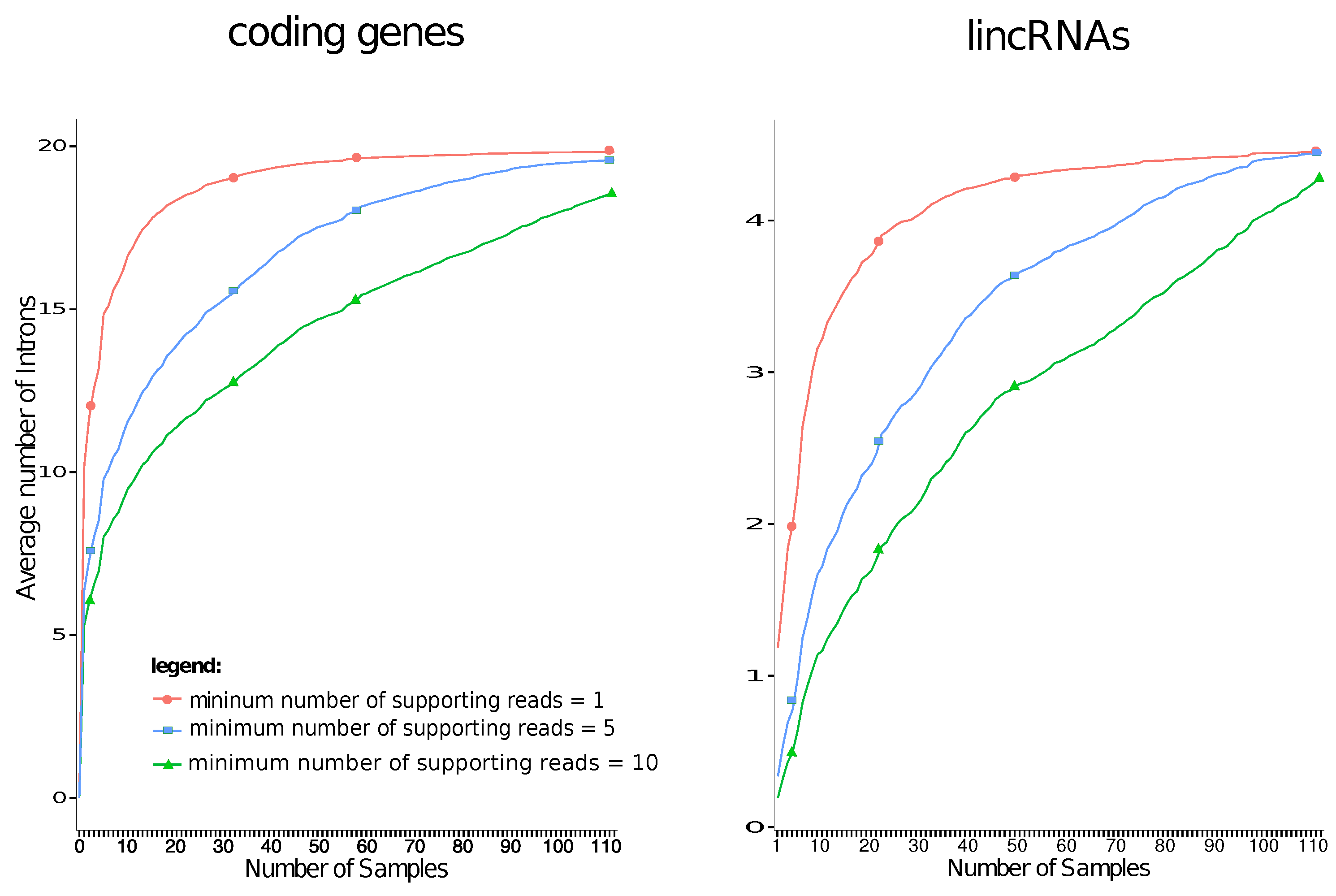
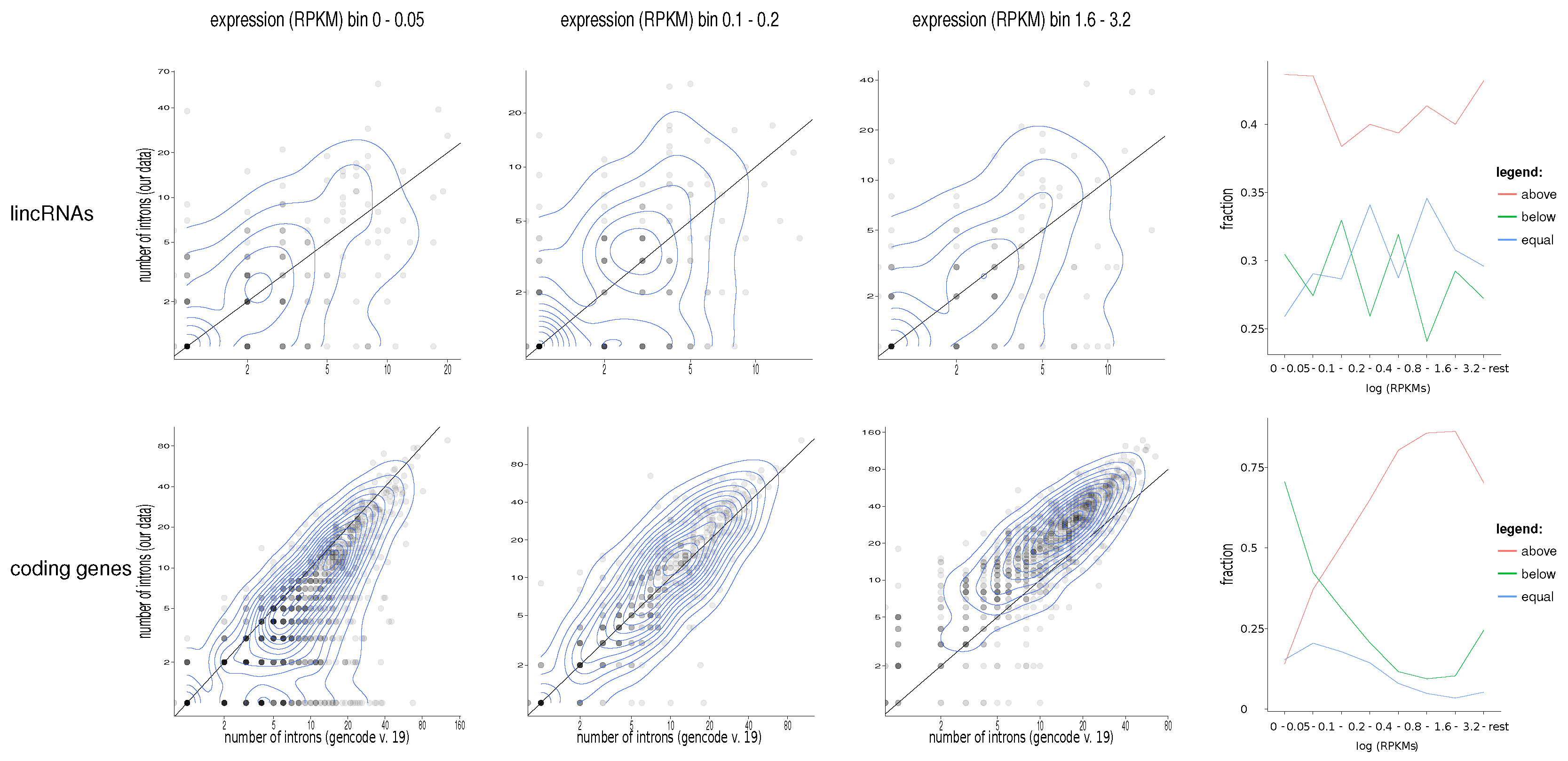
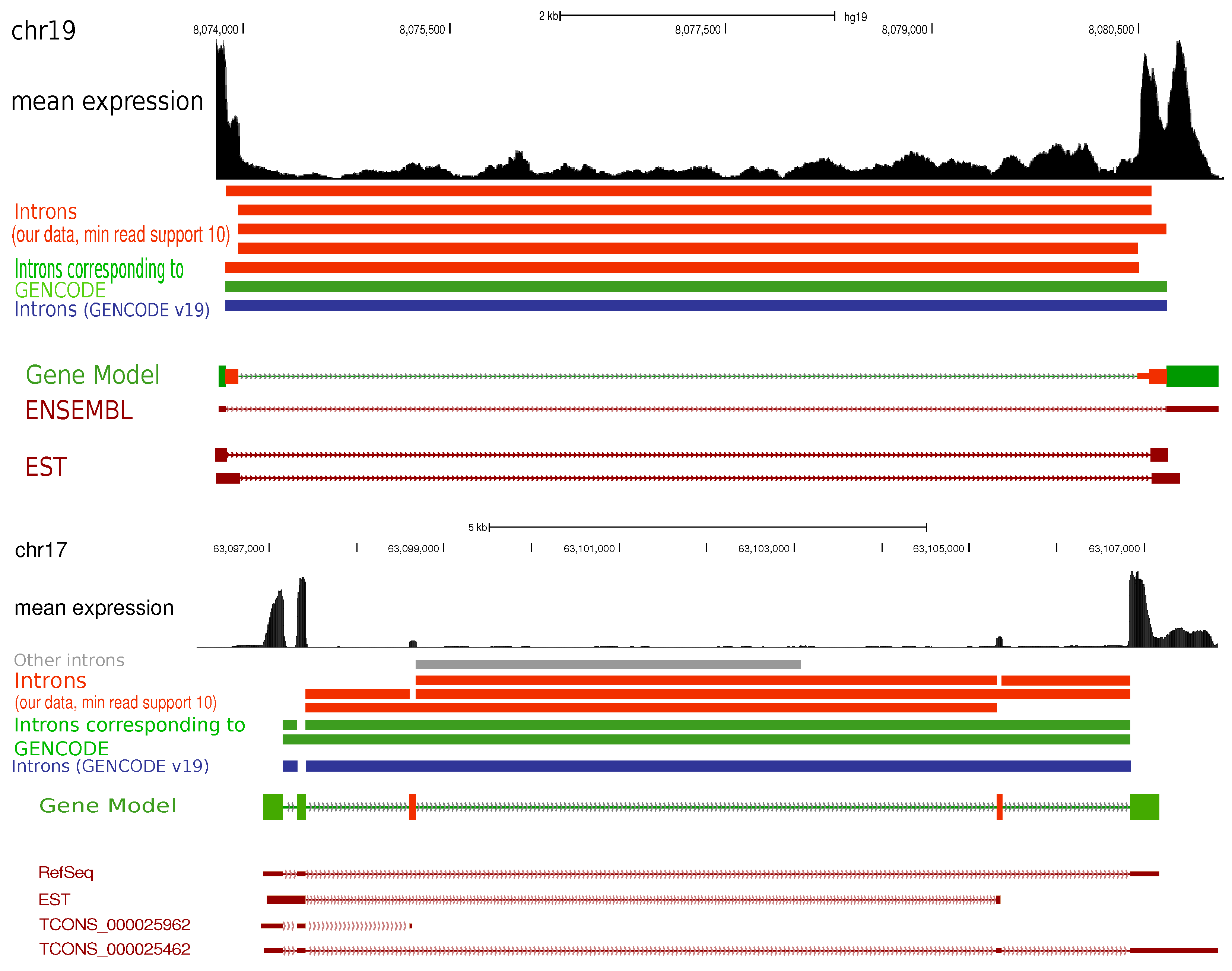
2. Results
3. Concluding Remarks
4. Materials and Methods
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| lncRNA | long non-coding RNA |
| lincRNA | long intergenic non-coding RNA |
| rRNA | ribosomal RNA |
| RPKM | reads per kilobase per million mapped reads |
| BL | Burkitt lymphoma |
| FL | follicular lymphoma |
| DLBCL | diffuse large B cell lymphoma |
References
- Clark, M.B.; Amaral, P.P.; Schlesinger, F.J.; Dinger, M.E.; Taft, R.J.; Rinn, J.L.; Ponting, C.P.; Stadler, P.F.; Morris, K.J.; Morillon, A.; et al. The reality of pervasive transcription. PLoS Biol. 2011, 9, e1000625. [Google Scholar] [CrossRef] [PubMed]
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar]
- Li, Z.; Yu, X.; Shen, J. ANRIL: A pivotal tumor suppressor long non-coding RNA in human cancers. Tumour Biol. 2016, 37, 5657–5661. [Google Scholar] [CrossRef] [PubMed]
- Aguilo, F.; Di Cecilia, S.; Walsh, M.J. Long Non-coding RNA ANRIL and Polycomb in Human Cancers and Cardiovascular Disease. Curr. Top. Microbiol. Immunol. 2016, 394, 29–39. [Google Scholar] [PubMed]
