
Investigating the Role of Non-Coding RNA in Non-Alcoholic Fatty Liver Disease

 , , ,
, , , 
Abstract
1. Introduction
2. Liver Cellular Function and Development of NAFLD/NASH
2.1. Pathogenesis and Clinical Aspects of NAFLD/NASH
2.2. Current Treatment Options for NAFLD/NASH
3. NcRNA and Intercellular Communication in Liver Cell Populations
3.1. MiRNA
3.2. LncRNA
3.3. CircRNA
3.4. PiRNA
4. NcRNA and the Dysregulated Pathways Related to NAFLD/NASH
4.1. NcRNA, Lipotoxicity and Oxidative Stress
4.2. NcRNA, Inflammation, and Fibrosis
5. The Role of ncRNA in NAFLD-Associated Metabolic Dysfunction
6. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| NAFLD | Non-alcoholic fatty liver disease |
| NASH | Non-alcoholic steatohepatitis |
| PNPLA3 | Patatin Like Phospholipase Domain Containing 3 |
| MBOAT7 | Membrane Bound O-Acyltransferase Domain Containing 7 |
| NcRNAs | Non-coding RNAs |
| MiRNA | Micro RNA |
| LncRNAs | Long non-coding RNAs |
| PIWI | P-element-induced wimpy testis |
| PiRNAs | Interacting RNAs |
| CircRNAs / ciRNAs | Circular RNAs |
| piRISC | piRNA-induced silencing complex |
| HCs | Hepatocytes |
| KCs | Kupffer cells |
| HSCs | Hepatic stellate cells |
| LSECs | Liver sinusoidal cells |
| ROS | Reactive Oxygen species |
| SS | Simple steatosis |
| HCC | Hepatocellular carcinoma |
| SREBP-1c | Sterol regulatory element–binding protein 1c |
| FFA | Free fatty acids |
| PPARs | Peroxisome proliferator–activated receptors |
| ACC | Acetyl-CoA carboxylase |
| FAS | Fatty acid synthase |
| TLR4 | Toll-like receptor 4 |
| NF-κB | Nuclear factor-κB |
| TGF-β | Transforming growth factor β |
| RNA-seq | RNA-sequencing |
| EVs | Extracellular vesicles |
| NOS | Nitric oxide synthase |
| TGM2 | Targeting transglutaminase 2 |
| SRA | Steroid receptor RNA activator |
| FOXO1 | Forkhead box protein O1 |
| Blnc1 | Brown fat-enriched lncRNA 1 |
| MALAT1 | Metastasis-associated lung adenocarcinoma transcript 1 |
| SCAR | Steatohepatitis-associated circRNA ATP5B Regulator |
| G3P | Glycerol-3-phosphate |
| FASN | Fatty acid synthase |
| AceCS | Acetyl-CoA synthetase |
| LXRs | Liver X receptors |
| mTOR | Mammalian target of rapamycin |
| TNF-R | Tumor necrosis factor receptor |
| MCD | Methionine-choline-deficient |
| Platr4 | Pluripotency-associated transcript 4 |
| RIP | RNA immunoprecipitation |
| ECM | Extra-cellular matrix |
| Cav1 | Caveolin-1 |
| OSBP | Oxysterol binding protein |
| FA2H | Fatty acid 2-hydroxylase |
References
- Huang, R.; Duan, X.; Fan, J.; Li, G.; Wang, B. Role of Noncoding RNA in Development of Nonalcoholic Fatty Liver Disease. Biomed. Res. Int. 2019, 2019, 8690592. [Google Scholar] [CrossRef]
- Riazi, K.; Azhari, H.; Charette, J.H.; Underwood, F.E.; King, J.A.; Afshar, E.E.; Swain, M.G.; Congly, S.E.; Kaplan, G.G.; Shaheen, A.A. The prevalence and incidence of NAFLD worldwide: A systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 2022, 7, 851–861. [Google Scholar] [CrossRef]
