1. Introduction
Recent strategies in cancer chemotherapy use a so-called multi-target approach, in which cancer cells are targeted while non-cancerous cells survive [
1]. This strategy also helps to reduce potential cancer resistance [
2]. The multiple-target therapy involves binding therapeutic molecules with a carrier such as nanoparticles (NPs), which increases their circulation time in plasma and enhances their delivery to cancer cells. Biologically produced gold nanoparticles (AuNPs) show great potential as carriers for the multi-target tumor therapy [
3]. The main advantage of AuNPs over non-biologically produced gold NPs (NB-AuNPs) is their lower toxicity and ability to easily conjugate with desired molecules due to the presence of capping agents on the AuNPs’ surface [
3]. The molecules of the AuNPs’ capping agents, such as peptides or amino acids [
4], can be attached to cargoes like proteins or nucleic acids through electrostatic interaction without the need for additional chemicals or procedures. The potential use of the AuNPs as drug carriers is also supported by their detection in various compartments of cancer cells following in vivo applications [
3].
In contrast to NB-AuNPs, the intracellular fate, cellular internalization, and trafficking of the AuNPs is unclear. The cellular uptake and passage through the cytoplasmic membrane of the AuNPs are influenced by the NPs’ size, charge, shape, and many other factors [
5]. The understanding of the AuNPs’ internalization route is important not only with respect to their safety, but also with respect to their integrity and ability to deliver intact cargoes to the cells. We need to know whether the AuNPs reach specific cells through any means of internalization and whether the conjugated cargo is still active enough to act on its target. We showed that changing the environmental pH to acidic levels, such as pH 2 or 4, caused AuNPs to agglomerate [
6]. In the current study, we did not attempt to change the surface charge of the AuNPs; instead, we aimed to investigate the mechanism of their cell membrane internalization while their conjugated cargo remains active. The AuNPs allowed us to directly conjugate desired molecules to their surface without altering their charge, thereby enabling passage through cell barriers. Therefore, rather than altering the surface charge of the AuNPs, it is preferable to conjugate them with molecules for targeting specific cells or the cell nucleus using various cell-penetrating peptides (CPPs) [
7].
So far, the main mechanism how the NPs enter mammalian cells is endocytosis, which is followed by NPs budding from endocytic vesicles and their aiming for intracellular location. Endocytosis is classified into two main types: (1) pinocytosis, which occurs in many types of cells and consists of caveolin-mediated, clathrin-mediated, and clathrin-/caveolae-independent endocytosis and macropinocytosis; and (2) phagocytosis, which mainly occurs in phagocyte cells [
5,
8,
9]. The other possible mechanism for AuNP entry is passive penetration, such as diffusion, which involves the interaction of the AuNPs with the lipid bilayer of the cell membrane followed by the direct penetration of the AuNPs without compromising the cell’s integrity [
5,
10].
A particular challenge is represented by the in vivo application of any NPs. Like many other carriers entering body, the AuNPs are usually surrounded by certain serum proteins, generally referred to as the “protein corona”. The protein corona can change properties of the AuNPs. Once AuNPs reach and penetrate a specific organ, they must interact with the cellular microenvironment, which possesses different proteins or pH milieu compared to the bloodstream. All of these factors can potentially have either positive or negative impact on the cellular uptake and fate of the AuNPs [
5]. Previously, we incubated the AuNPs with different concentrations of plasma and demonstrated that fibrinogen (Fg) was the main protein corona present in all concentrations used [
4]. Although coating the AuNPs’ surface with Fg to determine their cellular fate is an interesting topic, in this study, we have used the AuNPs alone to achieve the fundamental results.
