
Knockdown of the snoRNA-Jouvence Blocks the Proliferation and Leads to the Death of Human Primary Glioblastoma Cells

 , , and
, , and
Abstract
1. Introduction
2. Results
2.1. Knockdown of the snoRNA-Jou by Sh-Lentivirus Decreases Cancerous Cell Proliferation
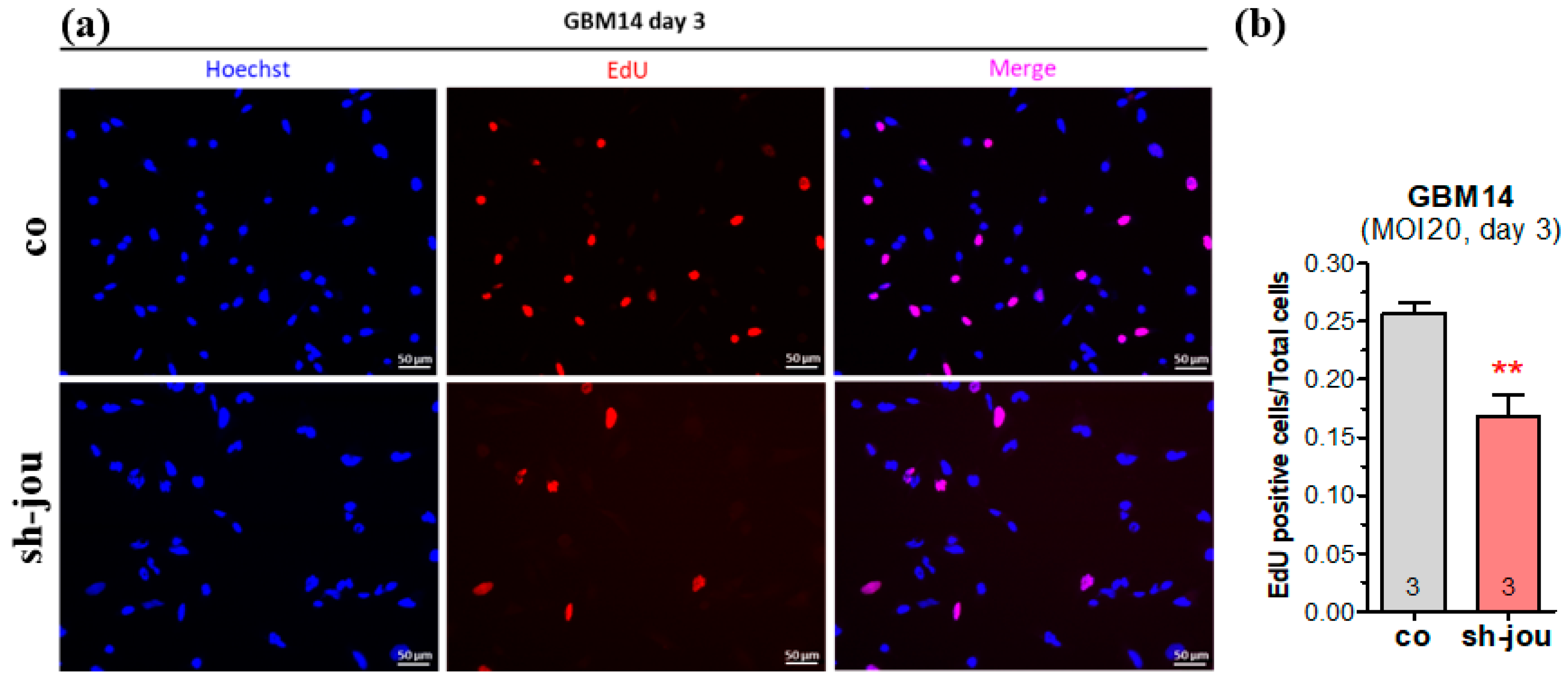
2.2. EdU Confirms the Decrease in Cell Proliferation
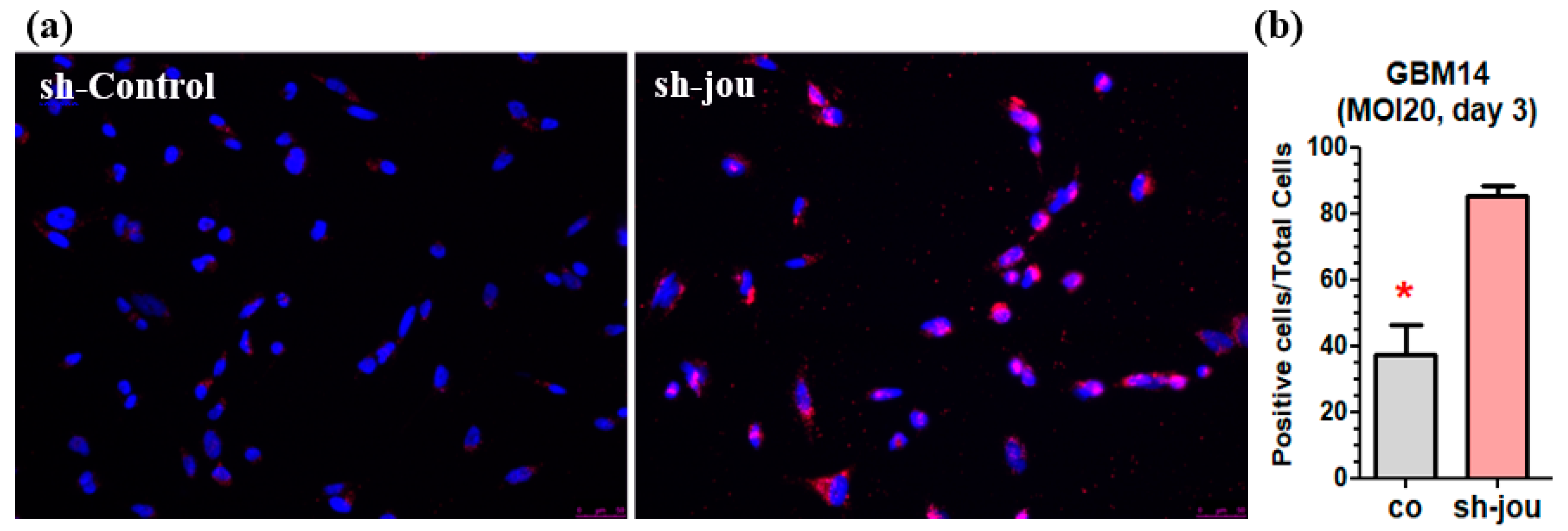
2.3. TUNEL Reveals the Presence of Apoptotic Cells
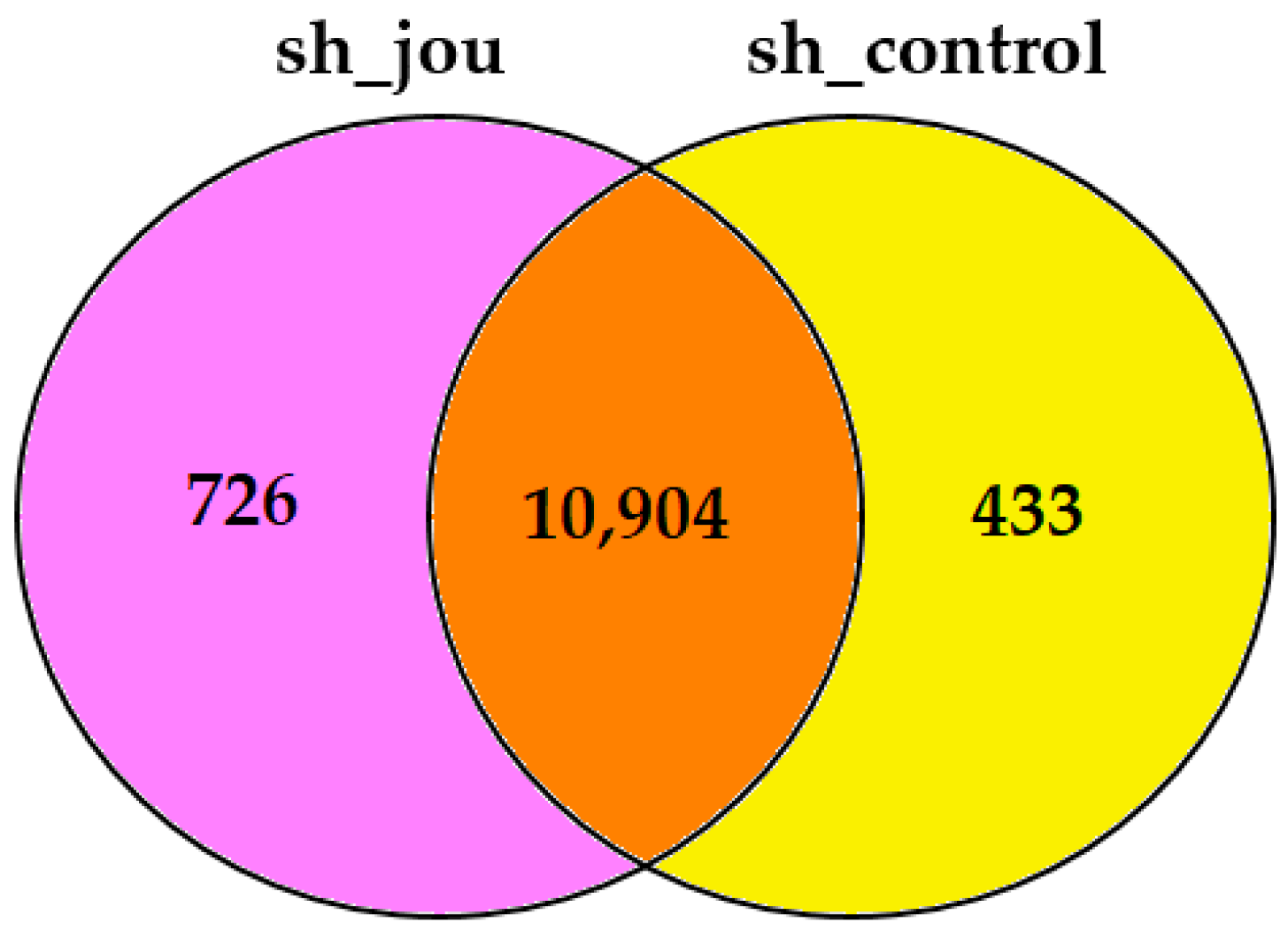
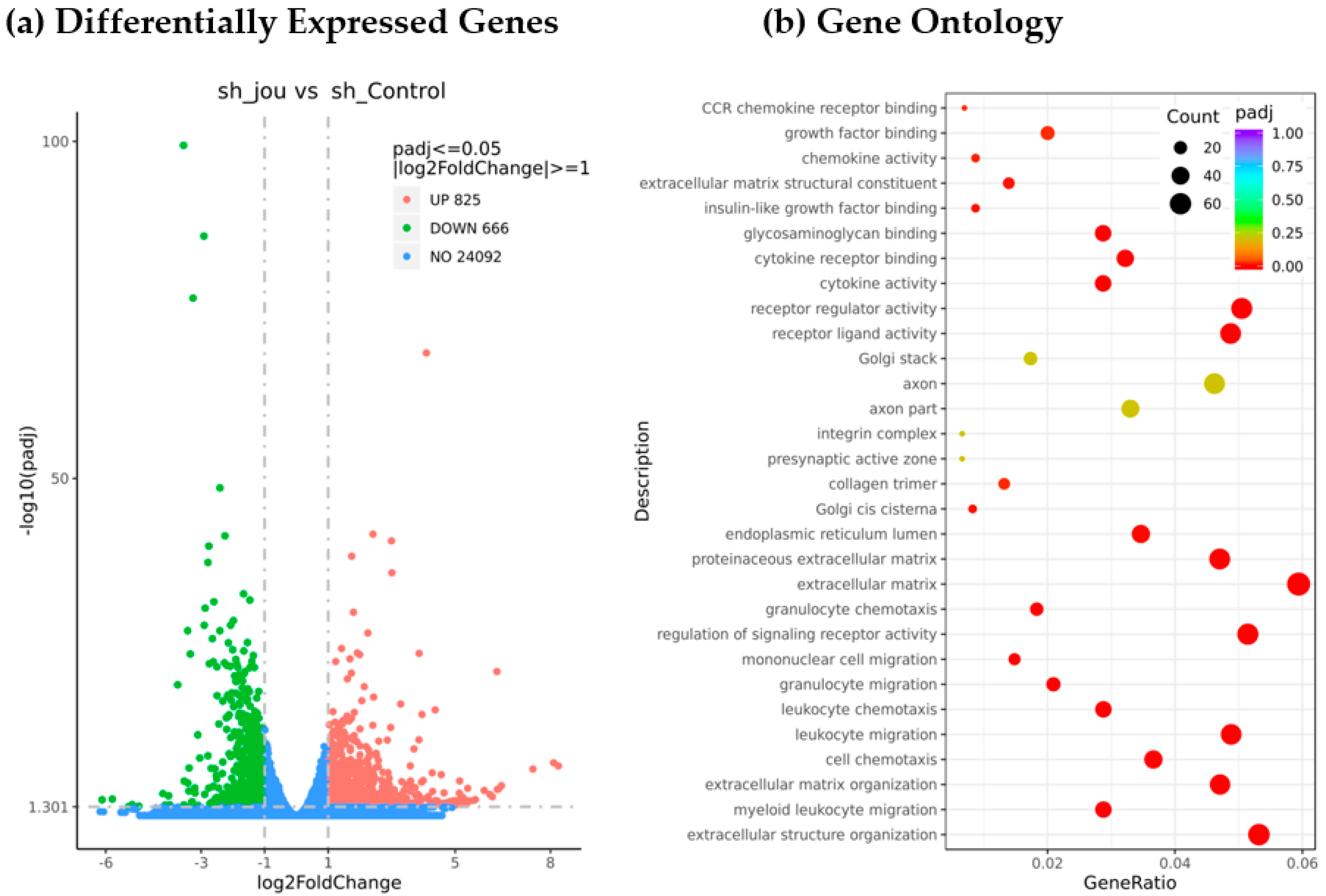
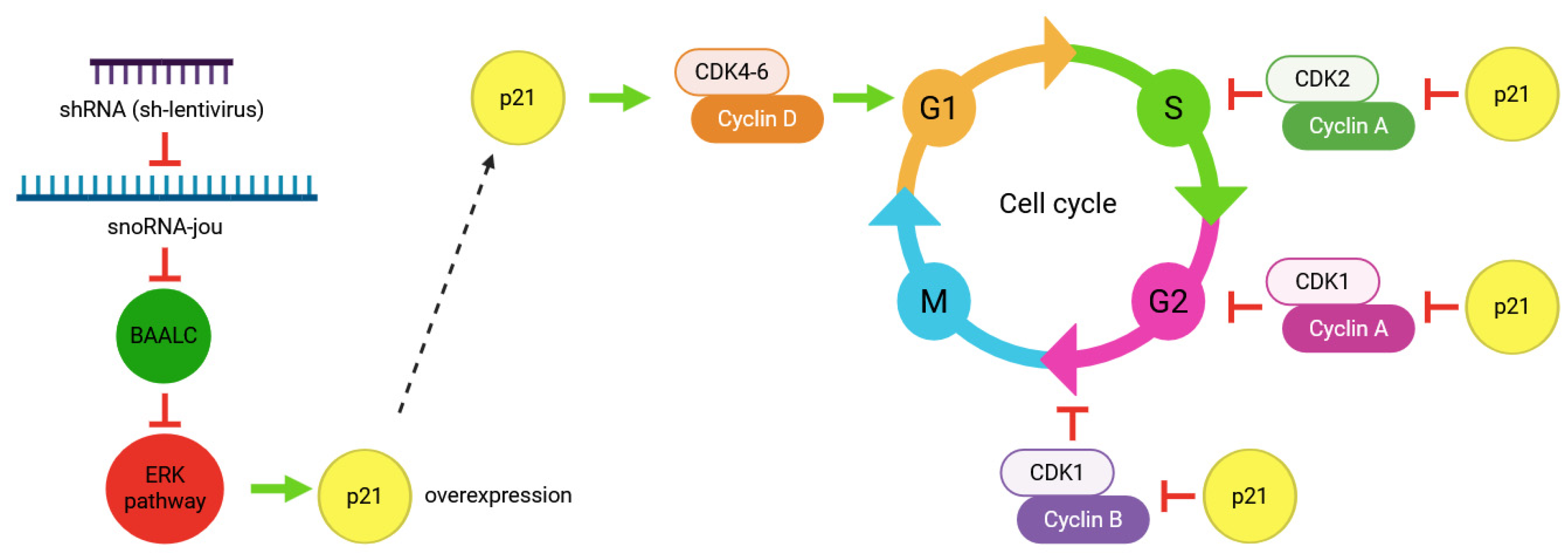
2.4. Transcriptomic Analysis Reveals the Decrease in Several Genes, Including BAALC
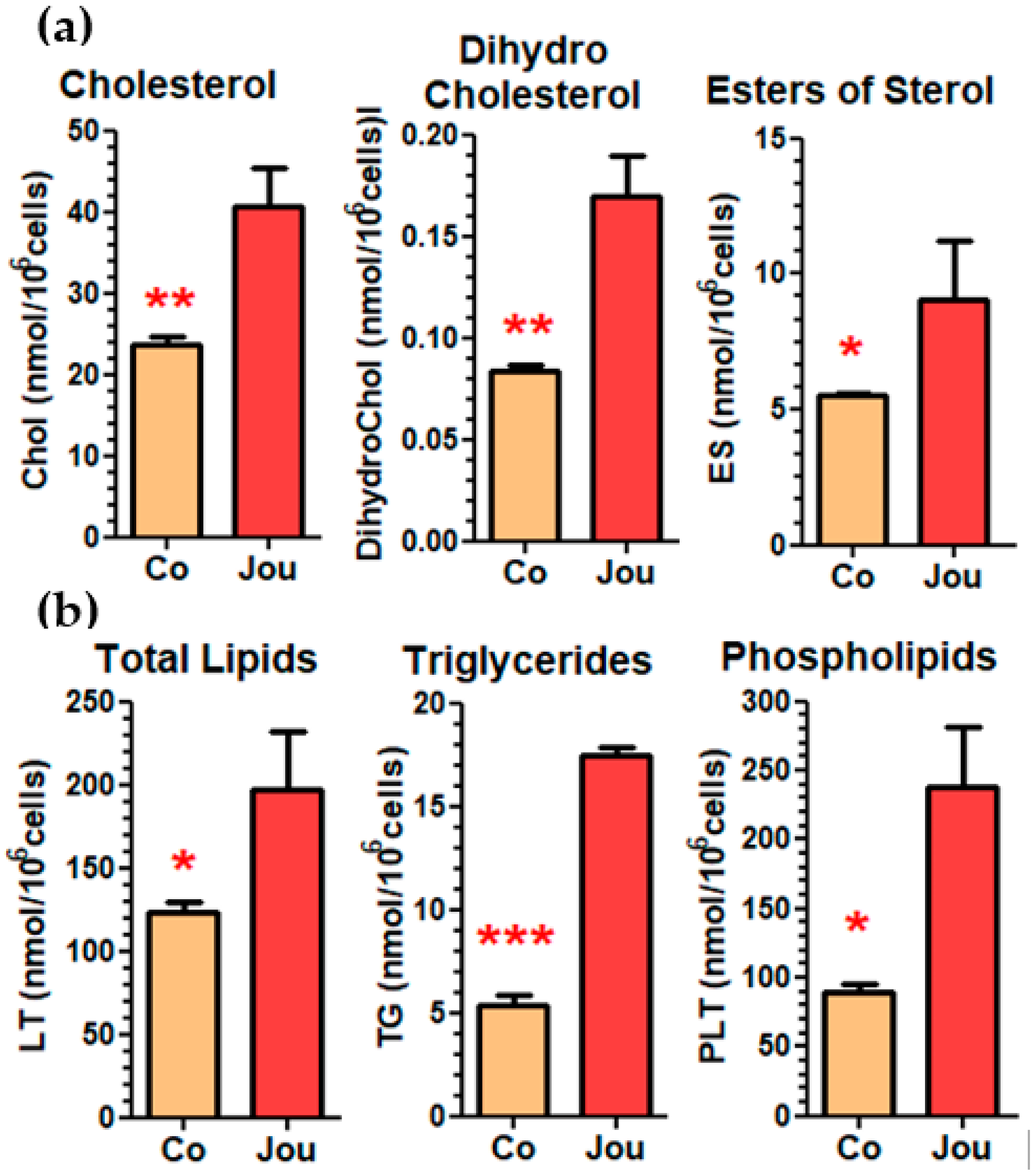
2.5. Lipidomic Analysis Reveals a Deregulation of Cholesterol and Other Lipids
3. Discussion
4. Methods
4.1. Cell Lines and Culture Conditions
4.1.1. Immortalized Cell Lines
4.1.2. GBM14-CHA Primary Cells
4.1.3. Culture Conditions
4.2. Lentivirus Design and Transduction
4.3. CellTiter-Glo Cell Viability Assay
4.4. Cell Counting
4.5. Transcriptomic Analysis (RNA-Seq)
4.6. Cell Proliferation ASSAY (EdU)
4.7. Apoptotic Cells Detection (TUNEL)
4.8. Lipidomic Analysis
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hanif, F.; Muzaffar, K.; Perveen, K.; Malhi, S.M.; Simjee, S.U. Glioblastoma Multiforme: A Review of its Epidemiology and Pathogenesis through Clinical Presentation and Treatment. Asian Pac. J. Cancer Prev. 2017, 18, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Li, Y.; Liu, C.-J.; Xiang, Y.; Li, C.; Ye, Y.; Zhang, Z.; Hawke, D.H.; Park, P.K.; Diao, L.; et al. A Pan-cancer Analysis of the Expression and Clinical Relevance of Small Nucleolar RNAs in Human Cancer. Cell Rep. 2017, 21, 1968–1981. [Google Scholar] [CrossRef] [PubMed]
- Thorenoor, N.; Slaby, O. Small nucleolar RNAs functioning and potential roles in cancer. Tumor Biol. 2014, 36, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Mannoor, K.; Liao, J.; Jiang, F. Small nucleolar RNAs in cancer. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2012, 1826, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.T.; Farzaneh, F. Are snoRNAs and snoRNA host genes new players in cancer? Nat. Rev. Cancer 2012, 12, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Wen, J.; Huang, Z.; Chen, X.-P.; Zhang, B.-X.; Chu, L. Small Nucleolar RNAs: Insight Into Their Function in Cancer. Front. Oncol. 2019, 9, 587. [Google Scholar] [CrossRef] [PubMed]
