Insights into the Function of Long Noncoding RNAs in Sepsis Revealed by Gene Co-Expression Network Analysis

Abstract
:1. Introduction
2. Results
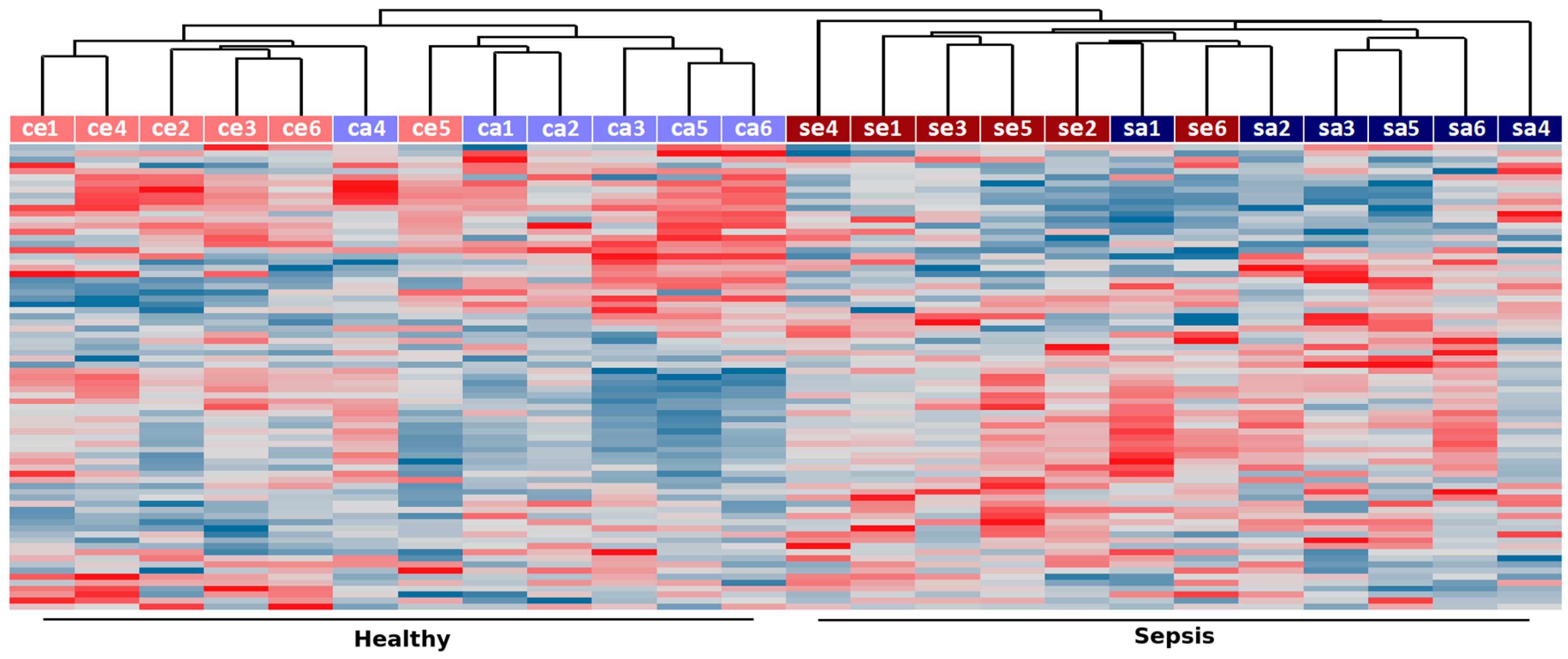
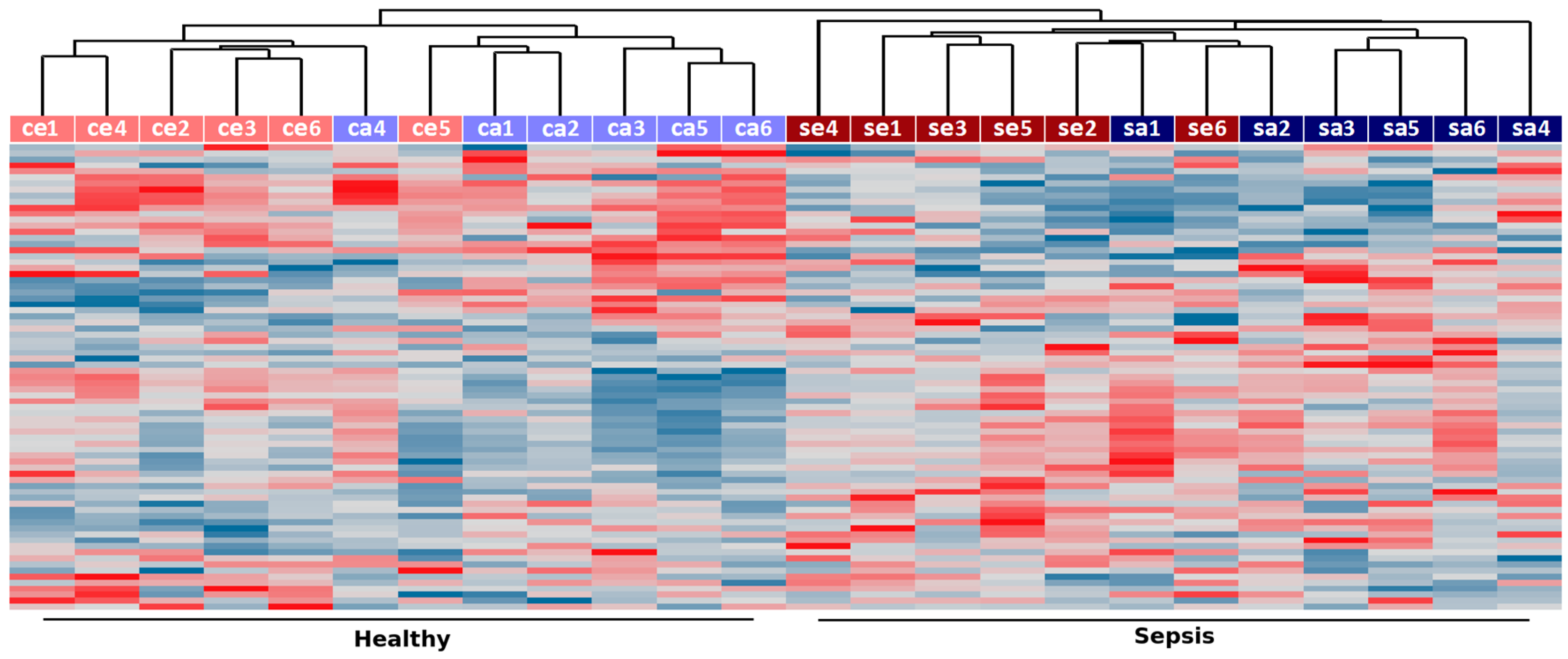
2.1. Identification of RNA Expression in the Innate Immune System in Sepsis
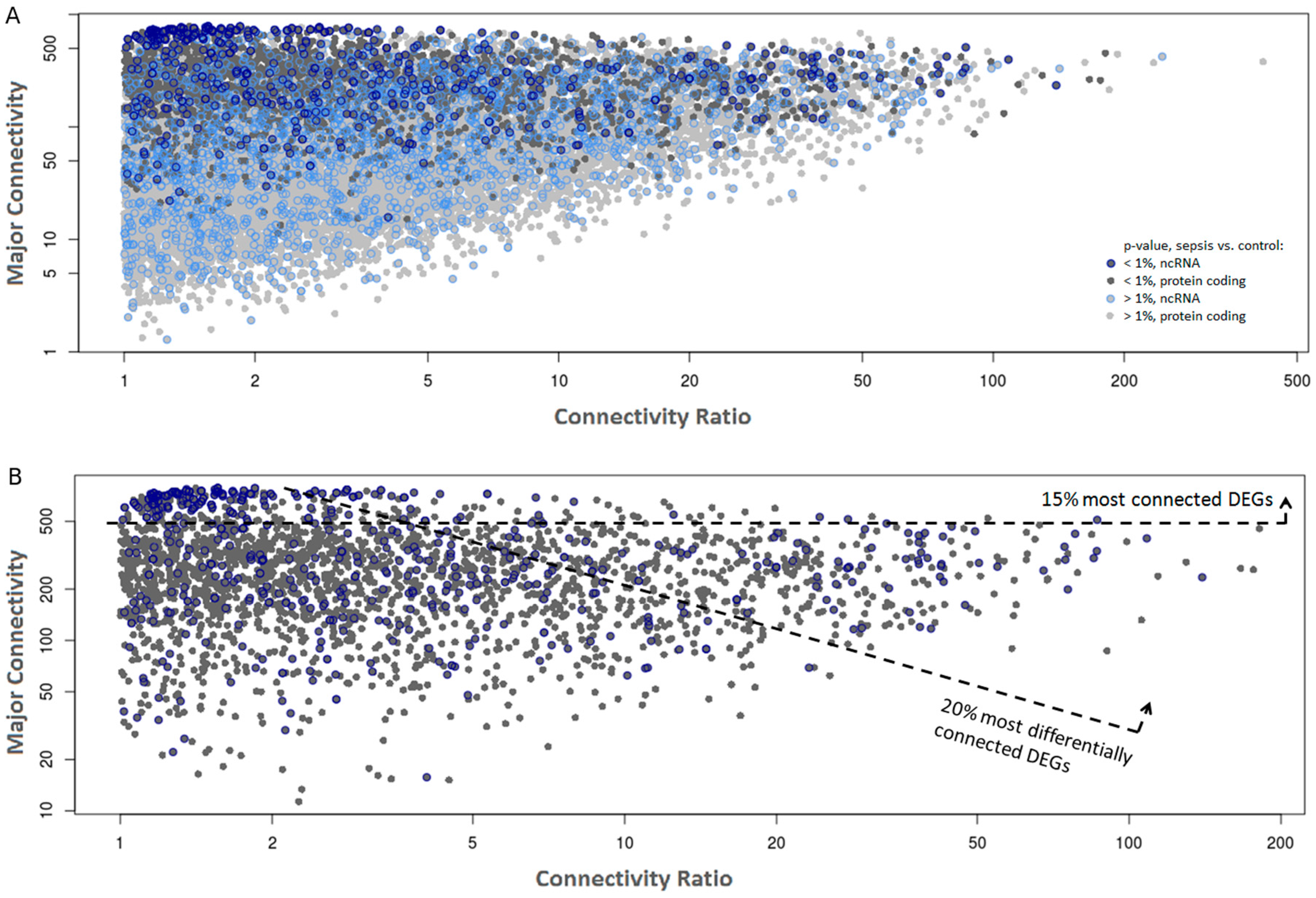
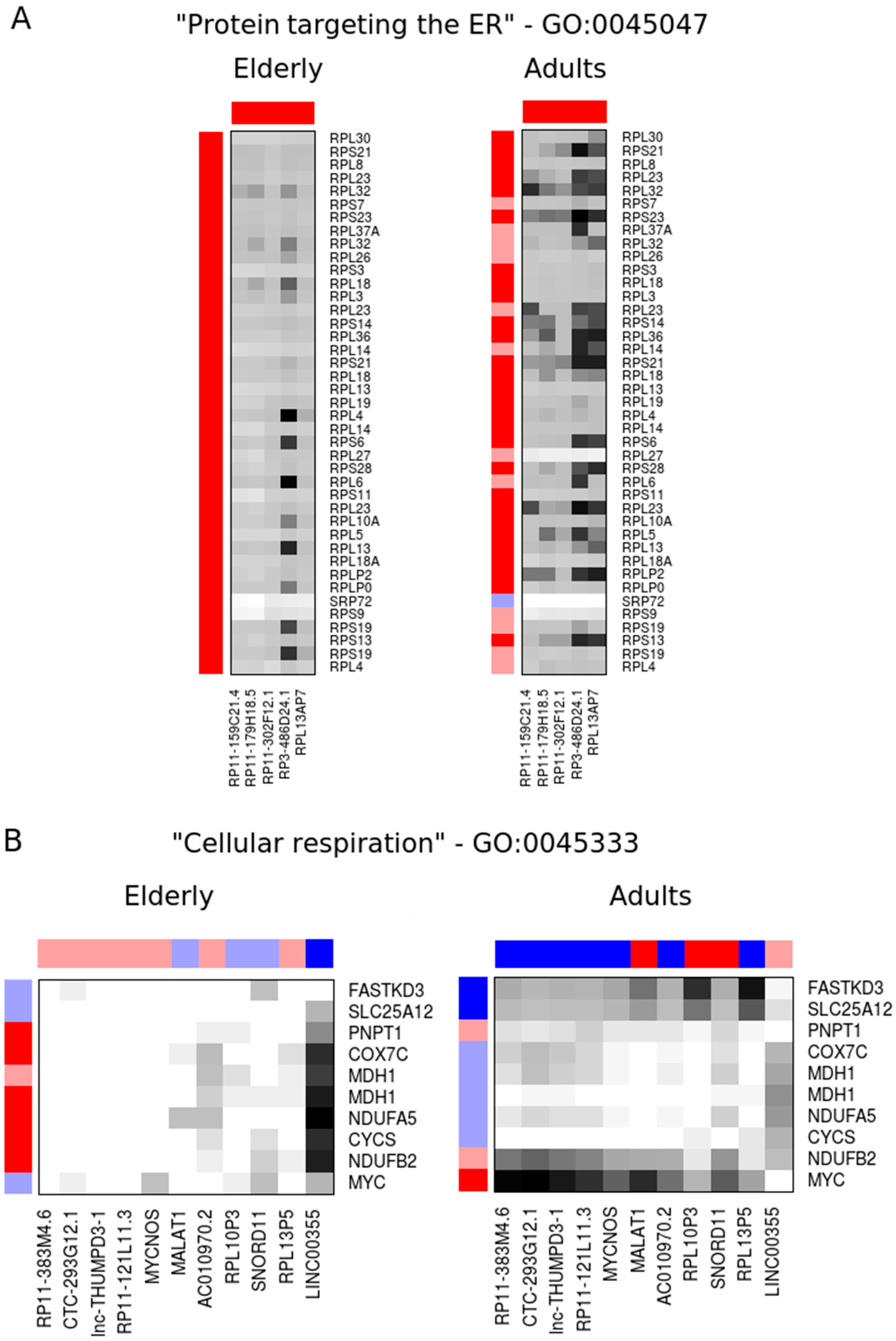
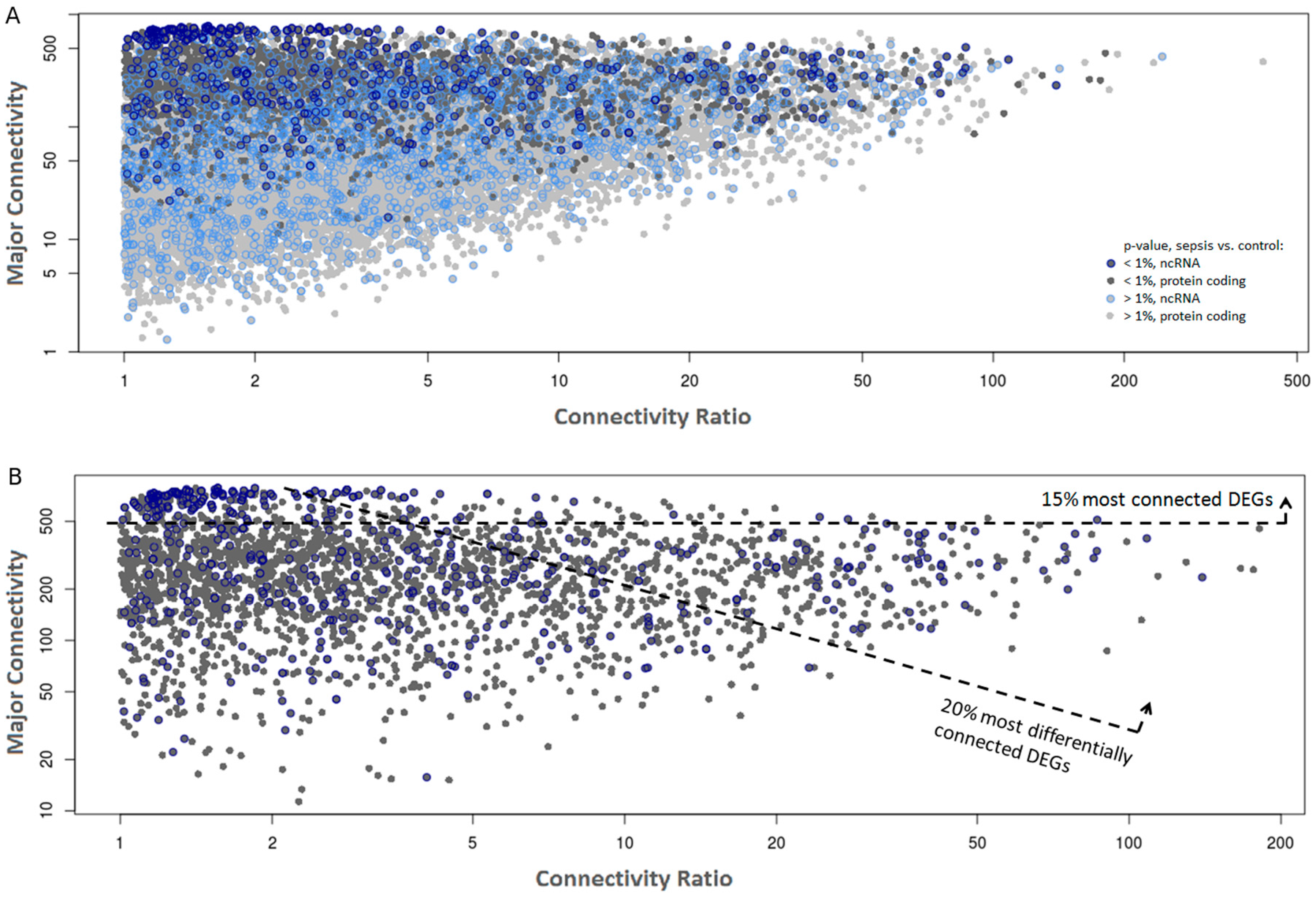
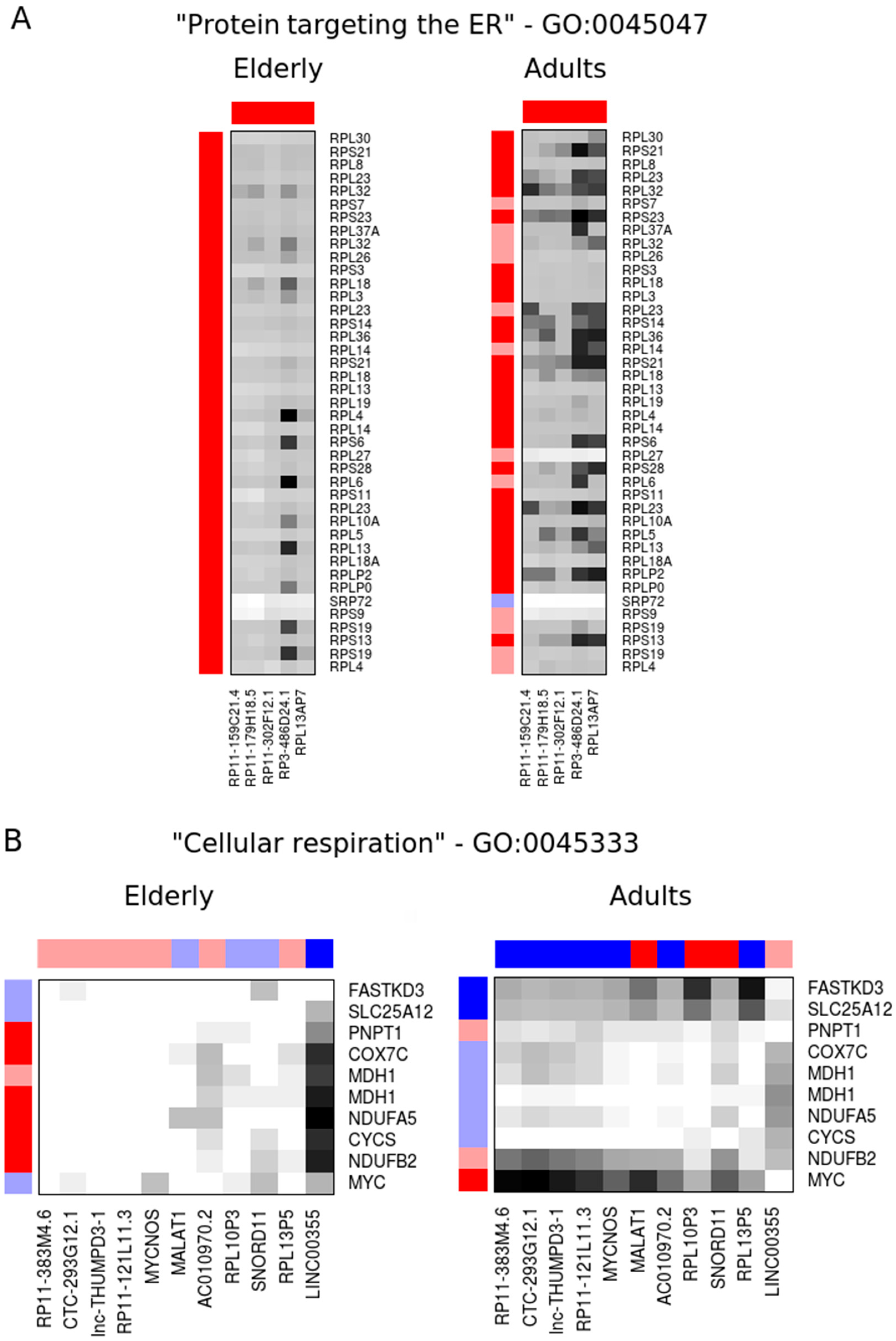
2.2. Co-Expression Networks of mRNAs and lncRNAs Are Perturbed in Sepsis
3. Discussion
4. Materials and Methods
4.1. Study Design
4.2. Oligoarray Reannotation for the lncRNA Analysis
- If a probe aligned to exons of protein-coding genes, it was annotated as “protein-coding”.
- If a probe aligned to annotated exons of RNAs classified as any pseudogene, and did not overlap protein-coding exons, it was annotated as “pseudogene”.
- If a probe aligned to annotated exons of lncRNAs and was not previously classified as a protein-coding or pseudogene, it was classified as “lncRNA”.
- If a probe aligned only to an intron of an annotated gene, to regions in the opposite strand of a known gene, or to regions without any gene annotations, in either strand, it was classified as “poorly annotated RNA”.
4.3. RNA Extraction, Oligoarray Hybridization, and Data Pre-Processing
4.4. Hierarchical Clustering of lncRNAs
4.5. Detection of Differentially-Expressed Genes
4.6. Building Co-Expression Networks in Sepsis
4.7. Functional Annotation and Pathway Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Xiao, W.; Mindrinos, M.N.; Seok, J.; Cuschieri, J.; Cuenca, A.G.; Gao, H.; Hayden, D.L.; Hennessy, L.; Moore, E.E.; Minei, J.P.; et al. A genomic storm in critically injured humans. J. Exp. Med. 2011, 208, 2581–2590. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.M.; Huang, S.J.; McLean, A.S. Genome-wide transcription profiling of human sepsis: A systematic review. Crit. Care 2010, 14, R237. [Google Scholar] [CrossRef] [PubMed]