- Yu, X.; Zheng, H.; Chan, M.T.; Wu, W.K. HULC: An oncogenic long non-coding RNA in human cancer. J. Cell. Mol. Med. 2017, 21, 410–417. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Peng, W.X.; Mo, Y.Y.; Luo, D. MALAT1-mediated tumorigenesis. Front. Biosci. 2017, 22, 66–80. [Google Scholar]
- Li, Z.; Shen, J.; Chan, M.T.; Wu, W.K. TUG1: A pivotal oncogenic long non-coding RNA of human cancers. Cell Prolif. 2016, 49, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Da Rocha, S.T.; Heard, E. Novel players in X inactivation: Insights into Xist-mediated gene silencing and chromosome conformation. Nat. Struct. Mol. Biol. 2017, 24, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Rinn, J.L. Modular regulatory principles of large non-coding RNAs. Nature 2012, 482, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.; Li, Y.; Zhou, L.; Cho, J.; Patel, B.; Terada, N.; Li, Y.; Bungert, J.; Qiu, Y.; Huang, S. HoxBlinc RNA Recruits Set1/MLL Complexes to Activate Hox Gene Expression Patterns and Mesoderm Lineage Development. Cell Rep. 2016, 14, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Luco, R.F. Retrotransposons jump into alternative-splicing regulation via a long noncoding RNA. Nat. Struct. Mol. Biol. 2016, 23, 952–954. [Google Scholar] [CrossRef] [PubMed]
