
miR-146 and miR-155: Two Key Modulators of Immune Response and Tumor Development



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
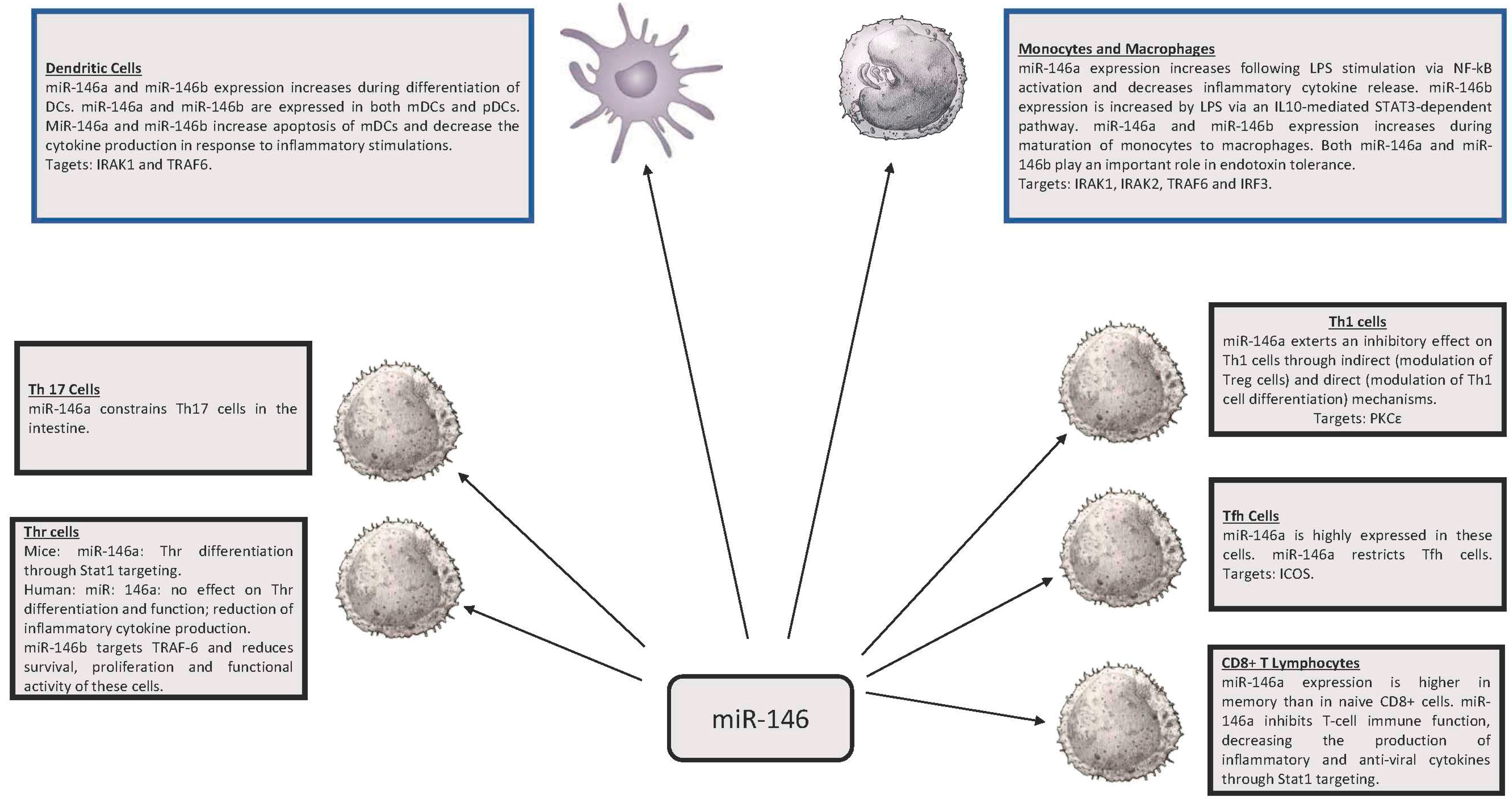
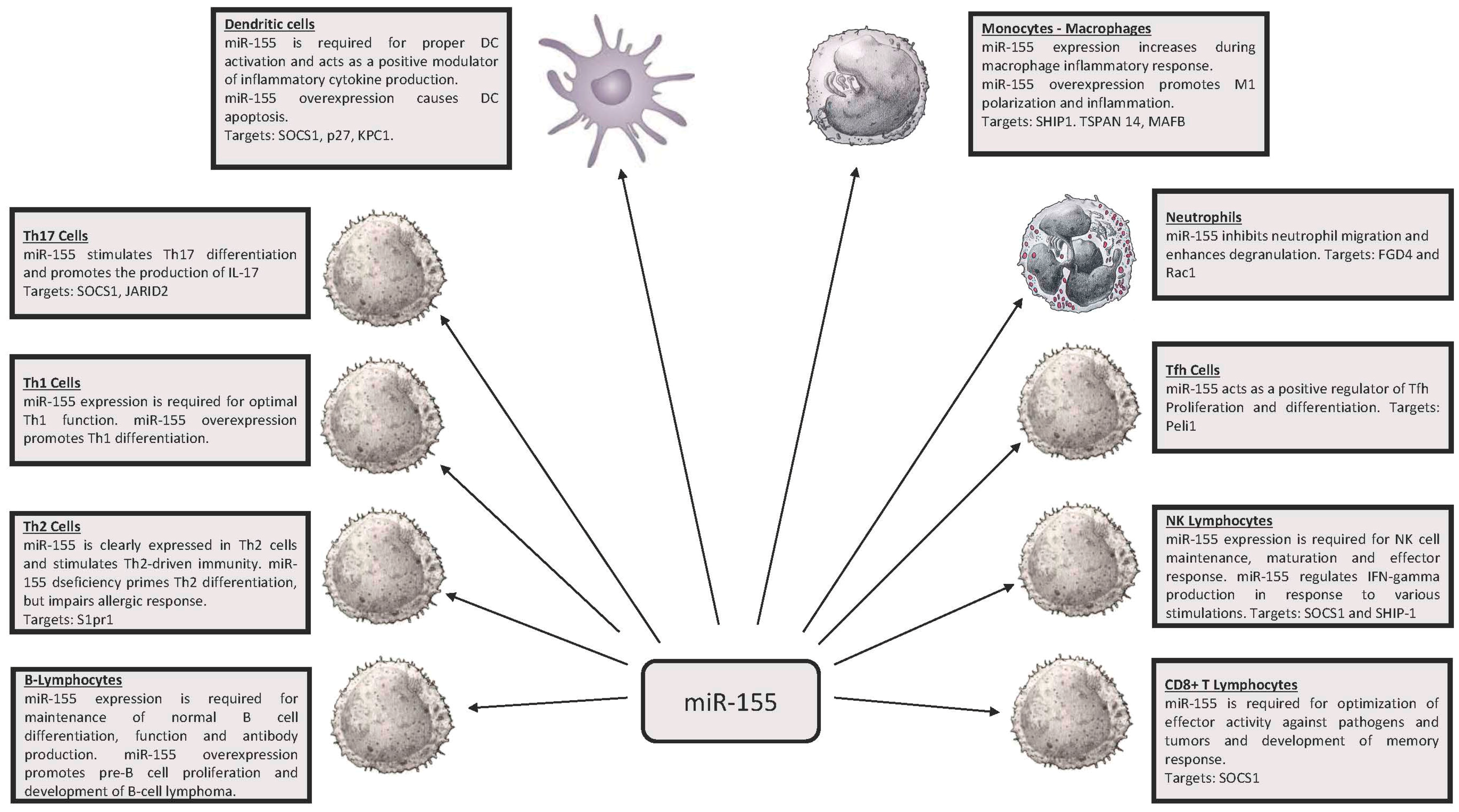
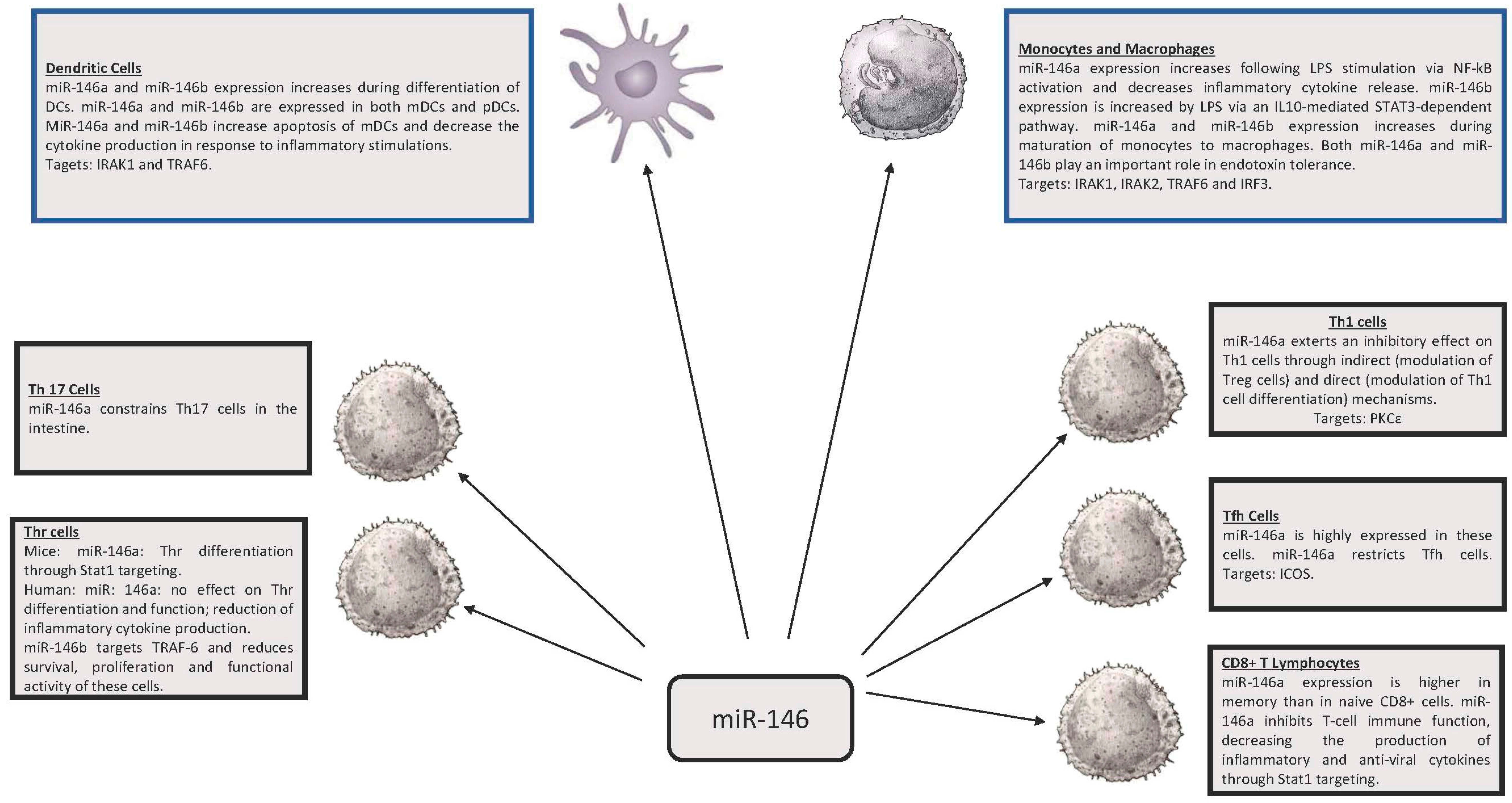
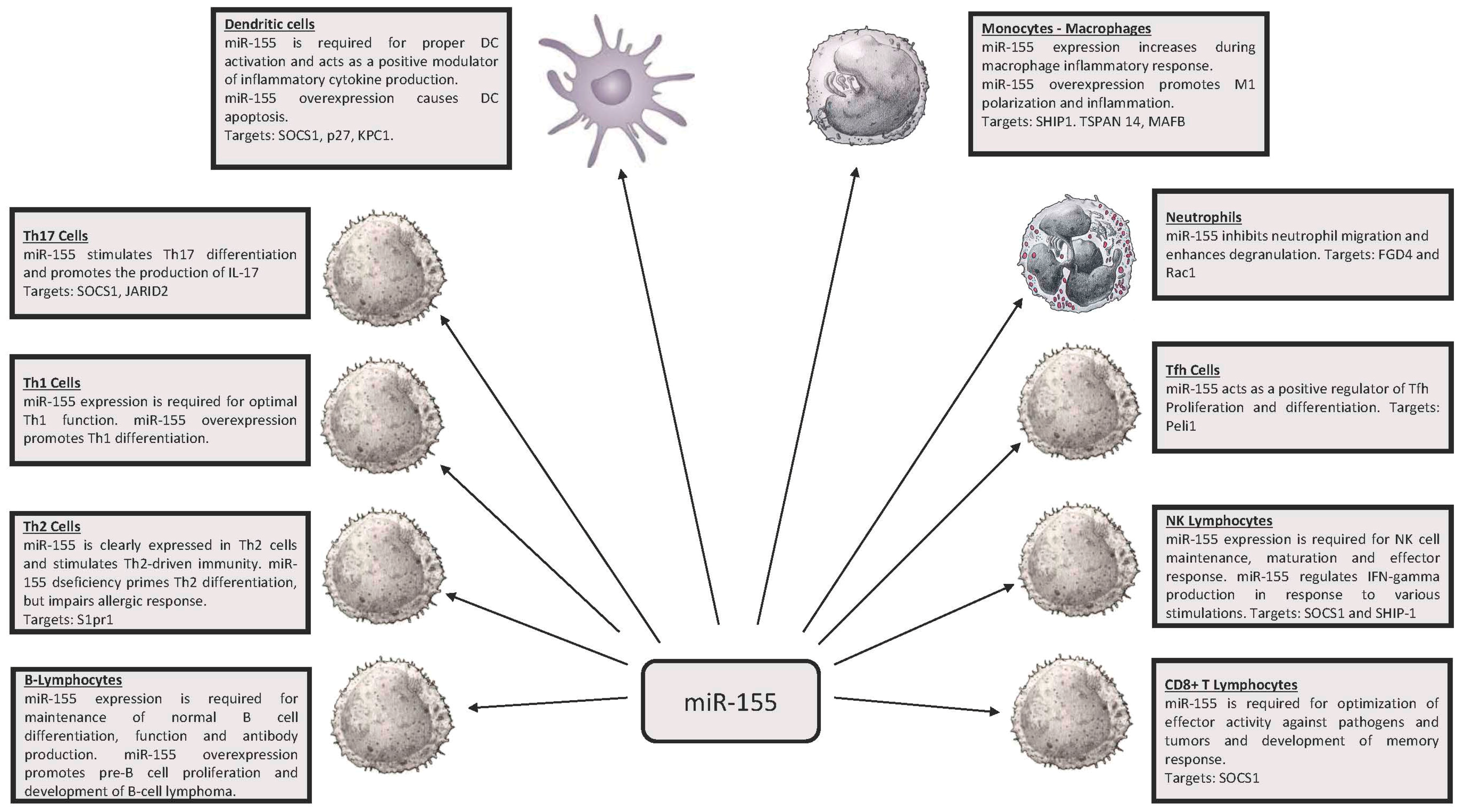
2. miR-146, miR-155 and Innate Immunity
3. miR-146 and miR-155 in Adaptive Immunity
3.1. Regulatory T Lymphocytes (Treg)
3.2. Th1 and Th2
3.3. T Follicular Helpers (Tfh) Cells
3.4. CD8+ Lymphocytes
3.5. T Helper 17 Lymphocytes (Th17)
4. miR-146 and miR-155 in Myeloid Neoplasia
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal. Transduct. Target. Ther. 2016. [Google Scholar] [CrossRef]
- Kim, Y.K.; Kim, B.; Kim, V.N. Re-evaluation of the roles of DROSHA, Export in 5, and DICER in microRNA biogenesis. Proc. Natl. Acad. Sci. USA 2016, 113, E1881–E1889. [Google Scholar] [CrossRef] [PubMed]
- Available online: http://www.mirbase.org (accessed on 1 June 2017).
