
Computational Approaches to tRNA-Derived Small RNAs


Abstract
:1. Introduction
2. tDRs
3. Identifying tDRs and Managing tDR Data
- identifying the tDRs;
- analyzing tDR functions;
- storing or managing and manipulating data.
3.1. Identifying tDRs
3.2. Databases of tDRs
4. Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lee, S.R.; Collins, K. Starvation-induced cleavage of the tRNA anticodon loop in Tetrahymena thermophila. J. Biol. Chem. 2005, 280, 42744–42749. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.L.; Wen, Y.Z.; Yang, J.H.; Liao, J.Y.; Shao, P.; Xu, H.; Zhou, H.; Wen, J.Z.; Lun, Z.R.; Ayala, F.J.; et al. Comparative transcriptome analysis of small noncoding RNAs in different stages of Trypanosoma brucei. RNA 2013, 19, 863–875. [Google Scholar] [CrossRef] [PubMed]
- Gebetsberger, J.; Zywicki, M.; Künzi, A.; Polacek, N. tRNA-derived fragments target the ribosome and function as regulatory non-coding RNA in Haloferax volcanii. Archaea 2012, 2012, 260909. [Google Scholar] [CrossRef] [PubMed]
- Heyer, R.; Dörr, M.; Jellen-Ritter, A.; Späth, B.; Babski, J.; Jaschinski, K.; Soppa, J.; Marchfelder, A. High throughput sequencing reveals a plethora of small RNAs including tRNA derived fragments in Haloferax volcanii. RNA Biol. 2012, 9, 1011–1018. [Google Scholar] [CrossRef] [PubMed]
- Nunes, C.C.; Gowda, M.; Sailsbery, J.; Xue, M.; Chen, F.; Brown, D.E.; Oh, Y.; Mitchell, T.K.; Dean, R.A. Diverse and tissue-enriched small RNAs in the plant pathogenic fungus, Magnaporthe oryzae. BMC Genom. 2011, 12, 288. [Google Scholar] [CrossRef] [PubMed]
- Åsman, A.K.; Vetukuri, R.R.; Jahan, S.N.; Fogelqvist, J.; Corcoran, P.; Avrova, A.O.; Whisson, S.C.; Dixelius, C. Fragmentation of tRNA in Phytophthora infestans asexual life cycle stages and during host plant infection. BMC Microbiol. 2014, 14, 308. [Google Scholar] [CrossRef] [PubMed]
- Couvillion, M.T.; Bounova, G.; Purdom, E.; Speed, T.P.; Collins, K. A Tetrahymena Piwi bound to mature tRNA 3’ fragments activates the exonuclease Xrn2 for RNA processing in the nucleus. Mol. Cell 2012, 48, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Soares, A.R.; Fernandes, N.; Reverendo, M.; Araújo, H.R.; Oliveira, J.L.; Moura, G.M.; Santos, M.A. Conserved and highly expressed tRNA derived fragments in zebrafish. BMC Mol. Biol. 2015, 16, 22. [Google Scholar] [CrossRef] [PubMed]
- Karaiskos, S.; Naqvi, A.S.; Swanson, K.E.; Grigoriev, A. Age-driven modulation of tRNA-derived fragments in Drosophila and their potential targets. Biol. Direct 2015, 10, 51. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Shi, J.; Zhang, Y.; Zhang, H.; Liao, S.; Li, W.; Lei, L.; Han, C.; Ning, L.; Cao, Y.; et al. A novel class of tRNA-derived small RNAs extremely enriched in mature mouse sperm. Cell Res. 2012, 22, 1609–1612. [Google Scholar] [CrossRef] [PubMed]