- Kale, S.S.; Yende, S. Effects of Aging on Inflammation and Hemostasis through the Continuum of Critical Illness. Aging Dis. 2011, 2, 501–511. [Google Scholar] [PubMed]
- Tang, B.M.; McLean, A.S.; Dawes, I.W.; Huang, S.J.; Lin, R.C. The use of gene-expression profiling to identify candidate genes in human sepsis. Am. J. Respir. Crit. Care Med. 2007, 176, 676–684. [Google Scholar] [CrossRef] [PubMed]
- Pellegrina, D.V.d.S.; Severino, P.; Barbeiro, H.V.; Andreghetto, F.M.; Velasco, I.T.; de Souza, H.P.; Machado, M.C.C.; Reis, E.M.; da Silva, F.P. Septic Shock in Advanced Age: Transcriptome Analysis Reveals Altered Molecular Signatures in Neutrophil Granulocytes. PLoS ONE 2015, 10, e0128341. [Google Scholar]
- Kung, J.T.Y.; Colognori, D.; Lee, J.T. Long Noncoding RNAs: Past, Present, and Future. Genetics 2013, 193, 651–669. [Google Scholar] [CrossRef] [PubMed]
- Rana, T.M. Illuminating the silence: Understanding the structure and function of small RNAs. Nat. Rev. Mol. Cell Biol. 2007, 8, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Vilanova, D.; Atalar, K.; Delfour, O.; Edgeworth, J.; Ostermann, M.; Hernandez-Fuentes, M.; Razafimahatratra, S.; Michot, B.; Persing, D.H.; et al. Genome-wide sequencing of cellular microRNAs identifies a combinatorial expression signature diagnostic of sepsis. PLoS ONE 2013, 8, e75918. [Google Scholar] [CrossRef] [PubMed]
- Tacke, F.; Roderburg, C.; Benz, F.; Cardenas, D.V.; Luedde, M.; Hippe, H.J.; Frey, N.; Vucur, M.; Gautheron, J.; Koch, A.; et al. Levels of Circulating miR-133a Are Elevated in Sepsis and Predict Mortality in Critically Ill Patients. Crit. Care Med. 2014, 42, 1096–1104. [Google Scholar] [CrossRef] [PubMed]