- Okada, H.; Kohanbash, G.; Lotze, M.T. MicroRNAs in immune regulation-opportunities for cancer immunotherapy. Int. J. Biochem. Cell. Biol. 2010, 42, 1256–1261. [Google Scholar] [CrossRef] [PubMed]
- Paladini, L.; Fabfris, L.; Bottai, G.; Raschioni, C.; Calin, G.A.; Santarpia, L. Targeting microRNAs as key modulators of tumor immune response. J. Exp. Clin. Cancer Res. 2016, 35, 103. [Google Scholar] [CrossRef] [PubMed]
- Labbaye, C.; Testa, U. The emerging role of miR-146a in the control of hematopoiesis, immune function and cancer. J. Hematol. Oncol. 2012, 5, 13. [Google Scholar] [CrossRef] [PubMed]
- Mashima, R. Physiological roles of miR-155. Immunology 2015, 145, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.L.; Rao, D.S.; Boldin, M.P.; Taganov, K.D.; O’Connell, R.M.; Baltimore, D. NF-kappaB dysregulation in microRNA-146a-deficient mice drives the development of myeloid malignancies. Proc. Natl. Acad. Sci. USA 2011, 108, 9184–9189. [Google Scholar] [CrossRef] [PubMed]
- Boldin, M.P.; Taganov, K.; Rao, D.; Yang, L.; Zhao, J.; Kalwani, M.; Garcia-Flores, Y.; Loung, M.; Devrekanli, A.; Xu, J.; et al. miR-146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice. J. Exp. Med. 2011, 208, 1189–1201. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.F.; Boldin, M.P.; Chaudry, A.; Liu, L.L.; Taganov, K.; Hanada, T.; Yoshimura, A.; Baltimore, D.; Rudenshy, A. Function of miR-146a in controlling Treg-mediated regulation of Th1 responses. Cell 2010, 142, 914–929. [Google Scholar] [CrossRef] [PubMed]
- Tam, W.; Ben-Yehuda, D.; Hayward, W.S. Bic, a novel gene activated by proviral insertions in avian leucosis virus-induced lymphomas, is likely to function through its noncoding RNA. Mol. Cell. Biol. 1997, 17, 1490–1502. [Google Scholar] [CrossRef] [PubMed]
- Tam, W. Identification and characterization of human BIC, a gene on chromosome 21 that encodes a noncoding RNA. Gene 2001, 274, 157–167. [Google Scholar] [CrossRef]
- Eis, P.S.; Tam, W.; Sun, L.; Chadburn, A.; Li, Z.; Gomez, M.F.; Lund, E.; Dahlberg, J.E. Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proc. Natl. Acad. Sci. USA 2005, 102, 3627–3632. [Google Scholar] [CrossRef] [PubMed]
- Cui, B.; Chen, L.; Zhang, S.; Mraz, M.; Fecteau, J.F.; Yu, J.; Ghia, E.M.; Zhang, L.; Bao, L.; Rassenti, L.Z.; et al. MicroRNA-155 influences B cell receptor signaling and associates with aggressive disease in chronic lymphocytic leukemia. Blood 2014, 124, 546–554. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.; Vigorito, E.; Clare, S.; Warren, V.; Couttet, P.; Soond, D.R.; van Dongen, S.; Grocock, R.J.; Das, P.P.; Miska, E.A.; et al. Requirement of bic/microRNA-155 for normal immune function. Science 2007, 316, 608–611. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Breton, T.Y.; Oliveira, Y.J.; Zhou, A.; Aljouli, S.; Pube, S.; Cameron, R.P.; Sekaly, M.C.; Nussenzweig, M.C.; Liu, K. Restricted dendritic cell and monocyte progenitors in human cord blood and bone marrow. J. Exp. Med. 2015, 212, 385–399. [Google Scholar] [CrossRef] [PubMed]
- Breton, G.; Lee, J.; Zhou, Y.J.; Schreiber, J.J.; Keler, T.; Puhr, N.; Anandasabapathy, S.; Schlesinger, M.; Caskey, K.; Liu, K.; et al. Circulating precursors of human CD1c+ and CD141+ dendritic cells. J. Exp. Med. 2015, 212, 401–413. [Google Scholar] [CrossRef] [PubMed]
- Breton, G.; Zheng, S.; Valeris, R.; Tojal da Silva, I.; Satija, R.; Nessenszweig, M.C. Human dendritic cells (DCs) are derived from distinct circulating precursors that are precommitted to become CD1c+ or CD141+ DCs. J. Exp. Med. 2016, 213, 2861–2870. [Google Scholar] [CrossRef] [PubMed]
- de Azevedo, M.T.; Saad, S.T.; Gilli, S.C. IL4 and IFN alpha generation of dendritic of dendritic cells reveals great migratory potential and NFĸB and cJun expression in IL4DCs. Immunol. Invest. 2013, 42, 711–725. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Huang, X.; Zhang, X.; Roensch, K.; Cao, Q.; Nakayama, K.I.; Blazar, B.R.; Zeng, Y.; Zhou, X. miR-221 and miR-155 regulate human dendritic cell development, apoptosis, and IL-12 production through targeting of p27kip1, kPC1, and SOCS-1. Blood 2011, 117, 4293–4303. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Huang, X.; Lu, C.; Cairo, M.S.; Zhou, X. MicroRNA-146a and MicroRNA-146b regulate human dendritic cell apoptosis and cytokine production by targeting TRAF6 and IRAK1 proteins. J. Biol. Chem. 2015, 290, 2831–2841. [Google Scholar] [CrossRef] [PubMed]
- Jurkin, J.; Schichl, Y.M.; Koeffel, R.; Bauer, T.; Richter, S.; Konradi, S.; Gesslbauer, B.; Starbl, H. miR-146a is differentially expressed by myeloid dendritic cell subsets and desensitizes cells to TLR2-dependent activation. J. Immunol. 2010, 184, 4955–4965. [Google Scholar] [CrossRef] [PubMed]
- Karrich, J.J.; Jachimowski, L.; Libouban, M.; Iyer, K.; Tanman-Kueter, E.W.; Nagasawa, M.; de Jong, E.C.; Uittebogaart, C.H.; Blom, B. MicroRNA-146a regulates survival and maturation of human plasmacytoid dendritic cells. Blood 2013, 122, 3001–3009. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Thai, T.H.; Calado, D.P.; Chaudry, A.; Kubo, M.; Tanaka, K.; Loeb, G.B.; Lee, H.; Yoshimura, A.; Rajewsky, K.; et al. Foxp3-dependent microRNA-155 confers competitive fitness to regulatory T cells by targeting SOCS1 protein. Immunity 2009, 30, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Ceppi, M.; Pereira, P.M.; Dunand-Sauthier, I.; Barras, E.; Reith, W.; Santos, M.A.; Pierre, P. MicroRNA-155 modulates the interleukin-1 signaling pathway in activated human monocyte-derived dendritic cells. Proc. Natl. Acad. Sci. USA 2009, 106, 2735–2740. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Nunez, R.T.; Louafi, F.; Friedmann, P.S.; Sanchez-Elsner, T. Micro RNA-155 modulates pathogen binding ability of dendritic cells by down-regulation of DC-specific intercellular adhesion molecule-3 grabbing non-integrin (DC-SIGN). J. Biol. Chem. 2009, 284, 16334–16342. [Google Scholar] [CrossRef] [PubMed]