- Harrow, J.; Frankish, A.; Gonzalez, J.M.; Tapanari, E.; Diekhans, M.; Kokocinski, F.; Aken, B.; Barrell, D.; Zadissa, A.; Searle, S.; et al. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res. 2012, 22, 1760–1774. [Google Scholar] [CrossRef] [PubMed]
- Cabili, M.N.; Trapnell, C.; Goff, L.; Koziol, M.; Tazon-Vega, B.; Regev, A.; John, L.R. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011, 25, 1915–1927. [Google Scholar] [CrossRef] [PubMed]
- Mas-Ponte, D.; Carlevaro-Fita, J.; Palumbo, E.; Hermoso Pulido, T.; Guigò, R.; Johnson, R. LncATLAS database for subcellular localisation of long noncoding RNAs. RNA 2017, 23, 1080–1087. [Google Scholar] [CrossRef] [PubMed]
- Hon, C.C.; Ramilowski, J.A.; Harshbarger, J.; Bertin, N.; Rackham, O.J.; Gough, J.; Denisenko, E.; Schmeier, S.; Poulsen, T.M.; Severin, J.; et al. An atlas of human long non-coding RNAs with accurate 5’ends. Nature 2017, 543, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Holdt, L.M.; Hoffmann, S.; Sass, K.; Langenberger, D.; Scholz, M.; Krohn, K.; Finstermeier, K.; Stahringer, A.; Wilfert, W.; Beutner, F.; et al. Alu Elements in ANRIL Non-Coding RNA at Chromosome 9p21 Modulate Atherogenic Cell Functions through Trans-Regulation of Gene Networks. PLoS Genet. 2013, 9, e1003588. [Google Scholar] [CrossRef] [PubMed]