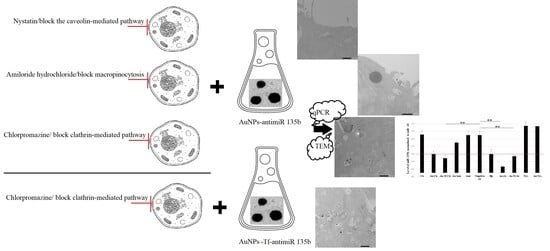
Our research aimed to understand the cellular uptake, intracellular transport, and fate of the AuNPs conjugated to specific cargo with respect to effective cargo delivery to cells. This process may differ from that of NB-AuNPs due to their specific surface coating, potentially impacting their medical applications. To achieve this goal, the AuNPs were bio-produced using
Fusarium oxysporum and used as carriers in the murine 4T1 cell line, since these cells express high amounts of miR 135b. Also, this model cell line was used in many of our previous studies, thus allowing more complex understanding [
3,
11,
12]. The AuNPs were conjugated with both antimiR 135b and transferrin (Tf). AntimiR 135b was used as an effector cargo here that needed to be delivered intact to the cells, find its target, and reduce the level of miR 135b. We showed previously that antimiR 135b can conjugate to the AuNPs directly and transfect the cells [
12]. Tf was employed to actively induce receptor-mediated endocytosis through uptake by the Tf receptor (TfR), which is part of the clathrin-mediated endocytosis pathway [
13]. Different chemicals were used to block various pathways of AuNP-antimiR 135b endocytosis, including the caveolin-mediated pathway, clathrin mediated pathway, and macropinocytosis, in both the Tf-conjugated and non-conjugated forms. To characterize the AuNPs-antimiR 135b (with or without the Tf) regarding their internalization efficacy and their ability to deliver the intact cargo to the cells, we employed two key methods: (1) TEM showing the distribution and amounts of AuNPs inside the cells and (2) qPCR of miR 135b demonstrating the effective internalization and delivery of the functional effector molecule into cell cytoplasm.
3. Discussion
After attachment to different molecules, AuNPs can exhibit differences in patterns of their spectra and shifts in maximum absorption peaks (MAPs). The surface plasmon resonance (SPR) shift of the AuNPs after attachment to different cargoes is known to occur due to changes in their size, charge, or polydispersity [
14]. It has been reported that an increase in the size of nanoparticles corresponds to a shift towards higher wavelength. Agglomeration, polydispersity, and the production of larger shapes result in a significant shift, exceeding 550 nm [
6].
The conjugation of the antimir 135b and/or Tf increased hydrodynamic diameter in comparison to source AuNPs, which was expected. Importantly, the zeta potential of the conjugates remained under −30 mV, which is important regarding conjugate stability.
Regarding the NB-AuNPs, there are studies demonstrating that different chemical and physical properties of the nanoparticles such as size, charge, shape, and coatings are responsible for their effect on cell internalization. On the other hand, there are no such data available on the AuNPs with potential use for gene therapy or drug delivery. The activity and the aforementioned properties of the AuNPs are altered by physiological conditions and the formation of a protein corona on the surface of the nanoparticles. In the current study, we used 4T1 cells as a model, since these cells express high amounts of miR 135b, and previous extensive studies on this model allow us a more complex overview [
3,
11,
12]. We are aware that, in different cell lines, the uptake of the AuNPs may occur via different internalization pathways, and a general statement cannot be made without more extensive studies. The current study is the first report on the uptake of the AuNPs and the determination of their internalization pathway in 4T1 cells. The murine 4T1 cells reflect well the behavior of Stage IV, Triple-Negative human breast cancer, including target metastatic sites [
15]. Given the similarities with human pathology, our findings might be later applicable in drug development for human use.
As demonstrated previously, different cell types vary in their uptake of the AuNPs [
11], and varying concentrations of plasma will result in different protein corona compositions on the surface of the AuNPs [
4]. Therefore, in order to mimic the same in vivo conditions, we utilized FBS in the cell culture to simulate the effects of the protein corona on the internalization of the AuNPs.