- Van der Werf, J.; Chin, C.V.; Fleming, N.I. SnoRNA in Cancer Progression, Metastasis and Immunotherapy Response. Biology 2021, 10, 809. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Cui, W.; Liu, M.; Zhang, F.; Zhao, Y.; Zhang, M.; Yin, Y.; Li, Y.; Che, Y.; Zhu, X.; et al. SnoRNAs: The promising targets for anti-tumor therapy. J. Pharm. Anal. 2024, 14, 101064. [Google Scholar] [CrossRef] [PubMed]
- Kiss, T. Small Nucleolar RNAs: An Abundant Group of Noncoding RNAs with Diverse Cellular Functions. Cell 2002, 109, 145–148. [Google Scholar] [CrossRef] [PubMed]
- Ge, J.; Yu, Y.-T. RNA pseudouridylation: New insights into an old modification. Trends Biochem. Sci. 2013, 38, 210–218. [Google Scholar] [CrossRef] [PubMed]
- van Riggelen, J.; Yetil, A.; Felsher, D.W. MYC as a regulator of ribosome biogenesis and protein synthesis. Nat. Rev. Cancer 2010, 10, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Hald, Ø.H.; Olsen, L.; Gallo-Oller, G.; Elfman, L.H.M.; Løkke, C.; Kogner, P.; Sveinbjörnsson, B.; Flægstad, T.; Johnsen, J.I.; Einvik, C. Inhibitors of ribosome biogenesis repress the growth of MYCN-amplified neuroblastoma. Oncogene 2018, 38, 2800–2813. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, K.; Kumazawa, T.; Kuroda, T.; Katagiri, N.; Tsuchiya, M.; Goto, N.; Furumai, R.; Murayama, A.; Yanagisawa, J.; Kimura, K. Perturbation of Ribosome Biogenesis Drives Cells into Senescence through 5S RNP-Mediated p53 Activation. Cell Rep. 2015, 10, 1310–1323. [Google Scholar] [CrossRef] [PubMed]
- Soulé, S.; Mellottée, L.; Arab, A.; Chen, C.; Martin, J.-R. Jouvence a small nucleolar RNA required in the gut extends lifespan in Drosophila. Nat. Commun. 2020, 11, 987. [Google Scholar] [CrossRef] [PubMed]
- Soulé, S.; Martin, J.R. ninaD regulates cholesterol homeostasis from the midgut which protects against neurodegeneration. BioRxiv 2020, 11, 401059. [Google Scholar]