- Lind, E.; Millar, D.; Dissanayake, D.; Savage, J.; Grimshaw, N.; Kerr, W.; Ohashi, P. miR-155 upregulation in dendritic cells is sufficient to break tolerance in vivo by negatively regulating SHIP1. J. Immunol. 2015, 195, 4632–4640. [Google Scholar] [CrossRef] [PubMed]
- Toganov, K.D.; Boldin, M.P.; Chang, K.J.; Baltimore, D. NF-kB-dependent induction of microRNA miR-146, an inhibitor targeted of signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12481–12486. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Wang, P.; Lin, L.; Ma, F.; An, H.; Wang, Z.; Cao, X. Micro-RNA-146a feedback inhibits RIG-1-dependent type I IFN production in macrophages by targeting TRAF6, IRAK1, and IRAK2. J. Immunol. 2009, 183, 2150–2158. [Google Scholar] [CrossRef] [PubMed]
- Eigsti, R.L.; Sudan, B.; Wilson, M.E.; Graff, J.W. Regulation of activation-associated microRNA accumulation rates during monocyte-to-macrophage differentiation. J. Biol. Chem. 2014, 289, 28433–28437. [Google Scholar] [CrossRef] [PubMed]
- Curtale, G.; Mirolo, M.; Renzi, T.A.; Rossato, M.; Bazzoni, F. Negative regulation of Toll-like receptor 4 signaling by IL10-dependent microRNA-146b. Proc. Natl. Acad. Sci. USA 2013, 110, 11499–11504. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Zhang, H.; Hao, Y.; Xu, F.; Yang, J.; Zhang, R.; Lu, G.; Zheng, Z.; Cui, M.; Qi, C.F.; et al. Reprogramming macrophage orientation by microRNA 146b targeting transcription factor IRF5. EBioMedicine 2016, 14, 83–96. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, R.M.; Taganov, K.D.; Boldin, M.P.; Cheng, G.; Baltimore, D. MicroRNA-155 is induced during the macrophage inflammatory response. Proc. Natl. Acad. Sci. USA 2007, 104, 1604–1609. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, R.M.; Chadhuri, A.A.; Rao, D.S.; Baltimore, D. Inositol phosphatase SHIP1 is a primary target of miR-155. Proc. Natl. Acad. Sci. USA 2009, 706, 1113–1118. [Google Scholar] [CrossRef] [PubMed]
- Graff, J.W.; Dickson, A.M.; Clay, G.; McCaffrey, A.P.; Wilson, M.E. Identifying functional microRNAs in macrophages with polarized phenotypes. J. Biol. Chem. 2012, 287, 21816–21825. [Google Scholar] [CrossRef] [PubMed]
- Jablonski, K.A.; Gaudet, A.D.; Amici, S.A.; Popovich, P.G.; Guereau-de-Arellano, M. Control of the inflammatory macrophage transcriptional signature by miR-155. PLoS ONE 2016, 11, e0159724. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Yin, Y.; Li, N.; Zhu, D.; Zhang, J.; Zhang, C.Y.; Zen, K. Re-polarization of tumor-associated macrophages to pro-inflammatory M1 macrophages by microRNA-155. J. Mol. Cell. Biol. 2012, 4, 341–343. [Google Scholar] [CrossRef] [PubMed]
- Androulidaki, A.; Iliopoulos, D.; Arranz, A.; Doxaki, C.; Schrower, S.; Zakarioudaki, V.; Margioris, A.N.; Tsichlis, P.N.; Tsatsanis, C. The kinase Akt1 controls macrophage response to lipopysaccharide by regulating microRNAs. Immunity 2009, 31, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Doxaki, C.; Kampranis, S.C.; Eliopoulos, A.G.; Spilianakis, C.; Tsatsanis, C. Coordinated regulation of miR-155 and miR-146a genes during induction of endotoxin tolerance in macrophage. J. Immunol. 2015, 195, 5750–5761. [Google Scholar] [CrossRef] [PubMed]
- Nahid, M.A.; Pauley, K.M.; Satoh, M.; Chan, E.K. miR-146a is critical for endotoxin-induced tolerance: Implication in innate immunity. J. Biol. Chem. 2009, 284, 34590–34599. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; John, C.M.; Jarvis, C.A. Induction of endotoxin tolerance by pathogenic Neisseria is correlated with the pathogenic potential of oligooligosaccharides and regulated by microRNA-146a. J. Immunol. 2014, 192, 1768–1777. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Meng, J.; Das, S.; Krishnan, A.; Haworth, J.; Charboneau, R.; Zeng, Y.; Ramakrishnan, S.; Roy, S. Morphine induced exacerbation of sepsis is mediated by tempering endotoxin tolerance through modulation of miR-146a. Sci. Rep. 2013, 3, 1977. [Google Scholar] [CrossRef] [PubMed]
- Chassin, C.; Kocur, M.; Pott, J.; Duerr, C.U.; Gutle, D.; Lotze, M.; Hornef, M.W. miR-146a mediates protective innate immune tolerance in the neonate intestine. Cell. Host Microbe 2010, 8, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Wang, X.; Zhang, X.; Ha, T.; Ma, H.; Liu, L.; Kalbfleisch, J.; Gao, X.; Kao, R.; Williams, D.; et al. Attenuation of cardiac dysfunction in polymicrobial sepsis bi microRNA-146a is mediated via targeting of IRAK1 and TRAF6 expression. J. Immunol. 2015, 195, 672–682. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wo, M.; Wen, J.; Li, M.; Zhen, X.; Feng, L.; Li, M.; Huang, X. MicroRNA-155 induction by mycobacterium bovic BCG enhances ROS production through targeting SHIP1. Mol. Immunol. 2014, 62, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wang, J.; Fang, Y.; Gong, S.; Li, M.; Wu, M.; Lai, X.; Zeng, G.; Wang, Y.; Yang, K.; Huang, X. micro-RNA-146a promotes mycobacterial survival in macrophages through suppressing nitric oxide production. Scient. Rep. 2016, 6, 23351. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Yue, Y.; Xu, W.; Xiong, S. MicroRNA-146° represses mycobasteria-induced inflammatory response and facilitates bacterial replication via targeting IRAK-1 and TRAF-6. PLoS ONE 2013, 8, e81438. [Google Scholar]