Regarding active cargo delivery, it was reported that the NB-AuNPs internalized through the clathrin-dependent pathway were destined for lysosomal degradation, while those internalized through other pathways (i.e., clathrin-independent pathways) were packaged in endosomes or caveosomes and sorted to a non-degradative pathway [
16,
17,
18,
19]. Using bio-produced AuNPs, we observed some internalized AuNPs in 4T1 cells pretreated with all three endocytosis inhibitors, although the quantity of internalized NPs varied. Important information was obtained studying the cargo functionality, which showed if the particular internalization pathway resulted in successful cargo delivery into cytoplasm. When the clathrin-dependent pathway was blocked, the AuNPs-antimiR 135b could enter via other simultaneous pathways. If the clathrin-dependent pathway is not blocked and the other pathways were blocked, the AuNPs-antimiR 135b or AuNPs-antimiR 135b-Tf were internalized through the clathrin-dependent pathway, and the antimiRs effects were likely diminished due to lysosomal digestion, as suggested in above-mentioned reports on NB-AuNPs. While the studies on NB-AuNPs reported that clathrin-mediated endocytosis is the primary method of NB-AuNPs’ internalization, our findings indicated that bio-produced AuNPs utilized both clathrin-dependent and -independent endocytosis pathways. However, a greater load of active cargo was delivered to the cells via clathrin-independent endocytosis. This difference may be due to the source of the coating on the surface of the AuNPs, which influences their cellular uptake and fate [
20].
Receptor-mediated endocytosis—specifically, clathrin- and caveolae-coated vesicles—is known to be the most prevalent method of NB-AuNP internalization [
21,
22]. Different in vitro studies have confirmed that the shape, size, and other physical properties such as the surface coating of NB-AuNPs determine the type of endocytosis. For example, clathrin-mediated endocytosis has been reported for Tf-coated NB-AuNPs of varying sizes, while 16 nm polyethylene glycol (PEG)-coated NB-AuNPs showed both clathrin- and caveolae-mediated endocytosis [
23]. In the case of 4.5 nm PEG-coated NB-AuNPs, only caveolae-mediated endocytosis was observed [
24], and, for 20 nm Fetal Bovine Serum (FBS)-coated NB-AuNPs, clathrin-mediated endocytosis was reported [
20]. The capping agent (coating) of the bio-produced AuNPs, which is secreted by the fungal cells, is an important factor for their internalization method, as well as for releasing the active cargo into the cell cytoplasm. The capping agent was shown to involve peptides and/or amino acids on the surface of the AuNPs, as reported in our previous study [
4]. There is a possibility of endosomal destabilization by a particular capping agent, promoting the cytoplasmic release of the cargo. The capping agents from various producers can also differently mask physical–chemical properties of the AuNPs core. There is a further need to study AuNPs with different physical properties (such as size, shape, zeta potential, etc.) to demonstrate and confirm their similarities in internalization with the NB-AuNPs. Even though the short antimiR sequences are generally more stable than longer RNA oligos, the AuNPs likely function as a further protection of ssRNAs [
12,
25] and hold promise as a safe and effective carrier for gene therapy. The AuNPs-antimiR 135b were not observed inside the nuclei, which may be attributed to the short incubation time (2 h) and the immediate fixation of the cells, or it is indeed the fate of the AuNP carriers to stay in cytoplasmic pools. This observation agrees with our previous study, where AuNPs were not found inside over 2000 nuclei of breast tumor tissue in mice using TEM [
3]. The long-term persistence of the AuNPs within cytoplasm and its subsequent effect on the particular cell still needs to be clarified.
Since the uptake studies are usually performed with the aim to prove that the carriers can deliver a particular cargo (i.e., drug) into cells, it is important to focus not only on the mean of uptake but also on the functionality of the carried molecule by a different approach. In the case of carriers for RNA-based therapy, used in this study, a parallel examination with microscopy (TEM) and functional study (qPCR) gave us valuable information.