- El-Khoury, F.; Bignon, J.; Martin, J.-R. jouvence, a new human snoRNA involved in the control of cell proliferation. BMC Genom. 2020, 21, 817. [Google Scholar] [CrossRef] [PubMed]
- Huh, H.D.; Kim, D.H.; Jeong, H.-S.; Park, H.W. Regulation of TEAD Transcription Factors in Cancer Biology. Cells 2019, 8, 600. [Google Scholar] [CrossRef] [PubMed]
- Tanner, S.M.; Austin, J.L.; Leone, G.; Rush, L.J.; Plass, C.; Heinonen, K.; Mrózek, K.; Sill, H.; Knuutila, S.; Kolitz, J.E.; et al. BAALC, the human member of a novel mammalian neuroectoderm gene lineage, is implicated in hematopoiesis and acute leukemia. Proc. Natl. Acad. Sci. USA 2001, 98, 13901–13906. [Google Scholar] [CrossRef] [PubMed]
- Morita, K.; Masamoto, Y.; Kataoka, K.; Koya, J.; Kagoya, Y.; Yashiroda, H.; Sato, T.; Murata, S.; Kurokawa, M. BAALC potentiates oncogenic ERK pathway through interactions with MEKK1 and KLF4. Leukemia 2015, 29, 2248–2256. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Lv, M.; Zhang, W.; Zhan, Q. Dysregulation of cholesterol metabolism in cancer progression. Oncogene 2023, 42, 3289–3302. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Deng, Y.; Deng, G.; Chen, P.; Wang, Y.; Wu, H.; Ji, Z.; Yao, Z.; Zhang, X.; Yu, B.; et al. High cholesterol induces apoptosis and autophagy through the ROS-activated AKT/FOXO1 pathway in tendon-derived stem cells. Stem Cell Res. Ther. 2020, 11, 131. [Google Scholar] [CrossRef] [PubMed]
- Jinn, S.; Brandis, K.A.; Ren, A.; Chacko, A.; Dudley-Rucker, N.; Gale, S.E.; Sidhu, R.; Fujiwara, H.; Jiang, H.; Olsen, B.N.; et al. snoRNA U17 Regulates Cellular Cholesterol Trafficking. Cell Metab. 2015, 21, 855–867. [Google Scholar] [CrossRef] [PubMed]
- McMahon, M.; Contreras, A.; Ruggero, D. Small RNAs with big implications: New insights into H/ACA snoRNA function and their role in human disease. Wiley Interdiscip. Rev. RNA 2015, 6, 173–189. [Google Scholar] [CrossRef] [PubMed]