- Vasilescu, C.; Rossi, S.; Shimizu, M.; Tudor, S.; Veronese, A.; Ferracin, M.; Nicoloso, M.S.; Barbarotto, E.; Popa, M.; Stanciulea, O.; et al. MicroRNA Fingerprints Identify miR-150 as a Plasma Prognostic Marker in Patients with Sepsis. PLoS ONE 2009, 4, e7405. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.C.; Ruan, Z.S.; Mao, Y.F.; Dong, W.W.; Zhang, Y.Q.; Yin, N.; Jiang, L. miR-27a is up regulated and promotes inflammatory response in sepsis. Cell Immunol. 2014, 290, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Fatica, A.; Bozzoni, I. Long non-coding RNAs: New players in cell differentiation and development. Nat. Rev. Genet. 2014, 15, 7–21. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.; Chang, H.Y. Uncovering the role of genomic “dark matter” in human disease. J. Clin. Investig. 2012, 122, 1589–1595. [Google Scholar] [CrossRef] [PubMed]
- Wilusz, J.E.; Sunwoo, H.; Spector, D.L. Long noncoding RNAs: Functional surprises from the RNA world. Gene Dev. 2009, 23, 1494–1504. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xuan, Z.; Liu, C. Long non-coding RNAs and complex human diseases. Int. J. Mol. Sci. 2013, 14, 18790–18808. [Google Scholar] [CrossRef] [PubMed]
- Lan, F.; Bayliss, P.E.; Rinn, J.L.; Whetstine, J.R.; Wang, J.K.; Chen, S.Z.; Iwase, S.; Alpatov, R.; Issaeva, I.; Canaani, E. A histone H3 lysine 27 demethylase regulates animal posterior development. Nature 2007, 449, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Geisler, S.; Coller, J. RNA in unexpected places: Long non-coding RNA functions in diverse cellular contexts. Nat. Rev. Mol. Cell Biol. 2013, 14, 699–712. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.; Maquat, L.E. lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3′ UTRs via Alu elements. Nature 2011, 470, 284–288. [Google Scholar] [CrossRef] [PubMed]
- Barabasi, A.L.; Oltvai, Z.N. Network biology: Understanding the cell’s functional organization. Nat. Rev. Genet. 2004, 5, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Liao, Q.; Liu, C.N.; Yuan, X.Y.; Kang, S.L.; Miao, R.Y.; Xiao, H.; Zhao, G.; Luo, H.; Bu, D.; Zhao, H.; et al. Large-scale prediction of long non-coding RNA functions in a coding-non-coding gene co-expression network. Nucleic Acids Res. 2011, 39, 3864–3878. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Heward, J.A.; Lindsay, M.A. Long non-coding RNAs in the regulation of the immune response. Trends Immunol. 2014, 35, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.A.; Caffrey, D.R. Long noncoding RNAs in innate and adaptive immunity. Curr. Opin. Immunol. 2014, 26, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Atianand, M.K.; Fitzgerald, K.A. Long non-coding RNAs and control of gene expression in the immune system. Trends Mol. Med. 2014, 20, 623–631. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, S.; Aiello, D.; Atianand, M.K.; Ricci, E.P.; Gandhi, P.; Hall, L.L.; Byron, M.; Monks, B.; Henry-Bezy, M.; Lawrence, J.B.; et al. A Long Noncoding RNA Mediates Both Activation and Repression of Immune Response Genes. Science 2013, 341, 789–792. [Google Scholar] [CrossRef] [PubMed]
- Rapicavoli, N.A.; Qu, K.; Zhang, J.J.; Mikhail, M.; Laberge, R.M.; Chang, H.Y. A mammalian pseudogene lncRNA at the interface of inflammation and anti-inflammatory therapeutics. eLife 2013, 2, e00762. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.H.; Chao, T.C.; Chang, K.Y.; Lin, N.W.; Patil, V.S.; Shimizu, C.; Head, S.R.; Burns, J.C.; Rana, T.M. The long noncoding RNA THRIL regulates TNFα expression through its interaction with hnRNPL. Proc. Natl. Acad. Sci. USA 2014, 111, 1002–1007. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, F.P.; Zampieri, F.G.; Barbeiro, D.F.; Barbeiro, H.V.; Goulart, A.C.; Filho, F.T.; Velasco, I.T.; da Cruz Neto, L.M.; de Souza, H.P.; Machado, M.C.C. Septic shock in older people: A prospective cohort study. Immun. Ageing 2013, 10, 21. [Google Scholar] [CrossRef] [PubMed]
- Angus, D.C.; Linde-Zwirble, W.T.; Lidicker, J.; Clermont, G.; Carcillo, J.; Pinsky, M.R. Epidemiology of severe sepsis in the United States: Analysis of incidence, outcome, and associated costs of care. Crit. Care Med. 2001, 29, 1303–1310. [Google Scholar] [CrossRef] [PubMed]