- Babiarz, J.E.; Ruby, J.G.; Wang, Y.; Bartel, D.P.; Blelloch, R. Mouse ES cells express endogenous shRNAs, siRNAs, and other Microprocessor-independent, Dicer-dependent small RNAs. Genes Dev. 2008, 22, 2773–2785. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.J.; Liu, Q.; Zhang, Y.C.; Qu, L.H.; Chen, Y.Q.; Gautheret, D. Genome-wide discovery and analysis of microRNAs and other small RNAs from rice embryogenic callus. RNA Biol. 2011, 8, 538–547. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, P.; Emara, M.M.; Villen, J.; Gygi, S.P.; Anderson, P. Angiogenin-induced tRNA fragments inhibit translation initiation. Mol. Cell 2011, 43, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Lee, I.; Ren, J.; Ajay, S.S.; Lee, Y.S.; Bao, X. Identification and functional characterization of tRNA-derived RNA fragments (tRFs) in respiratory syncytial virus infection. Mol. Ther. 2013, 21, 368–379. [Google Scholar] [CrossRef] [PubMed]
- Selitsky, S.R.; Baran-Gale, J.; Honda, M.; Yamane, D.; Masaki, T.; Fannin, E.E.; Guerra, B.; Shirasaki, T.; Shimakami, T.; Kaneko, S.; et al. Small tRNA-derived RNAs are increased and more abundant than microRNAs in chronic hepatitis B and C. Sci. Rep. 2015, 5, 7675. [Google Scholar] [CrossRef] [PubMed]
- Maute, R.L.; Schneider, C.; Sumazin, P.; Holmes, A.; Califano, A.; Basso, K.; Dalla-Favera, R. tRNA-derived microRNA modulates proliferation and the DNA damage response and is down-regulated in B cell lymphoma. Proc. Natl. Acad. Sci. USA 2013, 110, 1404–1409. [Google Scholar] [CrossRef] [PubMed]
- Honda, S.; Loher, P.; Shigematsu, M.; Palazzo, J.P.; Suzuki, R.; Imoto, I.; Rigoutsos, I.; Kirino, Y. Sex hormone-dependent tRNA halves enhance cell proliferation in breast and prostate cancers. Proc. Natl. Acad. Sci. USA 2015, 112, E3816–E3825. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, H.; Liu, X.; Nguyen, H.C.; Zhang, S.; Fish, L.; Tavazoie, S.F. Endogenous tRNA-derived fragments suppress breast cancer progression via YBX1 displacement. Cell 2015, 161, 790–802. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Shibata, Y.; Malhotra, A.; Dutta, A. A novel class of small RNAs: tRNA-derived RNA fragments (tRFs). Genes Dev. 2009, 23, 2639–2649. [Google Scholar] [CrossRef] [PubMed]
- Friedländer, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Abbott, J.A.; Francklyn, C.S.; Robey-Bond, S.M. Transfer RNA and human disease. In Molecular Biology of the Transfer RNA Revisited; Frontiers E-books: Lausanne, Switzerland, 2014; p. 142. [Google Scholar]