- Furci, L.; Schena, E.; Mitto, P.; Cirillo, D.M. Alteration of human macrophages microRNA expression profile upon infection with Mycobacterium tuberculosis. Int. J. Mycobacteriol. 2013, 2, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, S.V.; Diaz, A.; D’Attilio, L.; Marchesini, M.M.; Bogue, C.; Bay, C.; Boitasso, O.A. Altered microRNA expression levels in mononuclear cells of patients with pulmonary and pleural tuberculosis and their relation with components of the immune response. Mol. Immunol. 2013, 53, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhou, G.; Deng, X.; Yu, Q.; Sun, H.; Wang, Z.; Chen, H.; Jia, C.; Wang, D. Analysis of miRNA expression profiling in human macrophages responding to Mycobacterium infection: Induction of the immune regulator miR-146a. J. Infect. 2014, 68, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Li, F.; Jia, S.; Zhang, K.; Jiang, W.; Shang, Y.; Chang, K.; Deng, S.; Chen, M. Eraly secreted antigen ESAT-6 of Mycobacterium tuberculosis promotes apoptosis of macrophages via targeting the microRNA-155-SOCS1 interaction. Cell. Physiol. Biochem. 2015, 35, 1276–1288. [Google Scholar] [CrossRef] [PubMed]
- Ghorpade, D.S.; Leyland, R.; Kurowska-Stolarska, M.; Patil, S.A.; Balaji, K.N. MicroRNA-155 is required for Mycobacterium bovis BCG-mediated apoptosis of macrophages. Mol. Cell. Biol. 2012, 32, 2239–2253. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Halder, P.; Sahu, S.K.; Kumar, M.; Kumori, M.; Jana, K. Identification of a novel role of ESAT-6-dependent miR-155 induction during infection of macrophages with Mycobaterium tuberculosis. Cell. Microbiol. 2012, 14, 1620–1631. [Google Scholar] [CrossRef] [PubMed]
- Rotchild, A.C.; Sisson, J.R.; Shafiani, S.; Plaisier, C.; Min, D.; Mai, D.; Gilchrist, M.; Peschon, J.; Larson, R.P.; Bergthaler, A.; et al. MiR-155-regulated molecular network orchestrates cell fate in the innate and adoptive immune response to Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2016, 113, E6172–E6181. [Google Scholar] [CrossRef] [PubMed]
- Bhairavabholta, R.; Kim, Y.; Glass, D.; Escobar, T.; Patel, M.; Zahr, R.; Nguyen, C.; Kilaru, G.; Mulp, S.; Shevach, E. Transcriptome profiling of human FoxP3+ regulatory T cells. Hum. Immunol. 2016, 77, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Holstrom, K.; Pedersen, A.E.; Gad, M. Analysis of miR-146a and miR142–3p as potential markers of freshly isolated or in vitro-expanded human Treg cells. Scand. J. Immunol. 2017, 85, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Haupt, S.; Kreuzer, J.; Hammitizsch, A.; Proft, F.; Neumann, C.; Leipe, J.; Witt, M.; Schulze-Koops, H.; Sapenko, A. Decreased expression of miR-146a and miR-155 contributes to an abnormal phenotype in patients with rheumatoid arthritis. Ann. Rheum. Dis. 2015, 74, 1265–1274. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Hippen, K.; Lemire, A.; Cu, J.; Wang, W.; Ni, X.; Ranganathan, P.; Levine, B.; Riley, J.; June, C.; et al. miR-146b antagomir-treated human Tregs acquire increased GVHD inhibitory potency. Blood 2016, 128, 1424–1435. [Google Scholar] [CrossRef] [PubMed]
- Kohlhaas, S.; Garden, O.A.; Scudamore, C.; Turner, M.; Okkenhaug, K.; Vigorito, E. Cutting edge: The Foxp3 target miR-155 contributes to the development of regulatory T cells. J. Immunol. 2009, 182, 2578–2582. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Diaz, R.; Blanco-Dominguez, R.; Lasarte, S.; Tsilingiri, K.; Martin-Gayo, E.; Linilos-Pradilo, B.; de la Fuente, O.; Sanchez-Madrid, F.; Nagakawa, R.; Toribio, M.; et al. Thymus-derived regulatory T cell development is regulated by C-type lectin-mediated BIC/microRNA 155 expression. Mol. Cell. Biol. 2017, 37, e00341–e003416. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Boldin, M.P.; Yu, Y.; Liu, C.S.; Ea, C.K.; Ramakrishnan, P.; Taganov, K.D.; Zho, J.L.; Baltimore, D. miR-146a controls the resolution of T cell responses in mice. J. Exp. Med. 2012, 209, 1655–1670. [Google Scholar] [CrossRef] [PubMed]
- Mohnle, P.; Schutze, S.; van der Heide, V.; Hubner, M.; Luchting, B.; Sedlbauer, J.; Limbeck, E.; Hinske, L.; Briegel, J.; Kreth, S. Micro-RNA-146a controls Th1 cell differentiation of human CD4+ T lymphocytes by targeting PRKCε. Eur. J. Immunol. 2015, 45, 260–272. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Schambech, F.; De Jong, C.; Hammond, S.; Reiner, S. MicroRNA-155 inhibits IFN-γ signaling in CD4+ T cells. Eur. J. Immunol. 2010, 40, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Oertli, M.; Erigler, D.B.; Kohler, E.; Kock, M.; Meyer, T.F.; Mueller, A. MicroRNA-155 is essential for the T cell mediated control of Helicobacter pylori infection and for the induction of chronic gastritis and colitis. J. Immunol. 2011, 187, 3578–3586. [Google Scholar] [CrossRef] [PubMed]
- Okoye, I.S.; Czieso, S.; Ktistaki, E.; Roderick, K.; Coomes, S.; Pelly, V.S.; Kannan, Y.; Perez-Lloret, J.; Zhao, J.; Baltimore, D.; et al. Transcriptomics identified a critical role for Th2 cell-intrinsic miR-155 in mediating allergy and antihelminth immunity. Proc. Natl. Acad. Sci. USA 2014, 111, E3081–E3090. [Google Scholar] [CrossRef] [PubMed]