- Grubeck-Loebenstein, B.; Wick, G. The aging of the immune system. Adv. Immunol. 2002, 80, 243–284. [Google Scholar] [PubMed]
- Morrish, F.; Hockenbery, D. MYC and mitochondrial biogenesis. Cold Spring Harb. Perspect. Med. 2014, 4, a014225. [Google Scholar] [CrossRef] [PubMed]
- Simarro, M.; Gimenez-Cassina, A.; Kedersha, N.; Lazaro, J.B.; Adelmant, G.O.; Marto, J.A.; Rhee, K.; Tisdale, S.; Danial, N.; Benarafa, C.; et al. Fast kinase domain-containing protein 3 is a mitochondrial protein essential for cellular respiration. Biochem. Biophys. Res. Commun. 2010, 401, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Garrabou, G.; Morén, C.; López, S.; Tobías, E.; Cardellach, F.; Miró, O.; Casademont, J. The effects of sepsis on mitochondria. J. Infect. Dis. 2012, 205, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Galley, H.F. Oxidative stress and mitochondrial dysfunction in sepsis. Br. J. Anaesth. 2011, 107, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Jeger, V.; Djafarzadeh, S.; Jakob, S.M.; Takala, J. Mitochondrial function in sepsis. Eur. J. Clin. Investig. 2013, 43, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Huttemann, M. Energy crisis: The role of oxidative phosphorylation in acute inflammation and sepsis. Biochim. Biophys. Acta 2014, 1842, 1579–1586. [Google Scholar] [CrossRef] [PubMed]
- Singer, M. The role of mitochondrial dysfunction in sepsis-induced multi-organ failure. Virulence 2014, 5, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Puthanveetil, P.; Chen, S.; Feng, B.; Gautam, A.; Chakrabarti, S. Long non-coding RNA MALAT1 regulates hyperglycaemia induced inflammatory process in the endothelial cells. J. Cell Mol. Med. 2015, 19, 1418–1425. [Google Scholar] [CrossRef] [PubMed]