- Hopper, A.K.; Phizicky, E.M. tRNA transfers to the limelight. Genes Dev. 2003, 17, 162–180. [Google Scholar] [PubMed]
- Sobala, A.; Hutvagner, G. Transfer RNA-derived fragments: Origins, processing, and functions. Wiley Interdiscip. Rev. RNA 2011, 2, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Thompson, D.M.; Parker, R. The RNase Rny1p cleaves tRNAs and promotes cell death during oxidative stress in Saccharomyces cerevisiae. J. Cell Biol. 2009, 185, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Feng, J.; Liu, Q.; Sun, F.; Tie, Y.; Zhu, J.; Xing, R.; Sun, Z.; Zheng, X. Stress induces tRNA cleavage by angiogenin in mammalian cells. FEBS Lett. 2009, 583, 437–442. [Google Scholar] [CrossRef] [PubMed]
- MacIntosh, G.C.; Bariola, P.A.; Newbigin, E.; Green, P.J. Characterization of Rny1, the Saccharomyces cerevisiae member of the T2 RNase family of RNases: Unexpected functions for ancient enzymes? Proc. Natl. Acad. Sci. USA 2001, 98, 1018–1023. [Google Scholar] [CrossRef] [PubMed]
- Thompson, D.M.; Lu, C.; Green, P.J.; Parker, R. tRNA cleavage is a conserved response to oxidative stress in eukaryotes. RNA 2008, 14, 2095–2103. [Google Scholar] [CrossRef] [PubMed]
- Moroianu, J.; Riordan, J.F. Nuclear translocation of angiogenin in proliferating endothelial cells is essential to its angiogenic activity. Proc. Natl. Acad. Sci. USA 1994, 91, 1677–1681. [Google Scholar] [CrossRef] [PubMed]
- Thompson, D.M.; Parker, R. Stressing out over tRNA cleavage. Cell 2009, 138, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.Y.; Hopper, A.K. Multiple layers of stress-induced regulation in tRNA biology. Life 2016, 6, 16. [Google Scholar] [CrossRef] [PubMed]
- Haussecker, D.; Huang, Y.; Lau, A.; Parameswaran, P.; Fire, A.Z.; Kay, M.A. Human tRNA-derived small RNAs in the global regulation of RNA silencing. RNA 2010, 16, 673–695. [Google Scholar] [CrossRef] [PubMed]
- Telonis, A.G.; Loher, P.; Honda, S.; Jing, Y.; Palazzo, J.; Kirino, Y.; Rigoutsos, I. Dissecting tRNA-derived fragment complexities using personalized transcriptomes reveals novel fragment classes and unexpected dependencies. Oncotarget 2015, 6, 24797–24822. [Google Scholar] [CrossRef] [PubMed]
- Hanada, T.; Weitzer, S.; Mair, B.; Bernreuther, C.; Wainger, B.J.; Ichida, J.; Hanada, R.; Orthofer, M.; Cronin, S.J.; Komnenovic, V.; et al. CLP1 links tRNA metabolism to progressive motor-neuron loss. Nature 2013, 495, 474–480. [Google Scholar] [CrossRef] [PubMed]
- Loss-Morais, G.; Waterhouse, P.M.; Margis, R. Description of plant tRNA-derived RNA fragments (tRFs) associated with argonaute and identification of their putative targets. Biol. Direct 2013, 8, 6. [Google Scholar] [CrossRef] [PubMed]