- Bozgeyik, E.; Igci, Y.Z.; Sami Jacksi, M.F.; Arman, K.; Gurses, S.A.; Bozgeyik, I.; Pala, E.; Yumrutas, O.; Temiz, E.; Igci, M. A novel variable exonic region and differential expression of LINC00663 non-coding RNA in various cancer cell lines and normal human tissue samples. Tumour Biol. 2016, 37, 8791–8798. [Google Scholar] [CrossRef] [PubMed]
- Mercer, T.R.; Wilhelm, D.; Dinger, M.E.; Soldà, G.; Korbie, D.J.; Glazov, E.A.; Truong, V.; Schwenke, M.; Simons, C.; Matthaei, K.I.; et al. Expression of distinct RNAs from 3’ untranslated regions. Nucleic Acids Res. 2011, 39, 2393–2403. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, J.; Stadler, P.F. Evolution of the unspliced transcriptome. BMC Evol. Biol. 2015, 15, 166. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, S.; Otto, C.; Doose, G.; Tanzer, A.; Langenberger, D.; Christ, S.; Kunz, M.; Holdt, L.M.; Teupser, D.; Hackermüller, J.; et al. A multi-split mapping algorithm for circular RNA, splicing, trans-splicing, and fusion detection. Genome Biol. 2014, 15, R34. [Google Scholar] [CrossRef] [PubMed]
- Ensembl 83. Available online: ftp://ftp.ensembl.org/pub/release-83/gtf/ (accessed on 9 March 2016).
- GENCODE v.24. Available online: http://www.gencodegenes.org/releases/24.html (accessed on 21 March 2016).
- Ensembl 60. Available online: ftp://ftp.ensembl.org/pub/release-60/gtf/ (accessed on 12 June 2017).