- Vadie, N.; Saayman, S.; Lenox, A.; Ackley, A.; Clemson, M.; Burdach, J.; Hart, J.; Vogt, P.K.; Morris, K.V. MYCNOS functions as an antisense RNA regulating MYCN. RNA Biol. 2015, 12, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; Lyu, J.; Huang, J.; Xiang, D.; Xie, M.; Zeng, Q. Experimental treatments for mitochondrial dysfunction in sepsis: A narrative review. J. Res. Med. Sci. 2015, 20, 185–195. [Google Scholar] [PubMed]
- Bone, R.C.; Balk, R.A.; Cerra, F.B.; Dellinger, R.P.; Fein, A.M.; Knaus, W.A.; Schein, R.M.; Sibbald, W.J. Definitions for Sepsis and Organ Failure and Guidelines for the Use of Innovative Therapies in Sepsis. Chest 1992, 101, 1644–1655. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J. BLAT—The BLAST-like alignment tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Harrow, J.; Frankish, A.; Gonzalez, J.M.; Tapanari, E.; Diekhans, M.; Kokocinski, F.; Aken, B.L.; Barrell, D.; Zadissa, A.; Searle, S.; et al. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res. 2012, 22, 1760–1774. [Google Scholar] [CrossRef] [PubMed]
- Cabili, M.N.; Trapnell, C.; Goff, L.; Koziol, M.; Tazon-Vega, B.; Regev, A.; Rinn, J.L. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Gene Dev. 2011, 25, 1915–1927. [Google Scholar] [CrossRef] [PubMed]
- Volders, P.J.; Helsens, K.; Wang, X.; Menten, B.; Martens, L.; Gevaert, K.; Vandesompele, J.; Mestdagh, P. LNCipedia: A database for annotated human lncRNA transcript sequences and structures. Nucleic Acids Res. 2013, 41, D246–D251. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.Y.; Yuan, J.; Li, H.; Li, M.; Zhao, G.G.; Bu, D.C.; Zhu, W.M.; Wu, W.; Cheng, R.S.; Zhao, Y. NONCODEv4: Exploring the world of long non-coding RNA genes. Nucleic Acids Res. 2014, 42, D98–D103. [Google Scholar] [CrossRef] [PubMed]
- Pellegrina, D.V.; Severino, P.; Machado, M.C.; da Silva, F.P.; Reis, E.M. Microarray gene expression analysis of neutrophils from elderly septic patients. Genom. Data 2015, 6, 51–53. [Google Scholar] [CrossRef] [PubMed]
- Tusher, V.G.; Tibshirani, R.; Chu, G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. USA 2001, 98, 5116–5121. [Google Scholar] [CrossRef] [PubMed]