- Lalaouna, D.; Carrier, M.C.; Semsey, S.; Brouard, J.S.; Wang, J.; Wade, J.T.; Massé, E. A 3’ external transcribed spacer in a tRNA transcript acts as a sponge for small RNAs to prevent transcriptional noise. Mol. Cell 2015, 58, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.Y.; Guo, Y.H.; Zheng, L.L.; Li, Y.; Xu, W.L.; Zhang, Y.C.; Zhou, H.; Lun, Z.R.; Ayala, F.J.; Qu, L.H. Both endo-siRNAs and tRNA-derived small RNAs are involved in the differentiation of primitive eukaryote Giardia lamblia. Proc. Natl. Acad. Sci. USA 2014, 111, 14159–14164. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Luo, J.; Zhou, H.; Liao, J.Y.; Ma, L.M.; Chen, Y.Q.; Qu, L.H. Stress-induced tRNA-derived RNAs: A novel class of small RNAs in the primitive eukaryote Giardia lamblia. Nucleic Acids Res. 2008, 36, 6048–6055. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Yan, M.; Cao, Z.; Li, X.; Zhang, Y.; Shi, J.; Feng, G.H.; Peng, H.; Zhang, X.; Zhang, Y.; et al. Sperm tsRNAs contribute to intergenerational inheritance of an acquired metabolic disorder. Science 2016, 351, 397–400. [Google Scholar] [CrossRef] [PubMed]
- Sharma, U.; Conine, C.C.; Shea, J.M.; Boskovic, A.; Derr, A.G.; Bing, X.Y.; Belleannee, C.; Kucukural, A.; Serra, R.W.; Sun, F.; et al. Biogenesis and function of tRNA fragments during sperm maturation and fertilization in mammals. Science 2016, 351, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Kawaji, H.; Nakamura, M.; Takahashi, Y.; Sandelin, A.; Katayama, S.; Fukuda, S.; Daub, C.O.; Kai, C.; Kawai, J.; Yasuda, J.; et al. Hidden layers of human small RNAs. BMC Genom. 2008, 9, 157. [Google Scholar] [CrossRef] [PubMed]
- Cole, C.; Sobala, A.; Lu, C.; Thatcher, S.R.; Bowman, A.; Brown, J.W.; Green, P.J.; Barton, G.J.; Hutvagner, G. Filtering of deep sequencing data reveals the existence of abundant Dicer-dependent small RNAs derived from tRNAs. RNA 2009, 15, 2147–2160. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.Y.; Ma, L.M.; Guo, Y.H.; Zhang, Y.C.; Zhou, H.; Shao, P.; Chen, Y.Q.; Qu, L.H. Deep sequencing of human nuclear and cytoplasmic small RNAs reveals an unexpectedly complex subcellular distribution of miRNAs and tRNA 3’ trailers. PLoS ONE 2010, 5, e10563. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, L.C.; Lin, S.I.; Shih, A.C.C.; Chen, J.W.; Lin, W.Y.; Tseng, C.Y.; Li, W.H.; Chiou, T.J. Uncovering small RNA-mediated responses to phosphate deficiency in Arabidopsis by deep sequencing. Plant Physiol. 2009, 151, 2120–2132. [Google Scholar] [CrossRef] [PubMed]