- Malmhall, C.; Alawieh, S.; Lu, Y.; Sjostrand, M.; Bossios, A.; Eldh, M.; Radinger, M. MicroRNA-155 is essential for Th2-mediated allergen-induced eosiniphiliuc inflammation in the lung. J. Allergy Clin. Immunol. 2014, 133, 1429–1438. [Google Scholar] [CrossRef] [PubMed]
- Zech, A.; Ayata, C.K.; Pankratz, F.; Meyer, A.; Baudiss, K.; Cicko, S.; Yegutkin, G.G.; Grundmann, S.; Idzko, M. MicroRNA-155 modulates P2R signaling and Th2 priming of dendritic cells during allergic airway inflammation in mice. Allergy 2015, 70, 1121–1129. [Google Scholar] [CrossRef] [PubMed]
- Qayum, A.A.; Paranjape, A.; Abebayehu, D.; Kolawole, E.M.; Haque, T.T.; McLeod, J.J.; Spence, A.J.; Caslin, H.L.; Taruselli, M.T.; Chumanevick, A.P.; et al. Il-10-induced miR-155 targets SOCS1 to enhance IgE-mediated mast cell function. J. Immunol. 2016, 196, 4457–4467. [Google Scholar] [CrossRef] [PubMed]
- Pratama, A.; Srivastava, M.; Williams, N.; Papa, I.; Lee, S.; Dinh, X.; Hutloff, A.; Jordan, M.A.; Zhao, J.; Casellas, R.; et al. Micro-RNA146a regulates ICOS-ICSL signaling to limit accumulation of T follicular helper cells and germinal centres. Nat. Commun. 2015, 6, 6436. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Kagele, D.; Huffaker, T.; Runtsch, M.; Alexander, M.; Liu, J.; Bake, E.; Su, W.; Williams, M.; Rao, D.; et al. miR-155 promotes T follicular helper cell accumulation during chronic, low-grade inflammation. Immunity 2014, 41, 605–619. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.H.; Kang, S.G.; Huang, Z.; Wu, C.J.; Jin, H.Y.; Maine, C.; Liu, Y.; Shepherd, J.; Sahouri-Ghomi, M.; Gonzalez-Martin, A.; et al. A miR-155-Peli1-c-Rel pathway controls the generation and function of T follicular helper cells. J. Exp. Med. 2016, 213, 1901–1919. [Google Scholar] [CrossRef] [PubMed]
- Curtale, G.; Citarella, F.; Carissimi, C.; Goldoni, M.; Carucci, N.; Fulci, V.; Franceschini, D.; Meloni, F.; Barnaba, V.; Macino, G. An emerging player in the adaptive immune response: MicroRNA-146a is a modulator of IL-2 expression and activation-induced cell death in T lymphocytes. Blood 2010, 115, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhang, X.; Ju, Y.; Zhao, B.; Yan, X.; Hu, J.; Shi, L.; Yang, L.; Ma, Z.; Chen, L.; et al. MicroRNA-146a feedback suppresses T cell immune function by targeting Stat1 in patients with chronic hepatitis B. J. Immunol. 2013, 191, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Luo, X.; Cui, H.; Ni, X.; Yuan, M.; Guo, Y.; Huang, X.; Zhou, H.; de Vries, N.; Tak, P.P.; et al. MicroRNA-146a contributes to abnormal activation of the type I interferon pathway in human lupus by targeting the key signaling proteins. Arthritis Rheum. 2009, 60, 1065–1075. [Google Scholar] [CrossRef] [PubMed]
- Gracias, D.T.; Stelekati, E.; Hope, J.L.; Boesteanu, A.C.; Doering, T.A.; Norton, J.; Mueller, Y.M.; Fraietta, J.A.; Wherry, E.J.; Turner, M.; et al. The microRNA miR-155 controls CD8+ T cell responses by regulating interferon signaling. Nat. Immunol. 2013, 14, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Dudda, J.C.; Salaun, B.; Ji, Y.; Palmer, D.C.; Monnot, G.C.; Merck, E.; Boudousquie, C.; Utzschneider, D.T.; Escobar, T.M.; Perret, R. MicroRNA-155 is required for effector CD8+ T cell responses to virus infection and cancer. Immunity 2013, 38, 742–753. [Google Scholar] [CrossRef] [PubMed]
- Huffaker, T.B.; Hu, R.; Runtsch, M.C.; Bake, E.; Chen, X.; Zhao, J.; Round, J.L.; Baltimore, D.; O’Connell, R.M. Epistasis between microRNAs 155 and 146a during T cell-medaited antitumor immunity. Cell. Rep. 2012, 2, 1697–1709. [Google Scholar] [CrossRef] [PubMed]
- Runtsch, M.C.; Hu, R.; Alexander, M.; Wallace, J.; Kagele, D.; Petersen, C.; Valentine, J.F.; Welker, N.C.; Bronner, M.P.; Chen, X.; et al. MicroRNA-146a constrains multiple parameters of intestinal immunity and increases susceptibility to DSS colitis. Oncotarget 2015, 6, 28556–28572. [Google Scholar]
- Liu, L.; Hua, M.; Liu, C.; He, N.; Li, Z.; Ma, D. The aberrant expression of microRNAs and correlations with T cell subsets in patients with immune thrombocytopenia. Oncotarget 2016, 47, 76453–76463. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, R.; Kahn, D.; Gibson, W.; Round, T.; Schole, R.; Chaudhuri, A.; Kahn, M.; Rao, D.; Baltimore, D. MicroRNA-155 promotes autoimmune inflammation by enhancing inflammatory T cell development. Immunity 2010, 33, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Yao, R.; Ma, Y.L.; Liang, W.; Li, H.H.; Ma, Z.J.; Yu, X.; Liao, Y.H. MicroRNA-155 modulates Treg and Th17 cell differentiation and Th17 cell function by targeting SOCS1. PLoS ONE 2012, 7, e46082. [Google Scholar] [CrossRef] [PubMed]
- Ecsobar, T.M.; Kanellopoulou, C.; Kugler, D.G.; Kilaru, G.; Nguyen, G.K.; Nagarayan, V.; Bhairavabholta, R.; Northrup, D.; Zahr, R.; Burr, P.; et al. miR-155 activates cytokine gene expression in Th17 cells by regulating the DNA-binding protein Jarid2 to relieve polycomb-mediated repression. Immunity 2014, 40, 865–879. [Google Scholar]
- Hu, R.; Huffaker, T.; Kagele, D.; Runtsch, M.; Bake, E.; Chaudhuri, A.; Round, J.; O’Connell, R. MicroRNA-155 confers encephalogenic potential to Th17 cells by promoting effector gene expression. J. Immunol. 2013, 190, 5972–5980. [Google Scholar] [CrossRef] [PubMed]