- Hong, F.X.; Breitling, R.; McEntee, C.W.; Wittner, B.S.; Nemhauser, J.L.; Chory, J. RankProd: A bioconductor package for detecting differentially expressed genes in meta-analysis. Bioinformatics 2006, 22, 2825–2827. [Google Scholar] [CrossRef] [PubMed]
- Reimand, J.; Arak, T.; Adler, P.; Kolberg, L.; Reisberg, S.; Peterson, H.; Vilo, J. g:Profiler—A web server for functional interpretation of gene lists (2016 update). Nucleic Acids Res. 2016, 44, W83–W89. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Differentially Expressed Genes (DEGs) | ||||||
|---|---|---|---|---|---|---|
| Elderly vs. Adults | Sepsis vs. Controls | |||||
| Total | Detected | Sepsis | Control | Elderly | Adults | |
| In the array | 50,599 | 15,612 | 37 | 69 | 1677 | 1862 |
| Approved | 47,012 | 14,264 | 36 | 65 | 1411 | 1615 |
| Protein-coding | 26,542 | 11,895 | 27 | 54 | 1121 | 1353 |
| Pseudogenes | 2869 | 834 | 2 | 5 | 151 | 128 |
| lncRNAs | 14,832 | 1185 | 5 | 4 | 114 | 99 |
| Poorly annotated | 2781 | 350 | 2 | 2 | 25 | 36 |
| Sepsis vs. Control | |||||||
|---|---|---|---|---|---|---|---|
| Gene | Connectivity | Type | Elderly | Adults | |||
| Elderly | Adults | FC | p Value | FC | p Value | ||
| RP11-302F12.1 | 774 | 572 | pseudogene | 0.24 | 7.5 × 10−5 | 0.35 | 1.1 × 10−3 |
| RP3-486D24.1 | 746 | 581 | pseudogene | 0.36 | 6.0 × 10−4 | 0.43 | 3.0 × 10−3 |
| RPL13AP7 | 735 | 631 | pseudogene | 0.40 | 2.8 × 10−3 | 0.43 | 1.5 × 10−3 |
| RP11-159C21.4 | 676 | 483 | pseudogene | 0.39 | 2.1 × 10−3 | 0.42 | 8.8 × 10−3 |
| RP11-179H18.5 | 660 | 566 | pseudogene | 0.35 | 6.0 × 10−4 | 0.38 | 2.1 × 10−3 |
| Sepsis vs. Control | |||||||
|---|---|---|---|---|---|---|---|
| Gene | Connectivity | Type | Elderly | Adults | |||
| Elderly | Adults | FC | p Value | FC | p Value | ||
| RP11-383M4.6 | 15.0 | 450 | lincRNA | 0.97 | 7.4 × 10−1 | 1.86 | 3.2 × 10−3 |
| CTC-293G12.1 | 10.8 | 414 | lincRNA | 0.94 | 5.2 × 10−1 | 1.95 | 5.2 × 10−3 |
| lnc-THUMPD3-1 | 3.7 | 397 | ncRNA | 0.94 | 7.2 × 10−1 | 2.25 | 9.7 × 10−4 |