- Schubert, T.; Pusch, M.C.; Diermeier, S.; Benes, V.; Kremmer, E.; Imhof, A.; Längst, G. Df31 Protein and snoRNAs Maintain Accessible Higher-Order Structures of Chromatin. Mol. Cell 2012, 48, 434–444. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Alonso, D.; Malumbres, M. Mammalian cell cycle cyclins. Semin. Cell Dev. Biol. 2020, 107, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019, 26, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Baek, A.E.; Nelson, E.R. The Contribution of Cholesterol and Its Metabolites to the Pathophysiology of Breast Cancer. Horm. Cancer 2016, 7, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.-C.; Parajuli, K.R.; Duong, H.-Q.; Choi, J.E.; Han, S.I. Cholesterol induces autophagic and apoptotic death in gastric carcinoma cells. Int. J. Oncol. 2014, 44, 805–811. [Google Scholar] [CrossRef] [PubMed]
- Bazioti, V.; La Rose, A.M.; Maassen, S.; Bianchi, F.; de Boer, R.; Halmos, B.; Dabral, D.; Guilbaud, E.; Flohr-Svendsen, A.; Groenen, A.G.; et al. T cell cholesterol efflux suppresses apoptosis and senescence and increases atherosclerosis in middle aged mice. Nat. Commun. 2022, 13, 3799. [Google Scholar] [CrossRef] [PubMed]
- Lucken-Ardjomande, S.; Montessuit, S.; Martinou, J.-C. Bax activation and stress-induced apoptosis delayed by the accumulation of cholesterol in mitochondrial membranes. Cell Death Differ. 2008, 15, 484–493. [Google Scholar] [CrossRef] [PubMed]
- Freed-Pastor, W.A.; Mizuno, H.; Zhao, X.; Langerød, A.; Moon, S.-H.; Rodriguez-Barrueco, R.; Barsotti, A.; Chicas, A.; Li, W.; Polotskaia, A.; et al. Mutant p53 Disrupts Mammary Tissue Architecture via the Mevalonate Pathway. Cell 2012, 148, 244–258. [Google Scholar] [CrossRef] [PubMed]
- Moodbidri, M.S.; Shirsat, N.V. Induction of BAALC and down regulation of RAMP3 in astrocytes treated with differentiation inducers. Cell Biol. Int. 2006, 30, 210–213. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Liu, Y.; Ma, X.; Hu, H. The Influence of Cell Cycle Regulation on Chemotherapy. Int. J. Mol. Sci. 2021, 22, 6923. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.D.; Zhu, X.F. Small nucleolar RNAs (snoRNAs) as potential non-invasive biomarkers for early cancer detection. Chin. J. Cancer 2013, 32, 99–101. [Google Scholar] [CrossRef] [PubMed]