- Kluiver, J.; Poppema, S.; de Jong, D.; Blokxijl, T.; Harms, G.; Jacobs, S.; Kroesen, B.J.; van den Berg, A. BIC and miR-155 are highly expressed in Hodgkin, primary mediastinal and diffuse large B cell lymphomas. J. Pathol. 2005, 207, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Costinean, S.; Zanesi, N.; Pekarsky, Y.; Tili, E.; Volinia, S.; Heerema, N.; Croce, C.M. Pre-B cell proliferation and lymphoblastic leukemia/high-grade lymphoma in E(mu)-miR155 transgenic mice. Proc. Natl. Acad. Sci. USA 2006, 103, 7024–7029. [Google Scholar] [CrossRef] [PubMed]
- Costinean, S.; Sandhu, S.K.; Pedersen, I.M.; Tili, E.; Trotta, R.; Perrotti, D.; Ciarlariello, D.; Neviani, P.; Harb, J.; Kauffman, L.R.; et al. Src homology 2 domain-containing inositol-5-phosphatase and CCAAT enhancer-binding protein beta are targeted by miR-155 in B cells of Emicro-MiR-155 transgenic mice. Blood 2009, 114, 1374–1382. [Google Scholar] [CrossRef] [PubMed]
- Zu, F.; Zeng, L.; Tang, N.; Tang, Y.; Zhou, B.; Li, F.; Wu, W.; Zeng, X.; Peng, S. MicroRNA-155 downregulation promotes cell cycle arrest and apoptosis in diffuse large B cell lymphoma. Oncol. Res. 2016, 24, 415–427. [Google Scholar]
- Garzon, R.; Garofalo, M.; Martelli, M.P.; Briesewitz, R.; Wang, L.; Fernandez-Cymering, C.; Volinia, S.; Liu, C.G.; Schnittger, S.; Haferlach, T.; et al. Distinctive microRNA signature of acute myeloid leukemia bearing cytoplasmic mutated nucleophormin. Proc. Natl. Acad. Sci. USA 2008, 105, 3945–3950. [Google Scholar] [CrossRef] [PubMed]
- Faraoni, I.; Laterza, S.; Ardiri, D.; Ciardi, C.; Fazi, F.; Lo-Coco, F. MiR-424 and miR-155 deregulated expression in cytogenetically normal acute myeloid leukemia: Correlation with NPM1 and FLT3 mutation status. J. Hematol. Oncol. 2012, 5, 26. [Google Scholar] [CrossRef] [PubMed]
- Whitman, S.P.; Maharry, K.; Radmacher, M.D.; Becker, H.; Mrozek, K.; Margeson, D.; Holland, K.B.; Wu, Y.Z.; Schwind, S.; Metzeler, K.H.; et al. FLT3 internal tandem duplication associates with adverse outcome and gene- and microRNA-expression signatures in patients 60 years of age or older with primary cytogenetically normal acute myeloid leukemia: A cancer and Leukemia Group B study. Blood 2010, 116, 3622–3626. [Google Scholar] [CrossRef] [PubMed]
- Marcucci, G.; Maharry, K.; Metzler, K.; Volinia, S.; Wu, Y.Z.; Mrozek, K.; Nicolet, D.; Kohlscmidt, J.; Whitman, S.; Mendler, J.; et al. Clinical role of microRNAs in cytogenetically normal acute myeloid leukemia: MiR-155 upregulation independently identifies high-risk patients. J. Clin. Oncol. 2013, 31, 2086–2093. [Google Scholar] [CrossRef] [PubMed]
- Metzler, K.; Maharry, K.; Kohlswchmidt, J.; Volinia, S.; Mrozek, K.; Becker, H.; Nicolet, D.; Whitman, S.P.; Mendler, J.H.; Schwind, S.; et al. A stem cell-like gene expression signature associates with inferior outcomes and a distinct microRNA expression profile in adults with primary cytogenetically normal acute myeloid leukemia. Leukemia 2013, 27, 2023–2031. [Google Scholar] [CrossRef] [PubMed]
- Gerloff, D.; Wurm, A.A.; Hartmann, J.U.; Hilger, N.; Muller, A.M.; Katzerke, C.; Brauer-Hartmannm, D.; Namasu, C.Y.; Cross, M.; Schwind, S.; et al. Next generation sequencing and functional analysis of Mirna expression in acute myeloid leukemia patients with different FLT3 mutations: Block of MIR-155 in FLT3-ITD driven AML leads to downregulation of myeloid blasts in vivo. Blood 2015, 126, 2438. [Google Scholar]
- Gerloff, D.; Grundler, R.; Wurm, A.A.; Bauer-Hartmann, D.; Katrerke, C.; Hartmann, J.U.; Madan, V.; Muller-Tidow, C.; Duyster, J.; Tenen, D.G.; et al. NF-ĸB/Stat5/miR-155 network targets PU.1 in FLT3-ITD-driven acute myeloid leukemia. Leukemia 2015, 29, 535–547. [Google Scholar] [CrossRef] [PubMed]
- Khalife, J.; Radomska, H.S.; Santhanam, R.; Huang, X.; Neviani, P.; Saultz, J.; Wang, H.; Wu, Y.Z.; Alachkar, H.; Anghelina, M.; et al. Pharmacological targeting of miR-155 via NEDD8-activating enzyme inhibitor MLN4924 (Pevonedistat) in FLT3-ITD acute myeloid leukemia. Leukemia 2015, 29, 1981–1992. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Hua, M.; Guo, M.; Luo, J.M. SHIP1 is targeted by miR-155 in acute myeloid leukemia. Oncol. Rep. 2014, 32, 2253–2259. [Google Scholar] [CrossRef] [PubMed]
- Forrest, A.R.; Kanamori-Katayama, M.; Tomaru, Y.; Lasmann, T.; Ninomiya, N.; Takahashi, Y.; deHoon, M.J.; Kubosaki, A.; Kaiho, A.; Suzuki, M.; et al. Induction of microRNAs, miR-155, miR-222, miR-424 and miR-503, promotes monocytic differentiation through combinatorial regulation. Leukemia 2010, 24, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Palma, C.A.; Sheikha, D.; Lim, T.K.; Bryant, A.; Vu, T.T.; Jayeswal, V. MicroRNA-155 as an inducer of apoptosis and cell differentiation in acute myeloid leukemia. Mol. Cancer 2014, 13, 79. [Google Scholar] [CrossRef] [PubMed]
- O’ Connell, R.; Rao, D.; Chaudhuri, A.; Boldin, M.P.; Tagonov, K.; Nicoll, J.; Paquette, R.; Baltimore, D. Sustained expression of microRNA-155 in hematopoietic stem cells causes a myeloproliferative disorder. J. Exp. Med. 2008, 205, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Chu, S.H.; Heiser, D.; Li, L.; Kaplan, I.; Collector, M.; Huso, D.; Sharkis, S.J.; Civin, C.; Small, D. FLT3-ITD knockin impairs hematopoietic stem cell quiescence/homeostasis, leading to myeloproliferative neoplasm. Cell Stem Cell 2012, 11, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Zheng, R.; Friedman, A.D.; Levis, M.; Li, L.; Weir, E.G.; Small, D. Internal tandem duplication mutation of FLT3 blocks myeloid differentiation through suppression of C/EBP alpha expression. Blood 2004, 103, 1883–1890. [Google Scholar] [CrossRef] [PubMed]