- Hackenberg, M.; Huang, P.J.; Huang, C.Y.; Shi, B.J.; Gustafson, P.; Langridge, P. A comprehensive expression profile of microRNAs and other classes of non-coding small RNAs in barley under phosphorous-deficient and-sufficient conditions. DNA Res. 2013, 20, 109–125. [Google Scholar] [CrossRef] [PubMed]
- Alves, C.S.; Vicentini, R.; Duarte, G.T.; Pinoti, V.F.; Vincentz, M.; Nogueira, F.T. Genome-wide identification and characterization of tRNA-derived RNA fragments in land plants. Plant Mol. Biol. 2016, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Anaya, J.; Mudunuri, S.B.; Dutta, A. Meta-analysis of tRNA derived RNA fragments reveals that they are evolutionarily conserved and associate with AGO proteins to recognize specific RNA targets. BMC Biol. 2014, 12, 78. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Mudunuri, S.B.; Anaya, J.; Dutta, A. tRFdb: A database for transfer RNA fragments. Nucleic Acids Res. 2015, 43, D141–D145. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.L.; Xu, W.L.; Liu, S.; Sun, W.J.; Li, J.H.; Wu, J.; Yang, J.H.; Qu, L.H. tRF2Cancer: A web server to detect tRNA-derived small RNA fragments (tRFs) and their expression in multiple cancers. Nucleic Acids Res. 2016, 44, W185–W193. [Google Scholar] [CrossRef] [PubMed]
- Singleton, C.K.; Delude, R.L.; Ken, R.; Manning, S.S.; McPherson, C.E. Structure, expression, and regulation of members of the developmentally controlled V and H gene classes from Dictyostelium. Dev. Genet. 1991, 12, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Selitsky, S.R.; Sethupathy, P. tDRmapper: Challenges and solutions to mapping, naming, and quantifying tRNA-derived RNAs from human small RNA-sequencing data. BMC Bioinform. 2015, 16, 354. [Google Scholar] [CrossRef] [PubMed]
- Telonis, A.G.; Loher, P.; Kirino, Y.; Rigoutsos, I. Consequential considerations when mapping tRNA fragments. BMC Bioinform. 2016, 17, 123. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Xu, J.; Yang, D.; Tan, X.; Wang, H. Computational approaches for microRNA studies: A review. Mamm. Genome 2010, 21, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Telonis, A.G.; Kirino, Y.; Rigoutsos, I. Mitochondrial tRNA-lookalikes in nuclear chromosomes: Could they be functional? RNA Biol. 2015, 12, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Pliatsika, V.; Loher, P.; Telonis, A.G.; Rigoutsos, I. two types of: a framework for the interactive exploration of mitochondrial and nuclear tRNA fragments. Bioinformatics. 2016, 32, 2481–2489. [Google Scholar] [CrossRef] [PubMed]
- Olvedy, M.; Scaravilli, M.; Hoogstrate, Y.; Visakorpi, T.; Jenster, G.; Martens-Uzunova, E. A comprehensive repertoire of tRNA-derived fragments in prostate cancer. Oncotarget 2016, 7, 24766–24777. [Google Scholar] [CrossRef] [PubMed]