The methodology for NPs’ uptake, even in the case of more studied NB-AuNPs, is not unified, and every type of experimental focus or need for particular outputs has its own approach. For example, Dosumu et al. applied a coating of a red-luminescent ruthenium transition metal complex on the surface of NB-AuNPs and tracked the luminescence signal of the NB-AuNPs inside untreated A549 cells using confocal microscopy. They used a combination of Inductively Coupled Plasma Mass Spectrometry (ICP–MS) and TEM to complete their investigations [
26]. In another study, Yilmaz et al. utilized various endocytosis inhibitors against three cell lines (Beas-2b, A549, and PNT1A) and compared the internalization pathways of NB-AuNPs using two label-free methods: flow cytometry (due to the side scatter shift of the cells after AuNP internalization) and surface-enhanced Raman spectroscopy (SERS, by comparing different spectra) in the presence of controls [
16]. It was shown that the clathrin-mediated endocytosis of NB-AuNPs occurred in the presence of endocytosis blockers in two different cell lines (MRC5 lung fibroblasts and Chang liver cells), which was confirmed by TEM and Scanning Electron Microscopy (SEM) techniques, and quantified by ICP–MS and verified by confocal microscopy [
20]. We omitted the microscopy techniques such as fluorescent labeling for tracking the AuNPs due to issues with photobleaching, as well as the possibility that their attachment to the AuNPs may alter the surface charge, coating, size, and other properties of the AuNPs, which could affect their true route of internalization and cargo release. Also, the AuNPs possess some autofluorescence that may distort the actual results. Other available techniques, such as ICP–MS or GF-AAS, were not used because their detection limit for AuNPs is high. Instead, we employed TEM and qPCR as two different approaches that were the most suitable for our research focus (internalization and functionality of delivered cargo).
Using chemical inhibitors to study different internalization pathways may trigger off-target effects and potential cytotoxicity of the chemical inhibitors. In the current study, we employed three different chemical treatments: nystatin at a final concentration of 50 µM, amiloride hydrochloride hydrate at 0.5 mM, and chlorpromazine hydrochloride at 0.03 µM. The concentrations used for each chemical were below their respective cytotoxic levels [
17,
18]. Two of the chemicals mentioned above (nystatin and amiloride hydrochloride) exhibit known off-target activities. For instance, nystatin is primarily used as an antifungal agent, but it also exhibits off-target effects such as binding to cholesterol, disrupting cholesterol-enriched membrane microdomains (lipid rafts) and altering internalization pathways [
27]. We have leveraged this off-target activity of nystatin for our current research. Other off-target effects of nystatin, including nephrotoxicity, pro-inflammatory responses, and impacts on axon growth and regeneration, are observed mainly in vivo and are not relevant to our in vitro study. Amiloride hydrochloride hydrate is a pyridine compound used to treat hypertension and congestive heart failure. It functions as a potassium-sparing diuretic by blocking epithelial sodium channels (ENaC) in the distal nephron of the kidneys. Additionally, amiloride’s inhibition of Na
+/H
+ exchange may indirectly influence macropinocytosis by altering sub-membranous pH, which is essential for the activity of GTPases involved in actin remodeling—a critical step in macropinosome formation [
28]. While its primary mechanism of action as a drug is the modulation of renal ion channels, amiloride can also inhibit macropinocytosis. This effect is likely due to changes in sub-membranous pH resulting from its inhibition of Na+/H+ exchange, representing an off-target action that we leveraged in our study.
Another chemical we used in our study was chlorpromazine hydrochloride, an antipsychotic medication primarily used to treat psychotic disorders such as schizophrenia. As a cationic amphiphilic drug, chlorpromazine exerts its effects by binding to clathrin and AP2, thereby inhibiting their proper assembly into clathrin-coated vesicles at the plasma membrane. Additionally, it may impede dynamin, a GTPase essential for clathrin-mediated endocytosis. Chlorpromazine has also been shown to disrupt basolateral actin cytoskeleton dynamics, which are important for AQP2 endocytosis in kidney epithelial cells. However, detailed studies on the off-target effects of this drug are limited [
29].
4. Materials and Methods
4.1. AuNPs’ Source and Preparation
The AuNPs were produced by
F. oxysporum, according to our established protocol [
3]. Briefly,
F. oxysporum was cultured in Sabouraud Dextrose Broth (SDB, Sigma-Aldrich, Prague, Czech Republic) at 30 °C for one week, and the cell culture supernatant was used for AuNP production in the presence of a final concentration of 1 mmol HAuCl
4·3H
2O (Sigma-Aldrich, Prague, Czech Republic). After heating the supernatant at 80 °C for 5 min, in the presence of a control sample (i.e., SDB containing a final concentration of 1 mmol HAuCl
4·3H
2O), AuNPs were washed three times with ddH
2O and collected by centrifugation at 15,000×
g for 30 min.