- Del Campo, J.A.; Gallego-Durán, R.; Gallego, P.; Grande, L. Genetic and Epigenetic Regulation in Nonalcoholic Fatty Liver Disease (NAFLD). Int. J. Mol. Sci. 2018, 19, 911. [Google Scholar] [CrossRef] [PubMed]
- Jirtle, R.L.; Skinner, M.K. Environmental epigenomics and disease susceptibility. Nat. Rev. Genet. 2007, 8, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Slack, F.J.; Chinnaiyan, A.M. The Role of Non-coding RNAs in Oncology. Cell 2019, 179, 1033–1055. [Google Scholar] [CrossRef]
- Peterson, C.L.; Workman, J.L. Promoter targeting and chromatin remodeling by the SWI/SNF complex. Curr. Opin. Genet. Dev. 2000, 10, 187–192. [Google Scholar] [CrossRef]
- Krol, J.; Loedige, I.; Filipowicz, W. The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 2010, 11, 597–610. [Google Scholar] [CrossRef]
- Litwin, M.; Szczepańska-Buda, A.; Piotrowska, A.; Dzięgiel, P.; Witkiewicz, W. The meaning of PIWI proteins in cancer development. Oncol. Lett. 2017, 13, 3354–3362. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.N.; Li, Y.; Xia, S.Q.; Zhang, Y.Y.; Zheng, J.H.; Li, W. PIWI Proteins and PIWI-Interacting RNA: Emerging Roles in Cancer. Cell. Physiol. Biochem. 2017, 44, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Trefts, E.; Gannon, M.; Wasserman, D.H. The liver. Curr. Biol. 2017, 27, R1147–r1151. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Tu, W.; Liu, J.; Tian, D. Hepatocytes: A key role in liver inflammation. Front. Immunol. 2022, 13, 1083780. [Google Scholar] [CrossRef] [PubMed]
- Legaki, A.I.; Moustakas, I.I.; Sikorska, M.; Papadopoulos, G.; Velliou, R.I.; Chatzigeorgiou, A. Hepatocyte Mitochondrial Dynamics and Bioenergetics in Obesity-Related Non-Alcoholic Fatty Liver Disease. Curr. Obes. Rep. 2022, 11, 126–143. [Google Scholar] [CrossRef]
- Malhi, H.; Guicciardi, M.E.; Gores, G.J. Hepatocyte death: A clear and present danger. Physiol. Rev. 2010, 90, 1165–1194. [Google Scholar] [CrossRef]
- Chen, X.; Li, H.D.; Bu, F.T.; Li, X.F.; Chen, Y.; Zhu, S.; Wang, J.N.; Chen, S.Y.; Sun, Y.Y.; Pan, X.Y.; et al. Circular RNA circFBXW4 suppresses hepatic fibrosis via targeting the miR-18b-3p/FBXW7 axis. Theranostics 2020, 10, 4851–4870. [Google Scholar] [CrossRef]
- Chen, J.; Deng, X.; Liu, Y.; Tan, Q.; Huang, G.; Che, Q.; Guo, J.; Su, Z. Kupffer Cells in Non-alcoholic Fatty Liver Disease: Friend or Foe? Int. J. Biol. Sci. 2020, 16, 2367–2378. [Google Scholar] [CrossRef]
- Miyao, M.; Kotani, H.; Ishida, T.; Kawai, C.; Manabe, S.; Abiru, H.; Tamaki, K. Pivotal role of liver sinusoidal endothelial cells in NAFLD/NASH progression. Lab. Investig. 2015, 95, 1130–1144. [Google Scholar] [CrossRef]
- Wiering, L.; Subramanian, P.; Hammerich, L. Hepatic Stellate Cells: Dictating Outcome in Nonalcoholic Fatty Liver Disease. Cell. Mol. Gastroenterol. Hepatol. 2023, 15, 1277–1292. [Google Scholar] [CrossRef] [PubMed]
- Stout, R.D.; Suttles, J. Functional plasticity of macrophages: Reversible adaptation to changing microenvironments. J. Leukoc. Biol. 2004, 76, 509–513. [Google Scholar] [CrossRef]