- Chi, S.W.; Zang, J.B.; Mele, A.; Darnell, R.B. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature 2009, 460, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Zisoulis, D.G.; Lovci, M.T.; Wilbert, M.L.; Hutt, K.R.; Liang, T.Y.; Pasquinelli, A.E.; Yeo, G.W. Comprehensive discovery of endogenous Argonaute binding sites in Caenorhabditis elegans. Nat. Struct. Mol. Biol. 2010, 17, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Hafner, M.; Landthaler, M.; Burger, L.; Khorshid, M.; Hausser, J.; Berninger, P.; Rothballer, A.; Ascano, M.; Jungkamp, A.C.; Munschauer, M.; et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 2010, 141, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.H.; Li, J.H.; Shao, P.; Zhou, H.; Chen, Y.Q.; Qu, L.H. starBase: A database for exploring microRNA-mRNA interaction maps from Argonaute CLIP-Seq and Degradome-Seq data. Nucleic Acids Res. 2011, 39, D202–D209. [Google Scholar] [CrossRef] [PubMed]
- Li, J.H.; Liu, S.; Zhou, H.; Qu, L.H.; Yang, J.H. starBase v2. 0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014, 42, D92–D97. [Google Scholar] [CrossRef] [PubMed]
- Pillai, M.M.; Gillen, A.E.; Yamamoto, T.M.; Kline, E.; Brown, J.; Flory, K.; Hesselberth, J.R.; Kabos, P. HITS-CLIP reveals key regulators of nuclear receptor signaling in breast cancer. Breast Cancer Res. Treat. 2014, 146, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Dhahbi, J.M.; Spindler, S.R.; Atamna, H.; Yamakawa, A.; Boffelli, D.; Mote, P.; Martin, D.I. 5’ tRNA halves are present as abundant complexes in serum, concentrated in blood cells, and modulated by aging and calorie restriction. BMC Genom. 2013, 14, 298. [Google Scholar] [CrossRef] [PubMed]
- Dhahbi, J.M. Circulating small noncoding RNAs as biomarkers of aging. Ageing Res. Rev. 2014, 17, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Casas, E.; Cai, G.; Neill, J.D. Characterization of circulating transfer RNA-derived RNA fragments in cattle. Front. Genet. 2015, 6, 271. [Google Scholar] [CrossRef] [PubMed]
- Akat, K.M.; Moore-McGriff, D.; Morozov, P.; Brown, M.; Gogakos, T.; Da Rosa, J.C.; Mihailovic, A.; Sauer, M.; Ji, R.; Ramarathnam, A.; et al. Comparative RNA-sequencing analysis of myocardial and circulating small RNAs in human heart failure and their utility as biomarkers. Proc. Natl. Acad. Sci. USA 2014, 111, 11151–11156. [Google Scholar] [CrossRef] [PubMed]
- Cozen, A.E.; Quartley, E.; Holmes, A.D.; Hrabeta-Robinson, E.; Phizicky, E.M.; Lowe, T.M. ARM-seq: AlkB-facilitated RNA methylation sequencing reveals a complex landscape of modified tRNA fragments. Nat. Methods 2015, 12, 879–884. [Google Scholar] [CrossRef] [PubMed]