- Deng, T.; Gong, Y.; Liao, X.; Wang, X.; Zhou, X.; Zhu, G.; Mo, L. Integrative Analysis of a Novel Eleven-Small Nucleolar RNA Prognostic Signature in Patients With Lower Grade Glioma. Front. Oncol. 2021, 11, 650828. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Furumichi, M.; Morishima, K.; Tanabe, M. New approach for understanding genome variations in KEGG. Nucleic Acids Res. 2018, 47, D590–D595. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene_ID | Fold Ch. | padj | Name | Gene_Description |
|---|---|---|---|---|
| ERK1 and ERK2 cascade | ||||
| ENSG00000164929 | 0.09 | 0.000 | BAALC | BAALC, MAP3K1 and KLF4 binding |
| ENSG00000133048 | 0.37 | 0.000 | CHI3L1 | chitinase 3 like 1 |
| ENSG00000100234 | 0.36 | 0.000 | TIMP3 | TIMP metallopeptidase inhibitor 3 |
| ENSG00000204356 | 0.27 | 0.000 | NELFE | negative elong, factor complex member E |
| ENSG00000162545 | 0.27 | 0.000 | CAMK2N1 | Ca2+/calmodulin dep. Prot. kinase II inhib.1 |
| ENSG00000108691 | 0.23 | 0.000 | CCL2 | C-C motif chemokine ligand 2 |
| ENSG00000115414 | 0.37 | 0.000 | FN1 | fibronectin 1 |
| ENSG00000141736 | 0.32 | 0.000 | ERBB2 | erb-b2 receptor tyrosine kinase 2 |
| ENSG00000090339 | 0.45 | 0.000 | ICAM1 | intercellular adhesion molecule 1 |
| ENSG00000117228 | 0.39 | 0.000 | GBP1 | guanylate binding protein 1 |
| ENSG00000006210 | 0.32 | 0.000 | CX3CL1 | C-X3-C motif chemokine ligand 1 |
| ENSG00000152518 | 0.41 | 0.000 | ZFP36L2 | ZFP36 ring finger protein like 2 |
| ENSG00000003402 | 0.41 | 0.000 | CFLAR | CASP8 and FADD like apoptosis regulator |
| ENSG00000120875 | 0.49 | 0.000 | DUSP4 | dual specificity phosphatase 4 |
| ENSG00000154188 | 0.44 | 0.000 | ANGPT1 | angiopoietin 1 |
| ENSG00000136848 | 0.49 | 0.000 | DAB2IP | DAB2 interacting protein |
| ENSG00000006606 | 0.40 | 0.000 | CCL26 | C-C motif chemokine ligand 26 |
| ENSG00000113721 | 0.47 | 0.000 | PDGFRB | platelet derived growth factor receptor b |
| ENSG00000108688 | 0.20 | 0.000 | CCL7 | C-C motif chemokine ligand 7 |
| ENSG00000185650 | 0.41 | 0.000 | ZFP36L1 | ZFP36 ring finger protein like 1 |
| ENSG00000169220 | 0.46 | 0.000 | RGS14 | regulator of G protein signaling 14 |
| ENSG00000125845 | 0.48 | 0.000 | BMP2 | bone morphogenetic protein 2 |