To characterize the size and charge of the AuNPs, we employed dynamic light scattering (DLS) through a non-invasive backscattering method and electrophoretic light scattering (ELS), respectively, with a Zetasizer Ultra instrument (Malvern Panalytical, Malvern, UK). The measurements employed a refractive index of 0.18 and an absorbance of 3.43, and utilized double-distilled water (ddH
2O) as the dispersant. For size distribution assessment, an ultra-low-volume quartz cuvette (ZEN2112, Malvern Panalytical) was used, with data collected in backscatter mode. Zeta potential measurements were performed using a DTS1070 zeta cell with folded capillaries, all at a controlled temperature of 25 °C [
30]. Samples were measured in triplicates.
4.2. Preparation of AuNPs Conjugates
4.2.1. AuNP-AntimiR 135b and AuNP-AntimiR 135b-Tf Preparation
To prepare AuNP-antimiR 135b, the used sequence was antisense microRNA 135b with phosphorothioate bonds (PS) (5′ UpsCpsAps CAU AGG AAU GAA AAG CCpsAps UpsA 3′, Sigma Aldrich, Prague, Czech Republic) and followed optimized protocol reported earlier [
3]. Briefly, 200 µL of the washed and sterilized AuNPs (1.89 ± 0.08 mg/mL, according to our previous graphite furnace atomic absorption spectrometry (GF-AAS) data [
3]) was added to 100 µL of antimiR 135b (100 µmol), and the conjugate was incubated in a thermomixer at 1200 rpm at 4 °C for 24 h. AuNP-antimiR 135b-Tf was prepared in the same manner with the following amounts: 200 µL of AuNPs with 100 µL of antimiR 135b (100 µmol) and 5.4 µL of Tf (5 mg/mL, Life Technologies, Prague, Czech Republic). The samples were washed three times using RNase-free ddH
2O and centrifugation at 15,000 rcf for 30 min and resuspended in 200 µL of RNase-free ddH
2O. In order to ensure that the unbound antimiR 135b and Tf were washed from the sample, a Tecan spectrophotometer (Infinite 200 PRO UV-visible spectrophotometer, Tecan, Männedorf, Switzerland) was used and the fluorescence intensity of the supernatant from the final washing step was determined at excitation/emission wavelengths of 488/520 nm and 580/609 nm for determining the amounts of unbound antimir 135b and Tf, respectively. The free and diluted amounts of antimir 135b (1 µmol) and Tf (0.5 mg/mL) were used as positive controls [
3], and each measurement was conducted three times.
Determining the presence of antimiR 135b and Tf on the surface of the AuNPs, their amounts and conjugate characterizations involved using various methods, which are discussed below.
4.2.2. Absorption Spectra Measurement
After loading different molecules onto the AuNPs, we tested the spectra pattern and shift of maximum absorption peaks (MAPs) in comparison to the original AuNPs. An increase in AuNPs’ size, pointing to, for example, agglomeration, results in a significant spectrum shift towards higher wavelength. To see spectra patterns and their shifts after the formation of conjugates, we used the Tecan spectrophotometer with 1 nm step resolution and measured wavelength from 400 to 650 nm. The blank consisted of ddH2O used for the conjugate preparation and the measurements were conducted in triplicate.
4.2.3. Size and Surface Charge Characterization
To characterize the size and charge of the AuNP conjugates, triplicates of test samples (AuNPs-antimiR 135b and AuNPs-antimiR 135b-Tf), along with control triplicates (AuNPs), were assessed for their size and zeta potential using DLS and ELS with the Zetasizer, as detailed in
Section 4.1.
4.2.4. Tf and AntimiR Amount Calculations
First, standard curves of the known amounts of each molecule (Tf and antimiR 135b) were prepared based on the detection of specific excitation/emission wavelengths using a Tecan spectrophotometer, as described previously [
3]. The supernatant of the conjugate was analyzed prior to the addition of water and washing of the pellet, and the number of unbound molecules was determined using the standard curves. Finally, the number of bound molecules was calculated using the equations from each curve and an online tool available at
www.wolframalpha.com (accessed on 7 September 2025) [
3]. Tests were conducted in triplicate.