- Jou, J.; Choi, S.S.; Diehl, A.M. Mechanisms of disease progression in nonalcoholic fatty liver disease. Semin. Liver Dis. 2008, 28, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Fujii, H.; Kawada, N.; Japan Study Group Of Nafld, J.-N. The Role of Insulin Resistance and Diabetes in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2020, 21, 3863. [Google Scholar] [CrossRef] [PubMed]
- Kawano, Y.; Cohen, D.E. Mechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver disease. J. Gastroenterol. 2013, 48, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Aizawa, S.; Brar, G.; Tsukamoto, H. Cell Death and Liver Disease. Gut Liver 2020, 14, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Pradere, J.P.; Kluwe, J.; De Minicis, S.; Jiao, J.J.; Gwak, G.Y.; Dapito, D.H.; Jang, M.K.; Guenther, N.D.; Mederacke, I.; Friedman, R.; et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology 2013, 58, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Chai, J.; Wang, H.; Fu, L.; Peng, S.; Ni, X. Hepatic macrophages: Key players in the development and progression of liver fibrosis. Liver Int. 2021, 41, 2279–2294. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, M.; Seki, E. Hepatic Stellate Cell-Macrophage Crosstalk in Liver Fibrosis and Carcinogenesis. Semin. Liver Dis. 2020, 40, 307–320. [Google Scholar] [CrossRef]
- Jung, Y.; Diehl, A.M. Non-alcoholic steatohepatitis pathogenesis: Role of repair in regulating the disease progression. Dig. Dis. 2010, 28, 225–228. [Google Scholar] [CrossRef]
- Sanders, F.W.; Griffin, J.L. De novo lipogenesis in the liver in health and disease: More than just a shunting yard for glucose. Biol. Rev. Camb. Philos. Soc. 2016, 91, 452–468. [Google Scholar] [CrossRef]
- Olona, A.; Hateley, C.; Muralidharan, S.; Wenk, M.R.; Torta, F.; Behmoaras, J. Sphingolipid metabolism during Toll-like receptor 4 (TLR4)-mediated macrophage activation. Br. J. Pharmacol. 2021, 178, 4575–4587. [Google Scholar] [CrossRef] [PubMed]
- Hliwa, A.; Ramos-Molina, B.; Laski, D.; Mika, A.; Sledzinski, T. The Role of Fatty Acids in Non-Alcoholic Fatty Liver Disease Progression: An Update. Int. J. Mol. Sci. 2021, 22, 6900. [Google Scholar] [CrossRef]
- Kawamura, S.; Matsushita, Y.; Kurosaki, S.; Tange, M.; Fujiwara, N.; Hayata, Y.; Hayakawa, Y.; Suzuki, N.; Hata, M.; Tsuboi, M.; et al. Inhibiting SCAP/SREBP exacerbates liver injury and carcinogenesis in murine nonalcoholic steatohepatitis. J. Clin. Investig. 2022, 11, 132. [Google Scholar] [CrossRef]
- Rong, S.; Cortés, V.A.; Rashid, S.; Anderson, N.N.; McDonald, J.G.; Liang, G.; Moon, Y.A.; Hammer, R.E.; Horton, J.D. Expression of SREBP-1c Requires SREBP-2-mediated Generation of a Sterol Ligand for LXR in Livers of Mice. Elife 2017, 6, e25015. [Google Scholar] [CrossRef]
- Barnett, K.C.; Kagan, J.C. Lipids that directly regulate innate immune signal transduction. Innate Immun. 2020, 26, 4–14. [Google Scholar] [CrossRef]
- Bessone, F.; Razori, M.V.; Roma, M.G. Molecular pathways of nonalcoholic fatty liver disease development and progression. Cell. Mol. Life Sci. 2019, 76, 99–128. [Google Scholar] [CrossRef]