| ENSG00000148053 | 0.29 | 0.000 | NTRK2 | neurotrophic receptor tyrosine kinase 2 |
| ENSG00000145242 | 0.40 | 0.001 | EPHA5 | EPH receptor A5 |
| ENSG00000108700 | 0.06 | 0.001 | CCL8 | C-C motif chemokine ligand 8 |
| ENSG00000019582 | 0.44 | 0.003 | CD74 | CD74 molecule |
| ENSG00000271503 | 0.10 | 0.009 | CCL5 | C-C motif chemokine ligand 5 |
| ENSG00000183578 | 0.28 | 0.016 | TNFAIP8L3 | TNF alpha induced protein 8 like 3 |
| mitogen-activated protein kinase 11 | ||||
| ENSG00000006062 | 2.15 | 0.000 | MAP3K14 | mitogen-act. prot. kinase kin. Kin. 14 |
| ENSG00000185386 | 0.54 | 0.012 | MAPK11 | mitogen-activated protein kinase 11 |
| ENSG00000065559 | 1.66 | 0.000 | MAP2K4 | mitogen-activated protein kinase kinase 4 |
| ENSG00000071054 | 0.71 | 0.001 | MAP4K4 | mitogen-act. prot. kinase kin. Kin. Kin. 4 |
| ENSG00000100030 | 0.74 | 0.015 | MAPK1 | mitogen-activated protein kinase 1 |
| ENSG00000109339 | 0.64 | 0.055 | MAPK10 | mitogen-activated protein kinase 10 |
| ENSG00000138834 | 2.62 | 0.000 | MAPK8IP3 | mitogen-act. prot. kinase 8 interac. prot. 3 |
| ENSG00000006062 | 2.15 | 0.000 | MAP3K14 | mitogen-act. prot. kinase kinase kinase 14 |
| ENSG00000006432 | 1.77 | 0.000 | MAP3K9 | mitogen-act. prot. kinase kinase kinase 9 |
| ENSG00000143674 | 2.04 | 0.002 | MAP3K21 | mitogen-act. prot. kinase kinase kinase 21 |
| ENSG00000085511 | 1.33 | 0.019 | MAP3K4 | mitogen-act. prot. kinase kinase kinase 4 |
| ENSG00000107968 | 0.66 | 0.050 | MAP3K8 | mitogen-act. prot. kinase kinase kinase 8 |
| ENSG00000169967 | 1.27 | 0.066 | MAP3K2 | mitogen-act. prot. kinase kinase kinase 2 |
| ribosomal protein S6 kinase A1 | ||||
| ENSG00000117676 | 0.67 | 0.007 | RPS6KA1 | ribosomal protein S6 kinase A1 |
| ENSG00000177189 | 0.74 | 0.015 | RPS6KA3 | ribosomal protein S6 kinase A3 |
| ENSG00000162302 | 1.37 | 0.048 | RPS6KA4 | ribosomal protein S6 kinase A4 |
| ENSG00000108443 | 1.38 | 0.040 | RPS6KB1 | ribosomal protein S6 kinase B1 |
| Gene_ID | Fold Ch. | padj | Name | Gene_Description |
|---|---|---|---|---|
| cell cycle, cyclin, and CDK | ||||
| ENSG00000058091 | 2.33 | 0.000 | CDK14 | cyclin dependent kinase 14 |