4.2.5. Proof of AntimiR Conjugation by Electrophoretic Mobility
The conjugation of AuNPs to antimiR 135b changes the electrophoretic mobility of the antimiR 135b due to the heavier nature of the conjugate [
3,
12]. The 2%
w/
v agarose gel (Sigma Aldrich, Prague, Czech Republic) plus GelRed™ nucleic acid stain (Biotium, CA, USA) was prepared in Tris–acetate–EDTA buffer (TAE) and the samples (i.e., antimiR 135b and AuNPs-antimiR 135b) were mixed with MassRuler DNA loading dye (Thermo Fisher Scientific, Waltham, MA, USA) and loaded into the wells. One well was filled with MassRuler DNA ladder (Thermo Fisher Scientific, Prague, Czech Republic). The voltage used was 110 V and it ran for 45 min in TAE buffer [
12]. The gel and RNA bands were observed using a gel documentation system, and electrophoresis was repeated twice to confirm the results.
4.2.6. Proof of Tf Conjugation by a Liquid Chromatography–Mass Spectrometry (LC–MS)
Three independently prepared samples of AuNPs-antimiR 135b-Tf were collected and analyzed by LC–MS. Samples (AuNPs-antimiR 135b-Tf as test and Tf as control) were combined with 30 µL of 50 mM ammonium bicarbonate along with dithiothreitol (DTT) at a final concentration of 10 mM. This mixture was then incubated at 60 °C for 40 min. Once allowed to cool to room temperature, iodoacetamide was introduced to reach a final concentration of 30 mM, and the samples were incubated in darkness for 30 min. To halt the alkylation process, additional DTT was added to achieve a final concentration of 50 mM. Subsequently, trypsin was incorporated to a concentration of 0.1 µg/mL, and the mixture was allowed to incubate overnight at 37 °C. The resulting samples were subjected to analysis using a liquid chromatography system (Agilent 1200 series, Agilent Technologies, Santa Clara, CA, USA) linked to the timsToF Pro PASEF mass spectrometer, which featured a Captive spray (Bruker Daltonics, Fremont, CA, USA) operating in positive data-dependent mode. An autosampler injected 5 µL of the peptide mixture into a C18 trap column (UHPLC Fully Porous Polar C18 2.1 mm ID, Phenomenex, c/o Beckman Coulter, Prague, Czechia). After 5 min of trapping at a flow rate of 20 µL/min, the peptides were separated on a C18 column (Luna Omega 3 μm Polar C18 100 Å, 150 × 0.3 mm, Phenomenex) using a linear water–acetonitrile gradient over 35 min, transitioning from 5% (v/v) to 35% (v/v) acetonitrile at a flow rate of 4 µL/min. Both the trap and analytical columns were maintained at 50 °C.
For the timsTOF Pro settings, standard proteomics parameters from the PASEF method were applied. Specifically, the target intensity for each individual PASEF precursor was set at 6000, with an intensity threshold of 1500. The scan range was configured between 0.6 and 1.6 V s/cm2, with a ramp time of 100 ms. A total of 10 PASEF MS/MS scans were conducted. Precursor ions within an m/z range of 100 to 1700 with charge states ranging from ≥2+ to ≤6+ were selected for fragmentation, with active exclusion implemented for 0.4 min.
Raw data processing was carried out using PeaksStudio 10.0 software (Bioinformatics Solutions, Waterloo, ON, Canada). The search parameters included trypsin as the enzyme (specific), carbamidomethylation as a fixed modification, and the oxidation of methionine along with the acetylation of the protein N-terminus as variable modifications. The database utilized for searches was UniProt (all taxa, November 2021).