- Alemdehy, M.E.; de Looper, H.W.; Kavelaars, F.G.; Sanders, M.A.; Hoogenboeren, R.; Lowenberg, B.; Valk, P.J.; Touw, I.P.; Erkeland, S.J. MicroRNA-155 induces AML in combination with the loss of C/EBPA in mice. Leukemia 2016, 30, 2238–2241. [Google Scholar] [CrossRef] [PubMed]
- Zizter, N.C.; Ranganathan, P.; Dickinson, B.A.; Jackson, A.L.; Radman, D.M.; Croce, C.M.; Marcucci, G.; Garzon, R. Preclinical development of LNA Antimir-155 (MRG-106) in acute myeloid leukemia. Blood 2015, 126, 3802. [Google Scholar]
- Rialfkiaer, U.; Hagerdon, D.H.; Bangsgaard, N.; Lavendof, M.; Ahler, C.; Svensson, L.; Kopp, K.; Vannegaard, M.; Lanenborg, B.; Zibert, T.; et al. Diagnostic microRNA profiling in cutaneous T cell lymphoma (CTCL). Blood 2011, 118, 5891–5900. [Google Scholar] [CrossRef] [PubMed]
- Querfeld, C.; Pacheco, T.; Foss, F.M.; Halwani, A.S.; Porcu, P.; Seto, A.; Ruckman, J.; Landry, M.; Jackson, A.; Pestano, L.; et al. Preliminary results of a phase II trial evaluating MRG-106, a synthetic microRNA antagonist (LNA antimiR) of microRNA-155, in patients with CTCL. Blood 2016, 128, 1829. [Google Scholar]
- Spinello, I.; Quaranta, M.T.; Riccioni, R.; Riti, V.; Pasquini, L.; Pelosi, E.; Vitale, A.; Foà, R.; Testa, U.; Labbaye, C. MicroRNA-146a and AMD3100, two ways to control CXCR4 expression in acute myeloid leukemias. Blood Cancer J. 2011, 1, e26. [Google Scholar] [CrossRef] [PubMed]
- Lutherborrow, M.; Bryant, A.; Jayaswal, V.; Agapion, D.; Palma, C.; Yang, Y.H.; Ma, D. Expression profiling of cytogenetically normal acute myeloid leukemia identifies microRNAs that target genes involved in monocytic differentiation. Am. J. Hematol. 2011, 86, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Li, X.; Cai, J.; Zhang, Y.; Xu, L.; Xiao, F.; Liu, J.; Hu, J.; Chen, F. Up-regulated miR-146a expression in leukemia cells induced by G-CSF correlates with enhanced sensitivity to cytabarine treatment: A putative model predictive biomarker for chemotherapy. Blood 2016, 128, 1661. [Google Scholar]
- Zhong, X.; Xu, L.; Li, X.; Sheng, X.; Hu, J. Granulocyte colony-stimulating factor inhibits CXZCR4/SDF-1 signaling and overcomes stromal-mediated drug resistance in acute myeloid leukemia via miR-146a. Blood 2015, 126, 4763. [Google Scholar]
- Li, Z.; Lu, J.; Sun, M.; Mi, S.; Zhang, H.; Luo, R.; Chen, P.; Wang, Y.; Yan, M.; Qian, Z.; et al. Distinct microRNA expression profiles in acute myeloid leukemia with common translocations. Proc. Natl. Acad. Sci. USA 2008, 105, 15535–15540. [Google Scholar] [CrossRef] [PubMed]
- Mohr, S.; Doebele, C.; Comoglio, F.; Berg, T.; Beck, J.; Bohenberger, H.; Alexe, G.; Corso, J.; Strobel, P.; Wachter, A.; et al. HoxA9 and Meis1 cooperatively induce addiction to Syk signaling by suppressing miT-146a in acute myeloid leukemia. Cancer Cell. 2017, 31, 549–562. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, T.; Nakatake, M.; Kuwata, T.; Couzinet, A.; Goitsuka, R.; Tsutsumi, S.; Aburatani, H.; Valk, P.; Delwel, R.; Nakamura, T. MEIS1-mediated transactivation of synaptotagmin-like 1 promotes CXCL12/CXCR4 signaling and leukemogenesis. J. Clin. Invest. 2016, 126, 1664–1678. [Google Scholar] [CrossRef] [PubMed]
- Starczynowski, D.T.; Kuchenbauer, P.; Argiropoulos, B.; Sung, S.; Morin, R.; Muranyi, A.; Hirst, M.; Hogge, D.; Marra, M.; Wells, R.A.; et al. Identification of miR-145 and miR-146a as mediators of the 5q- syndrome phenotype. Nat. Med. 2010, 16, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.L.; Starczynowski, D.T. Role of microRNA-146a in normal and malignant hematopoietic stem cell function. Front. Genet. 2014, 5, 219. [Google Scholar] [CrossRef] [PubMed]
- Varney, M.E.; Niuederkorn, M.; Konno, H.; Matsumura, T.; Gohda, J.; Yoshida, N.; Akiyama, T.; Christie, S.; Fang, J.; Miller, D.; et al. Loss of Tifab, a del(5q) MDS gene, alters hematopoiesis through derepression of Toll-like receptor-TRAF6 signaling. J. Exp. Med. 2015, 212, 1967–1985. [Google Scholar] [CrossRef] [PubMed]
- Varney, M.E.; Choi, K.; Bolanos, L.; Christie, S.; Fang, J.; Grimes, L.H.; Maciejewski, J.P.; Inoue, J.I.; Starczynowski, D.T. Epistasis between TIFAB and miR-146a: Neighboring genes in del(5q) myelodysplastic syndrome. Leukemia 2017, 31, 491–495. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.F.; Gasteiger, G.; Yu, I.S.; Chaudry, A.; Hsin, J.P.; Lu, Y.; Bos, P.D.; Lin, L.L.; Zawislak, C.I.; Cho, S.; et al. A single miRNA-mRNA interaction affects the immune response in a context- and cell-type-specific manner. Immunity 2015, 43, 52–64. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Testa, U.; Pelosi, E.; Castelli, G.; Labbaye, C. miR-146 and miR-155: Two Key Modulators of Immune Response and Tumor Development. Non-Coding RNA 2017, 3, 22. https://doi.org/10.3390/ncrna3030022
Testa U, Pelosi E, Castelli G, Labbaye C. miR-146 and miR-155: Two Key Modulators of Immune Response and Tumor Development. Non-Coding RNA. 2017; 3(3):22. https://doi.org/10.3390/ncrna3030022
Chicago/Turabian StyleTesta, Ugo, Elvira Pelosi, Germana Castelli, and Catherine Labbaye. 2017. "miR-146 and miR-155: Two Key Modulators of Immune Response and Tumor Development" Non-Coding RNA 3, no. 3: 22. https://doi.org/10.3390/ncrna3030022
APA StyleTesta, U., Pelosi, E., Castelli, G., & Labbaye, C. (2017). miR-146 and miR-155: Two Key Modulators of Immune Response and Tumor Development. Non-Coding RNA, 3(3), 22. https://doi.org/10.3390/ncrna3030022