| ENSG00000108465 | 3.71 | 0.000 | CDK5RAP3 | CDK5 regulatory subunit ass. prot. 3 |
| ENSG00000111328 | 0.63 | 0.001 | CDK2AP1 | cyclin dependent kinase 2 ass. prot. 1 |
| ENSG00000138395 | 3.49 | 0.001 | CDK15 | cyclin dependent kinase 15 |
| ENSG00000129355 | 0.57 | 0.002 | CDKN2D | cyclin dependent kinase inhibitor 2D |
| ENSG00000164885 | 0.68 | 0.014 | CDK5 | cyclin dependent kinase 5 |
| ENSG00000156345 | 0.61 | 0.017 | CDK20 | cyclin dependent kinase 20 |
| ENSG00000100865 | 0.69 | 0.012 | CINP | cyclin dependent kinase 2 inter. prot. |
| ENSG00000129757 | 0.63 | 0.023 | CDKN1C | cyclin dependent kinase inhibitor 1C |
| ENSG00000124762 | 1.35 | 0.038 | CDKN1A | cyclin dependent kinase inhibitor 1A |
| ENSG00000167258 | 1.38 | 0.061 | CDK12 | cyclin dependent kinase 12 |
| ENSG00000185862 | 3.69 | 0.000 | EVI2B | ecotropic viral integration site 2B |
| ENSG00000198625 | 1.74 | 0.001 | MDM4 | MDM4, p53 regulator |
| ENSG00000136807 | 1.49 | 0.004 | CDK9 | cyclin dependent kinase 9 |
| ENSG00000123374 | 1.38 | 0.007 | CDK2 | cyclin dependent kinase 2 |
| ENSG00000009950 | 2.54 | 0.061 | MLXIPL | MLX interacting protein like |
| ENSG00000163660 | 2.62 | 0.000 | CCNL1 | cyclin L1 |
| ENSG00000112576 | 0.58 | 0.000 | CCND3 | cyclin D3 |
| ENSG00000221978 | 2.15 | 0.002 | CCNL2 | cyclin L2 |
| ENSG00000147082 | 9.28 | 0.003 | CCNB3 | cyclin B3 |
| ENSG00000118971 | 2.88 | 0.022 | CCND2 | cyclin D2 |
| ENSG00000113328 | 0.74 | 0.024 | CCNG1 | cyclin G1 |
| ENSG00000134480 | 1.41 | 0.041 | CCNH | cyclin H |
| ENSG00000107443 | 1.56 | 0.042 | CCNJ | cyclin J |
| TP53 and associated protein | ||||
| ENSG00000078804 | 0.41 | 0.000 | TP53INP2 | tumor prot. p53 inducible nuclear prot. 2 |
| ENSG00000167543 | 0.55 | 0.009 | TP53I13 | tumor prot. p53 inducible protein 13 |
| ENSG00000172315 | 0.65 | 0.034 | TP53RK | TP53 regulating kinase |
| ENSG00000164938 | 1.64 | 0.069 | TP53INP1 | tumor protein p53 inducible nuclear prot. 1 |
| ENSG00000175274 | 0.69 | 0.070 | TP53I11 | tumor protein p53 inducible protein 11 |
| ENSG00000246640 | 4.46 | 0.001 | PICART1 | p53-inducible cancer-ass. RNA transcript 1 |
| ENSG00000120279 | 4.03 | 0.002 | MYCT1 | MYC target 1 |