4.3. Cell Culture and Blocker Dosages
The 4T1 mouse mammary adenocarcinoma (ATCC CRL-2539) was used in this study [
11], and a working medium, consisting of Roswell Park Memorial Institute Medium 1640 (RPMI-1640, Sigma Aldrich, Prague, Czech Republic), 10% fetal bovine serum (FBS, Gibco, MA, USA), 4.5 g/L glucose (Sigma Aldrich, Prague, Czech Republic), and 44 µg/mL gentamicin (Sandoz, Novartis Company, Prague, Czech Republic), was used for culturing the cells. The MTT assay for the toxicity assessment of the AuNPs was not conducted according to the IC
50 determined from the previous study, which showed that AuNPs with an initial dose of 61.1 µg/100 µL did not induce toxicity higher than the IC
50 in the cell culture [
3]. Therefore, the same amounts were used in this experiment.
In order to block the caveolin-mediated pathway, we used nystatin (0.5 mg/mL, Sigma Aldrich, Prague, Czech Republic) at a final concentration of 50 µM [
31]. For blocking macropinocytosis, we used amiloride hydrochloride hydrate (10 mM, Sigma Aldrich, Prague, Czech Republic) at a final concertation of 0.5 mM [
32]. For blocking the clathrin-mediated pathway, we used chlorpromazine hydrochloride (1 mg/mL, Sigma Aldrich, Prague, Czech Republic) at a final concertation of 0.03 µM [
32].
4.4. TEM and Energy Dispersive X-Ray Spectroscopy (EDS) Analyses
Additionally, 4T1 cells (2 × 10 3 cells/mL) were cultured in the same working medium as mentioned above, but on round cover slips (12 mm) in a 24-well plate. After 48 h of incubation, the cells reached 60–70% confluence in 500 µL of fresh culture medium. The wells containing cover slips were then divided into 7 groups (each group in triplicate). Two groups were treated with the above-mentioned amounts of nystatin or amiloride hydrochloride, and two groups after treatment with chlorpromazine. After 30 min of incubation, the chemicals were washed twice with PBS, and the wells were refilled with 500 µL of the working medium. In parallel, 3 groups (each in triplicate) remained untreated and served as controls. After pretreatment with chemicals, two groups (nystatin and amiloride hydrochloride) were incubated with 2.5 µL of AuNPs-antimiR 135b. The third and fourth groups (chlorpromazine treatment) were incubated either with AuNP-antimiR 135b or AuNP-antimiR 135b-Tf. The control groups included cells without any treatment, and two groups incubated with 2.5 µL of AuNP-antimiR 135b or AuNP-antimiR 135b-Tf. The incubation time with the AuNPs was 2 h; then, cells were washed with PBS and fixed with 1% glutaraldehyde (GA) containing 2.5% Paraformaldehyde/Sodium cacodylate buffer (PFA/SB buffer). After 1 h of fixation at 4 °C and three washings with SB buffer, 1% OsO4 in SB was added, followed by another hour of incubation in the dark at room temperature. The plate was then washed with SB buffer, rinsed with water three times, and incubated with various concentrations of acetone in water (30%, 50%, 70%, 90%, and 95%) before finally using water-free acetone. The samples were embedded and polymerized using Epon-Durcupan. The resin blocks were cut into 85 nm slices using an ultramicrotome (Leica EM UC6, Prague, Czech Republic) and placed on copper grids (square, 200 mesh, Agar Scientific, Essex, UK). Images were acquired using a transmission electron microscope Jeol JEM 1400 Flash (120 kV, JEOL, Ltd., Tokyo, Japan), operated at 80 kV, and equipped with a tungsten cathode and bottom-mounted FLASH 2kx2k CMOS camera (TVIPS GmbH, Gilching, Germany). The same AuNPs used for cell experiments were also analyzed alone using TEM. For this purpose, 2 mL of the sample was placed on a glow-discharge-activated grid (30 s, 1 kV, 10 mA). After air drying, the samples were analyzed by TEM (JEOL JEM-F200), operated at 200 kV and equipped with a Cold FEG and TVIPS XF416 camera (TVIPS GmbH).
To prove the presence of elemental Au in the samples, the control cells incubated with AuNPs were checked using a JED 2300 X-ray spectrometer (JEOL, Ltd.) to obtain EDS spectra from the zones containing AuNPs. The spectra were then analyzed using Jeol AnalysisStation software JEM-F200.