- Li, B.; Zhang, C.; Zhan, Y.T. Nonalcoholic Fatty Liver Disease Cirrhosis: A Review of Its Epidemiology, Risk Factors, Clinical Presentation, Diagnosis, Management, and Prognosis. Can. J. Gastroenterol. Hepatol. 2018, 2018, 2784537. [Google Scholar] [CrossRef]
- Hardy, T.; Oakley, F.; Anstee, Q.M.; Day, C.P. Nonalcoholic Fatty Liver Disease: Pathogenesis and Disease Spectrum. Annu. Rev. Pathol. 2016, 11, 451–496. [Google Scholar] [CrossRef]
- Wu, Y.; Ding, Y.; Tanaka, Y.; Zhang, W. Risk factors contributing to type 2 diabetes and recent advances in the treatment and prevention. Int. J. Med. Sci. 2014, 11, 1185–1200. [Google Scholar] [CrossRef]
- Stern, C.; Castera, L. Identification of high-risk subjects in nonalcoholic fatty liver disease. Clin. Mol. Hepatol. 2023, 29, S196–S206. [Google Scholar] [CrossRef] [PubMed]
- Brunner, K.T.; Henneberg, C.J.; Wilechansky, R.M.; Long, M.T. Nonalcoholic Fatty Liver Disease and Obesity Treatment. Curr. Obes. Rep. 2019, 8, 220–228. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N. Engl. J. Med. 2010, 362, 1675–1685. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, A.; Boyon, N.; Shao, A. Physicians and nurses use and recommend dietary supplements: Report of a survey. Nutr. J. 2009, 8, 29. [Google Scholar] [CrossRef]
- Belfort, R.; Harrison, S.A.; Brown, K.; Darland, C.; Finch, J.; Hardies, J.; Balas, B.; Gastaldelli, A.; Tio, F.; Pulcini, J.; et al. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N. Engl. J. Med. 2006, 355, 2297–2307. [Google Scholar] [CrossRef]
- Byrne, C.D. Banting memorial lecture 2022: ‘Type 2 diabetes and nonalcoholic fatty liver disease: Partners in crime’. Diabet Med. 2022, 39, e14912. [Google Scholar] [CrossRef] [PubMed]
- Dutta, P.; Kumar, Y.; Babu, A.T.; Giri Ravindran, S.; Salam, A.; Rai, B.; Baskar, A.; Dhawan, A.; Jomy, M. Tirzepatide: A Promising Drug for Type 2 Diabetes and Beyond. Cureus 2023, 15, e38379. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.Y.; Xie, L.; Zhang, N.P.; Zhou, D.; Liu, T.T.; Wu, J. Updates on novel pharmacotherapeutics for the treatment of nonalcoholic steatohepatitis. Acta Pharmacol. Sin. 2022, 43, 1180–1190. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.J.; Lopez, P.; Lawitz, E.J.; Lucas, K.J.; Loeffler, J.; Kim, W.; Goh, G.B.B.; Huang, J.F.; Serra, C.; Andreone, P.; et al. Tropifexor for nonalcoholic steatohepatitis: An adaptive, randomized, placebo-controlled phase 2a/b trial. Nat. Med. 2023, 29, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Luo, Z.; Gao, H.; Dos Reis, F.C.G.; Bandyopadhyay, G.; Jin, Z.; Manda, K.A.; Isaac, R.; Yang, M.; Fu, W.; et al. Hepatocyte-derived exosomes from early onset obese mice promote insulin sensitivity through miR-3075. Nat. Metab. 2021, 3, 1163–1174. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.L.; Pan, Q.; Cao, H.X.; Xin, F.Z.; Zhao, Z.H.; Yang, R.X.; Zeng, J.; Zhou, H.; Fan, J.G. Lipotoxic Hepatocyte-Derived Exosomal MicroRNA 192-5p Activates Macrophages Through Rictor/Akt/Forkhead Box Transcription Factor O1 Signaling in Nonalcoholic Fatty Liver Disease. Hepatology 2020, 72, 454–469. [Google Scholar] [CrossRef]