| Cholesterol and sterol pathways | ||||
| ENSG00000116133 | 3.53 | 0.000 | DHCR24 | 24-dehydrocholesterol reductase |
| ENSG00000138135 | 0.32 | 0.000 | CH25H | cholesterol 25-hydroxylase |
| ENSG00000198911 | 0.40 | 0.000 | SREBF2 | sterol regul. element binding TF 2 |
| ENSG00000160285 | 0.57 | 0.000 | LSS | lanosterol synthase |
| ENSG00000072310 | 0.56 | 0.001 | SREBF1 | sterol regulatory element binding TF 1 |
| ENSG00000109929 | 1.74 | 0.009 | SC5D | sterol-C5-desaturase |
| ENSG00000079156 | 1.84 | 0.006 | OSBPL6 | oxysterol binding protein like 6 |
| ENSG00000021762 | 1.43 | 0.014 | OSBPL5 | oxysterol binding protein like 5 |
| ENSG00000141447 | 0.70 | 0.044 | OSBPL1A | oxysterol binding protein like 1A |
| ENSG00000117859 | 1.26 | 0.068 | OSBPL9 | oxysterol binding protein like 9 |
| ENSG00000015520 | 3.29 | 0.084 | NPC1L1 | NPC1 like intra. cholesterol transporter 1 |
| ENSG00000172893 | 1.37 | 0.094 | DHCR7 | 7-dehydrocholesterol reductase |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jaque-Cabrera, L.; Buggiani, J.; Bignon, J.; Daira, P.; Bernoud-Hubac, N.; Martin, J.-R. Knockdown of the snoRNA-Jouvence Blocks the Proliferation and Leads to the Death of Human Primary Glioblastoma Cells. Non-Coding RNA 2025, 11, 54. https://doi.org/10.3390/ncrna11040054
Jaque-Cabrera L, Buggiani J, Bignon J, Daira P, Bernoud-Hubac N, Martin J-R. Knockdown of the snoRNA-Jouvence Blocks the Proliferation and Leads to the Death of Human Primary Glioblastoma Cells. Non-Coding RNA. 2025; 11(4):54. https://doi.org/10.3390/ncrna11040054
Chicago/Turabian StyleJaque-Cabrera, Lola, Julia Buggiani, Jérôme Bignon, Patricia Daira, Nathalie Bernoud-Hubac, and Jean-René Martin. 2025. "Knockdown of the snoRNA-Jouvence Blocks the Proliferation and Leads to the Death of Human Primary Glioblastoma Cells" Non-Coding RNA 11, no. 4: 54. https://doi.org/10.3390/ncrna11040054
APA StyleJaque-Cabrera, L., Buggiani, J., Bignon, J., Daira, P., Bernoud-Hubac, N., & Martin, J.-R. (2025). Knockdown of the snoRNA-Jouvence Blocks the Proliferation and Leads to the Death of Human Primary Glioblastoma Cells. Non-Coding RNA, 11(4), 54. https://doi.org/10.3390/ncrna11040054