- Richter, J.; Schlesner, M.; Hoffmann, S.; Kreuz, M.; Leich, E.; Burkhardt, B.; Rosolowski, M.; Ammerpohl, O.; Wagener, R.; Bernhart, S.H.; et al. Recurrent mutation of the ID3 gene in Burkitt Lymphoma identified by integrated genome, exome and transcriptome sequencing. Nat. Genet. 2012, 44, 1316–1320. [Google Scholar] [CrossRef] [PubMed]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef] [PubMed]
- Ellis, J.D.; Barrios-Rodiles, M.; Çolak, R.; Irimia, M.; Kim, T.; Calarco, J.A.; Wang, X.; Pan, Q.; O’Hanlon, D.; Kim, P.M.; et al. Tissue-specific alternative splicing remodels protein-protein interaction networks. Mol. Cell 2012, 46, 884–892. [Google Scholar] [CrossRef] [PubMed]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed]
- Washietl, S.; Kellis, M.; Garber, M. Evolutionary dynamics and tissue specificity of human long noncoding RNAs in six mammals. Genome Res. 2014, 24, 616–628. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, S.; Otto, C.; Kurtz, S.; Sharma, C.; Khaitovich, P.; Vogel, J.; Stadler, P.F.; Hackermüller, J. Fast mapping of short sequences with mismatches, insertions and deletions using index structures. PLoS Comput. Biol. 2009, 5, e1000502. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- GENCODE v.7. Available online: http://www.gencodegenes.org/releases/7.html (accessed on 20 March 2016).
- Flicek, P.; Amode, M.R.; Barrell, D.; Beal, K.; Billis, K.; Brent, S.; Carvalho-Silva, D.; Clapham, P.; Coates, G.; Fitzgerald, S.; et al. Ensembl 2014. Nucleic Acids Res. 2014, 42, 749–755. [Google Scholar] [CrossRef] [PubMed]
- Flicek, P.; Amode, M.R.; Barrell, D.; Beal, K.; Brent, S.; Carvalho-Silva, D.; Clapham, P.; Coates, G.; Fairley, S.; Fitzgerald, S.; et al. Ensembl 2012. Nucleic Acids Res. 2012, 40, D84–D90. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Li, H.; Fang, S.; Kang, Y.; Wu, W.; Hao, Y.; Li, Z.; Bu, D.; Sun, N.; Zhang, M.Q.; et al. NONCODE 2016: An informative and valuable data source of long non-coding RNAs. Nucleic Acids Res. 2016, 44, D203–D208. [Google Scholar] [CrossRef] [PubMed]
- NONCODE 2016. Available online: http://www.noncode.org/ (accessed on 20 July 2016).
- Supplemental Material from Integrative Annotation of Human Large Intergenic Noncoding RNAs Reveals Global Properties and Specific Subclasses. Available online: http://genesdev.cshlp.org/content/25/18/1915/suppl/DC1 (accessed on 11 May 2016).

{kind=link}
{kind=link}
{kind=link}
| Genes | Transcripts | Exons | (Avg.) | Introns | (Avg.) | |
|---|---|---|---|---|---|---|
| Ensembl 60 | 1443 | 1703 | 4921 | 2.89 | 3218 | 1.88 |
| Cabili 2011 | 8263 | 14,353 | 33,045 | 2.30 | 18,607 | 1.30 |
| NONCODE 2016 | 160,376 | 233,696 | 536,111 | 2.29 | 305,771 | 1.31 |
| GENCODE v7 | 9580 | 14,984 | 42,060 | 2.81 | 28,998 | 1.94 |
| GENCODE v24 | 15,941 | 28,031 | 68,457 | 2.44 | 45,016 | 1.61 |
| Genes | Transcripts | Exons | (Avg.) | Introns | (Avg.) | |
|---|---|---|---|---|---|---|
| v7 | 3296 | 4563 | 12,584 | 2.76 | 8394 | 1.84 |
| v19 | 5257 | 7487 | 18,774 | 2.51 | 12,010 | 1.60 |
| v24 | 4961 | 7318 | 18,685 | 2.55 | 12,202 | 1.67 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sen, R.; Doose, G.; Stadler, P.F. Rare Splice Variants in Long Non-Coding RNAs. Non-Coding RNA 2017, 3, 23. https://doi.org/10.3390/ncrna3030023
Sen R, Doose G, Stadler PF. Rare Splice Variants in Long Non-Coding RNAs. Non-Coding RNA. 2017; 3(3):23. https://doi.org/10.3390/ncrna3030023
Chicago/Turabian StyleSen, Rituparno, Gero Doose, and Peter F. Stadler. 2017. "Rare Splice Variants in Long Non-Coding RNAs" Non-Coding RNA 3, no. 3: 23. https://doi.org/10.3390/ncrna3030023
APA StyleSen, R., Doose, G., & Stadler, P. F. (2017). Rare Splice Variants in Long Non-Coding RNAs. Non-Coding RNA, 3(3), 23. https://doi.org/10.3390/ncrna3030023