- Sharma, J.N.; Al-Omran, A.; Parvathy, S.S. Role of nitric oxide in inflammatory diseases. Inflammopharmacology 2007, 15, 252–259. [Google Scholar] [CrossRef]
- Pourhoseini, S.; Seth, R.K.; Das, S.; Dattaroy, D.; Kadiiska, M.B.; Xie, G.; Michelotti, G.A.; Nagarkatti, M.; Diehl, A.M.; Chatterjee, S. Upregulation of miR21 and repression of Grhl3 by leptin mediates sinusoidal endothelial injury in experimental nonalcoholic steatohepatitis. PLoS ONE 2015, 10, e0116780. [Google Scholar] [CrossRef]
- Liu, H.; Niu, Q.; Wang, T.; Dong, H.; Bian, C. Lipotoxic hepatocytes promote nonalcoholic fatty liver disease progression by delivering microRNA-9-5p and activating macrophages. Int. J. Biol. Sci. 2021, 17, 3745–3759. [Google Scholar] [CrossRef]
- Luo, X.; Luo, S.Z.; Xu, Z.X.; Zhou, C.; Li, Z.H.; Zhou, X.Y.; Xu, M.Y. Lipotoxic hepatocyte-derived exosomal miR-1297 promotes hepatic stellate cell activation through the PTEN signaling pathway in metabolic-associated fatty liver disease. World J. Gastroenterol. 2021, 27, 1419–1434. [Google Scholar] [CrossRef]
- Jopling, C. Liver-specific microRNA-122: Biogenesis and function. RNA Biol. 2012, 9, 137–142. [Google Scholar] [CrossRef]
- Lagos-Quintana, M.; Rauhut, R.; Yalcin, A.; Meyer, J.; Lendeckel, W.; Tuschl, T. Identification of tissue-specific microRNAs from mouse. Curr. Biol. 2002, 12, 735–739. [Google Scholar] [CrossRef]
- Esau, C.; Davis, S.; Murray, S.F.; Yu, X.X.; Pandey, S.K.; Pear, M.; Watts, L.; Booten, S.L.; Graham, M.; McKay, R.; et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006, 3, 87–98. [Google Scholar] [CrossRef]
- Cheung, O.; Puri, P.; Eicken, C.; Contos, M.J.; Mirshahi, F.; Maher, J.W.; Kellum, J.M.; Min, H.; Luketic, V.A.; Sanyal, A.J. Nonalcoholic steatohepatitis is associated with altered hepatic MicroRNA expression. Hepatology 2008, 48, 1810–1820. [Google Scholar] [CrossRef]
- Pirola, C.J.; Fernández Gianotti, T.; Castaño, G.O.; Mallardi, P.; San Martino, J.; Mora Gonzalez Lopez Ledesma, M.; Flichman, D.; Mirshahi, F.; Sanyal, A.J.; Sookoian, S. Circulating microRNA signature in non-alcoholic fatty liver disease: From serum non-coding RNAs to liver histology and disease pathogenesis. Gut 2015, 64, 800–812. [Google Scholar] [CrossRef] [PubMed]
- Inomata, Y.; Oh, J.W.; Taniguchi, K.; Sugito, N.; Kawaguchi, N.; Hirokawa, F.; Lee, S.W.; Akao, Y.; Takai, S.; Kim, K.P.; et al. Downregulation of miR-122-5p Activates Glycolysis via PKM2 in Kupffer Cells of Rat and Mouse Models of Non-Alcoholic Steatohepatitis. Int. J. Mol. Sci. 2022, 23, 5230. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Basu, S.; Bandyopadhyay, D.; Mukherjee, K.; Datta, D.; Chakraborty, S.; Jana, S.; Adak, M.; Bose, S.; Chakrabarti, S.; et al. Inhibition of extracellular vesicle-associated MMP2 abrogates intercellular hepatic miR-122 transfer to liver macrophages and curtails inflammation. iScience 2021, 24, 103428. [Google Scholar] [CrossRef]