- Motorin, Y.; Muller, S.; Behm-Ansmant, I.; Branlant, C. Identification of modified residues in RNAs by reverse transcription-based methods. Methods Enzymol. 2007, 425, 21–53. [Google Scholar] [PubMed]
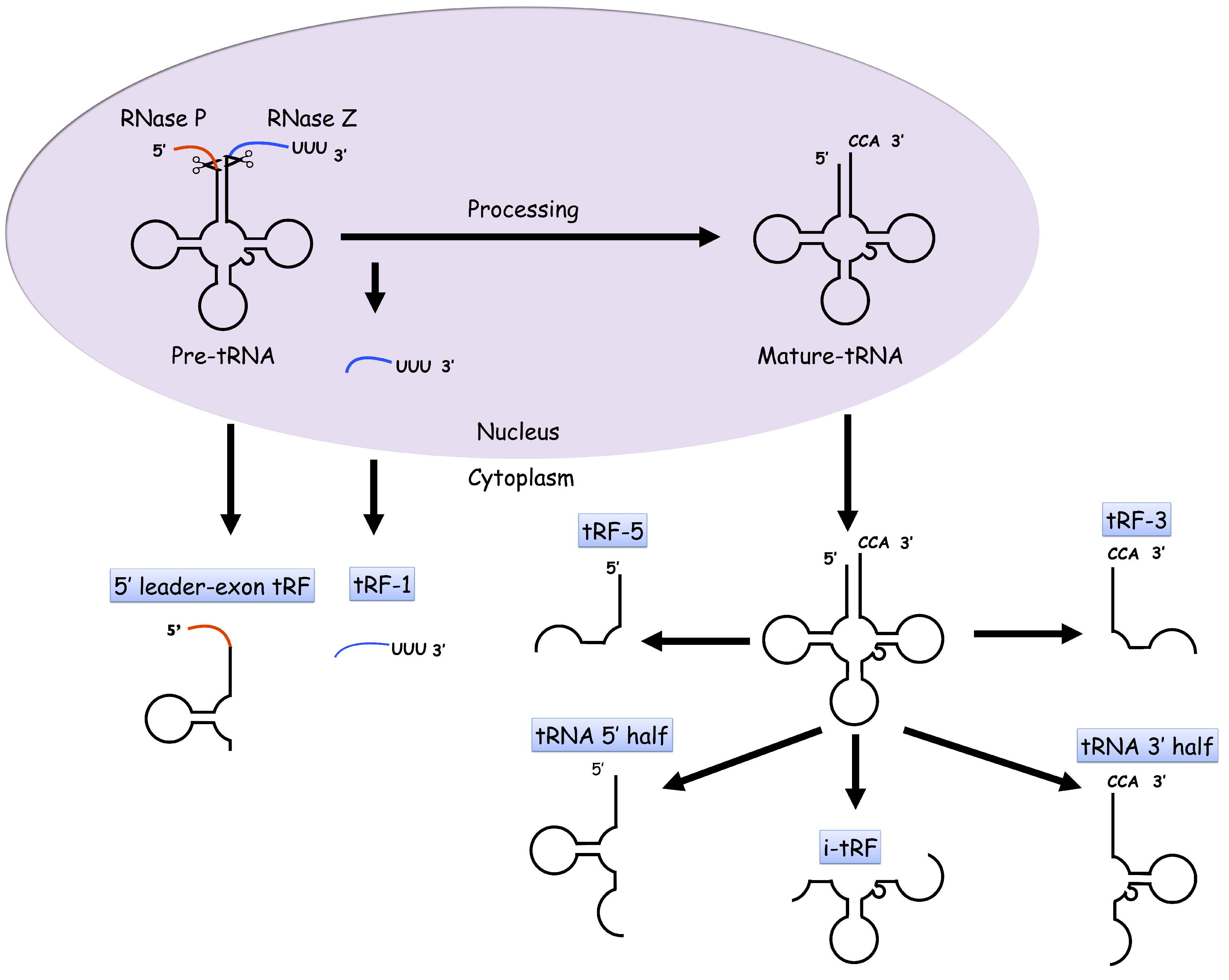

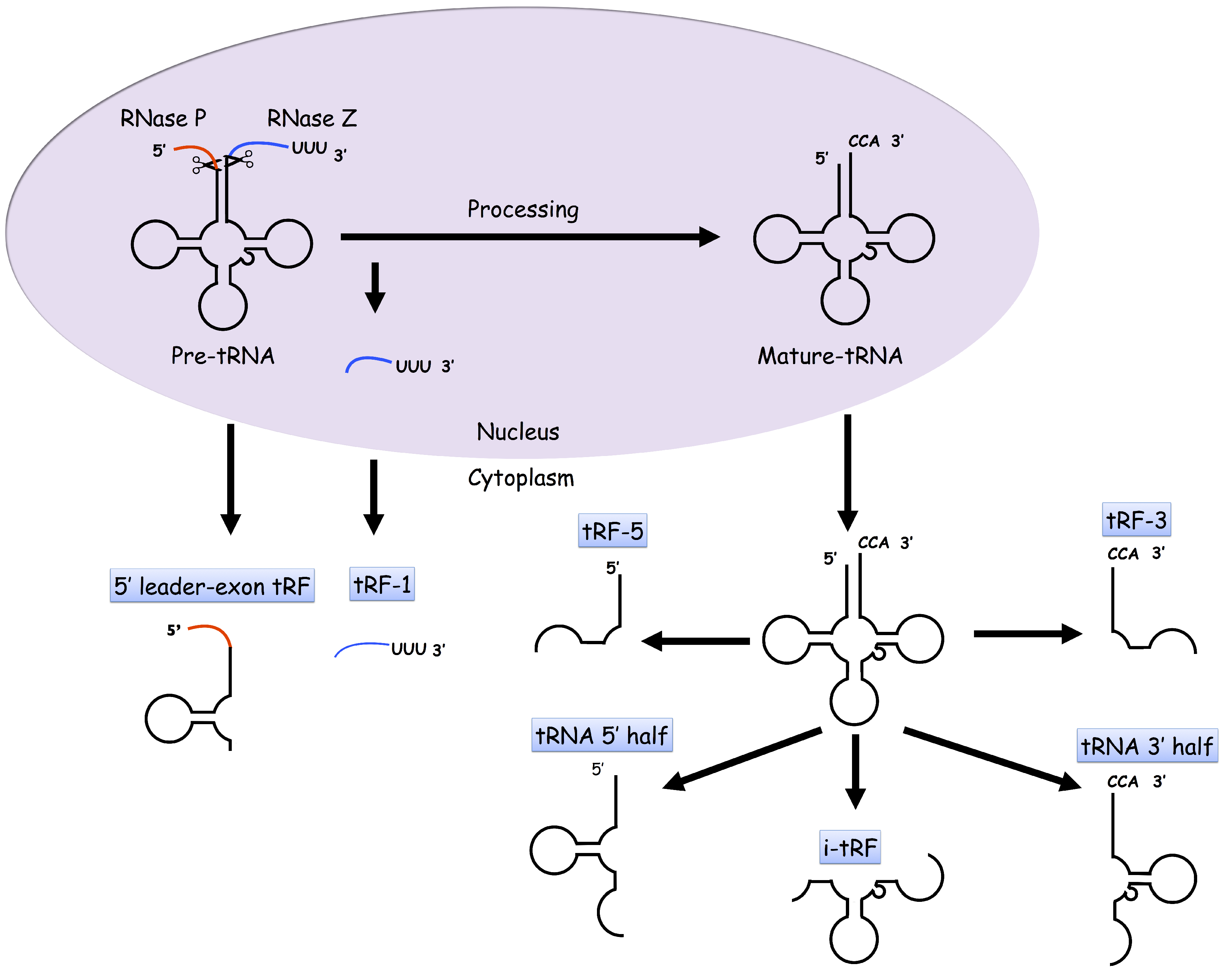{kind=link}
| Name | Mapping to | Potential Effects | Mapping Tools | Mismatches, Indels, and Chemical Modification | tDR Types Detectable | User Interface |
|---|---|---|---|---|---|---|
| Telonis et al., [32,50] | Genome to exclude reads mappable outside tRNAs | Potential false negative | – | Mismatches/indels not allowed | All four tRF types | No packaged pipelines provided |
| tDRmapper | tRNAs alone | Potential false positive | Built-in algorithms | Error type hierarchy | Multiple tRFs and tRNA halves | Command line |
| tRFfinder | First to genome and known transcripts, then to tRNAs | Moderate false positive & negative | bowtie/bowtie2 | Allowed (with options), but given different weights (scores) in result page | Multiple tDR including all tRF types | Website interface |
| Name | Records | Methods | Species | User Interface |
|---|---|---|---|---|
| tRFdb | tRF-3s, -5s, -1s from nucleus | Bioinformatic prediction | Eight species | A website to search and view tRFs |
| tRFinCancer | All four types from nucleus across cancer types | Bioinformatic prediction | 32 human cancer types | A website to view tRFs expression across cancers |
| MINTbase | All types from mature, nuclear and mitochondrial tRNAs | Bioinformatic prediction | Human | Website to search and view tRFs |
| Olvedy et al. repertoire | All types in prostate cancer | Bioinformatic prediction, with qPCR quantification to validate differential expression of selected tRFs | Human prostate cancer | A catalogue |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, W.-L.; Yang, Y.; Wang, Y.-D.; Qu, L.-H.; Zheng, L.-L. Computational Approaches to tRNA-Derived Small RNAs. Non-Coding RNA 2017, 3, 2. https://doi.org/10.3390/ncrna3010002
Xu W-L, Yang Y, Wang Y-D, Qu L-H, Zheng L-L. Computational Approaches to tRNA-Derived Small RNAs. Non-Coding RNA. 2017; 3(1):2. https://doi.org/10.3390/ncrna3010002
Chicago/Turabian StyleXu, Wei-Lin, Ye Yang, Yi-Dan Wang, Liang-Hu Qu, and Ling-Ling Zheng. 2017. "Computational Approaches to tRNA-Derived Small RNAs" Non-Coding RNA 3, no. 1: 2. https://doi.org/10.3390/ncrna3010002
APA StyleXu, W.-L., Yang, Y., Wang, Y.-D., Qu, L.-H., & Zheng, L.-L. (2017). Computational Approaches to tRNA-Derived Small RNAs. Non-Coding RNA, 3(1), 2. https://doi.org/10.3390/ncrna3010002