TEM photomicrographs were used to roughly evaluate the amount of internalized AuNPs per cell in each group (i.e., AuNPs-antimiR-135b and AuNPs-antimiR-135b-Tf).
For this purpose, the cells (n = 3/group) were first examined at a magnification of 5 µm, and then AuNPs were counted in at least three random areas of each cell at a magnification of 500 nm. The number of AuNPs was recorded, and the average total number of nanoparticles was calculated for each group. Additionally, colocalization and the quantities of AuNPs within early endosomes and late endosomes/lysosomes were assessed. To compare the results, data were compared using analysis of variance (ANOVA) in ANOVA Calculator—One Way ANOVA and Tukey HSD tests available at
https://www.statskingdom.com/180Anova1way.html (accessed on 7 September 2025).
p-values of ≤0.05 were considered as significant.
4.5. Quantitation of miR 135b Knock-Down Using qPCR
Independent duplicates of all the samples for qPCR were prepared. Eight plates were cultured with the same amounts of cells (2 × 10
3 cells/mL) and, after overnight incubation in the cell culture condition, the plates were incubated with different chemicals; one plate incubated with nystatin, one plate with amiloride hydrochloride, and two plates incubated with chlorpromazine. The concentrations used were based on previous studies involving 4T1 cells [
31,
32]. The incubation time was 60 min, after which the chemicals were removed and cells were washed with PBS. The plates were then refilled with 1 mL of the working medium. In the first two sets (plates treated with nystatin and amiloride hydrochloride), each plate was incubated with 10 µL of AuNPs-antimiR 135b. The plates in the third set (plates treated with chlorpromazine) were each incubated with 10 µL of either AuNP-antimiR 135b or AuNP-antimiR 135b-Tf. Additionally, seven plates were used as controls: two were filled with 10 µL of either AuNP-antimiR 135b or AuNP-antimiR 135b-Tf, one plate was filled with X-tremeGENE™ HP DNA Transfection Reagent (Sigma Aldrich, Prague, Czech Republic and in this article is named HP)-antimiR 135b serving as a positive transfection control, one untreated plate served as a negative control (cell without any pretreatment and no AuNPs conjugates), and three pretreated plates, each containing the same amounts of chemicals (i.e., nystatin, amiloride hydrochloride, and chlorpromazine), were only refilled with the working medium and used as chemical-treated controls. The plates were then incubated under cell culture conditions, washed with PBS after 2 h, refilled with the working medium, and incubated overnight.
MicroRNA was extracted using the High Pure miRNA Isolation Kit (Roche, Prague, Czech Republic). The High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific, Prague, Czech Republic) was used to generate cDNA with miR 16 (assay ID 000391) and miR 135b (assay ID 002261) RT primers (Thermo Fisher Scientific, Prague, Czech Republic). The level of miR 135b relative to miR 16 (as an internal control) was determined using real-time polymerase chain reaction (qPCR) and an iQ5 Real-Time PCR Detection System (BioRad, Prague, Czech Republic). The qPCR reaction mixture consisted of TaqMan Universal PCR Master Mix (No AmpErase, Thermo Fisher Scientific, Prague, Czech Republic) with an appropriate mix of primers/probes (miR 135b assay ID 002261 and miR 16 assay ID 000391). Each reaction was run in triplicate. Results were analyzed using the iQ5 Optical System Software 2.1 (BioRad, Prague, Czech Republic), and changes in the level of miR 135b were calculated based on the 2
−ddCt method. Remarkable change was considered using cut-off = 0.5. Differences with
p-value < 0.01 were considered as significant [
3,
33].
Since we use an optimized ratio and incubation time for loading antimiR onto AuNPs, we expect a similar load of the antimiR per each AuNP. The successful delivery of the antimiR is demonstrated by the effective but not complete knock-down of the target miR 135b after the cells were treated with the AuNPs-antimiR 135b only. Using the non-treated cells as a reference sample, we can quantify the efficacy of miR 135b knock-down reflecting also the number of internalized AuNPs in cell cytoplasm.
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}