| RP11-121L11.3 | 16.9 | 372 | lincRNA | 0.98 | 7.8 × 10−1 | 1.86 | 4.0 × 10−3 |
| MYCNOS | 4.0 | 298 | antisense | 0.95 | 6.7 × 10−1 | 1.76 | 9.6 × 10−3 |
| MALAT1 | 6.6 | 281 | lincRNA | 1.21 | 5.0 × 10−1 | 0.37 | 2.0 × 10−4 |
| AC010970.2 | 7.3 | 280 | pseudogene | 0.92 | 5.8 × 10−1 | 1.86 | 4.1 × 10−3 |
| RPL10P3 | 14.9 | 274 | pseudogene | 1.11 | 8.4 × 10−1 | 0.41 | 8.0 × 10−4 |
| SNORD11 | 5.6 | 237 | snoRNA | 1.25 | 4.0 × 10−1 | 0.38 | 3.5 × 10−4 |
| RPL13P5 | 1.7 | 235 | pseudogene | 0.91 | 8.6 × 10−1 | 2.37 | 6.0 × 10−4 |
| LINC00355 | 291 | 11.0 | lincRNA | 1.79 | 5.3 × 10−3 | 0.96 | 7.3 × 10−1 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pellegrina, D.V.d.S.; Severino, P.; Barbeiro, H.V.; De Souza, H.P.; Machado, M.C.C.; Pinheiro-da-Silva, F.; Reis, E.M. Insights into the Function of Long Noncoding RNAs in Sepsis Revealed by Gene Co-Expression Network Analysis. Non-Coding RNA 2017, 3, 5. https://doi.org/10.3390/ncrna3010005
Pellegrina DVdS, Severino P, Barbeiro HV, De Souza HP, Machado MCC, Pinheiro-da-Silva F, Reis EM. Insights into the Function of Long Noncoding RNAs in Sepsis Revealed by Gene Co-Expression Network Analysis. Non-Coding RNA. 2017; 3(1):5. https://doi.org/10.3390/ncrna3010005
Chicago/Turabian StylePellegrina, Diogo Vieira da Silva, Patricia Severino, Hermes Vieira Barbeiro, Heraldo Possolo De Souza, Marcel Cerqueira César Machado, Fabiano Pinheiro-da-Silva, and Eduardo Moraes Reis. 2017. "Insights into the Function of Long Noncoding RNAs in Sepsis Revealed by Gene Co-Expression Network Analysis" Non-Coding RNA 3, no. 1: 5. https://doi.org/10.3390/ncrna3010005
APA StylePellegrina, D. V. d. S., Severino, P., Barbeiro, H. V., De Souza, H. P., Machado, M. C. C., Pinheiro-da-Silva, F., & Reis, E. M. (2017). Insights into the Function of Long Noncoding RNAs in Sepsis Revealed by Gene Co-Expression Network Analysis. Non-Coding RNA, 3(1), 5. https://doi.org/10.3390/ncrna3010005