- Lanz, R.B.; McKenna, N.J.; Onate, S.A.; Albrecht, U.; Wong, J.; Tsai, S.Y.; Tsai, M.J.; O'Malley, B.W. A steroid receptor coactivator, SRA, functions as an RNA and is present in an SRC-1 complex. Cell 1999, 97, 17–27. [Google Scholar] [CrossRef]
- Chen, G.; Yu, D.; Nian, X.; Liu, J.; Koenig, R.J.; Xu, B.; Sheng, L. LncRNA SRA promotes hepatic steatosis through repressing the expression of adipose triglyceride lipase (ATGL). Sci. Rep. 2016, 6, 35531. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.Y.; Xiong, X.; Liu, T.; Mi, L.; Peng, X.; Rui, C.; Guo, L.; Li, S.; Li, X.; Lin, J.D. Long noncoding RNA licensing of obesity-linked hepatic lipogenesis and NAFLD pathogenesis. Nat. Commun. 2018, 9, 2986. [Google Scholar] [CrossRef]
- Nakamuta, M.; Fujino, T.; Yada, R.; Yada, M.; Yasutake, K.; Yoshimoto, T.; Harada, N.; Higuchi, N.; Kato, M.; Kohjima, M.; et al. Impact of cholesterol metabolism and the LXRalpha-SREBP-1c pathway on nonalcoholic fatty liver disease. Int. J. Mol. Med. 2009, 23, 603–608. [Google Scholar] [CrossRef]
- Leti, F.; Legendre, C.; Still, C.D.; Chu, X.; Petrick, A.; Gerhard, G.S.; DiStefano, J.K. Altered expression of MALAT1 lncRNA in nonalcoholic steatohepatitis fibrosis regulates CXCL5 in hepatic stellate cells. Transl. Res. 2017, 190, 25–39. [Google Scholar] [CrossRef]
- Zhang, K.; Shi, Z.; Zhang, M.; Dong, X.; Zheng, L.; Li, G.; Han, X.; Yao, Z.; Han, T.; Hong, W. Silencing lncRNA Lfar1 alleviates the classical activation and pyoptosis of macrophage in hepatic fibrosis. Cell Death Dis. 2020, 11, 132. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, R.; Wang, Y.; Zhu, W.; Zhao, D.; Wang, X.; Yang, H.; Gurley, E.C.; Chen, W.; Hylemon, P.B.; et al. Cholangiocyte-Derived Exosomal lncRNA H19 Promotes Macrophage Activation and Hepatic Inflammation under Cholestatic Conditions. Cells 2020, 9, 190. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Li, L.; Xu, Y.; Du, J.; Ling, C. Roles of hepatic stellate cells in NAFLD: From the perspective of inflammation and fibrosis. Front. Pharmacol. 2022, 13, 958428. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Liu, C.H.; Wu, D.; Jiang, W.; Zhang, N.; Tang, H. LncRNA and circRNA in Patients with Non-Alcoholic Fatty Liver Disease: A Systematic Review. Biomolecules 2023, 13, 560. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Liang, Y.; Song, R.; Yang, G.; Han, J.; Lan, Y.; Pan, S.; Zhu, M.; Liu, Y.; Wang, Y.; et al. Long non-coding RNA NEAT1-modulated abnormal lipolysis via ATGL drives hepatocellular carcinoma proliferation. Mol. Cancer 2018, 17, 90. [Google Scholar] [CrossRef] [PubMed]
- Wang, X. Down-regulation of lncRNA-NEAT1 alleviated the non-alcoholic fatty liver disease via mTOR/S6K1 signaling pathway. J. Cell Biochem. 2018, 119, 1567–1574. [Google Scholar] [CrossRef]
- Yuan, X.; Diao, J.; Du, A.; Wen, S.; Zhou, L.; Pan, Y. Circular RNA expression profiles and features in NAFLD mice: A study using RNA-seq data. J. Transl. Med. 2020, 18, 476. [Google Scholar] [CrossRef]
- Zhao, Q.; Liu, J.; Deng, H.; Ma, R.; Liao, J.Y.; Liang, H.; Hu, J.; Li, J.; Guo, Z.; Cai, J.; et al. Targeting Mitochondria-Located circRNA SCAR Alleviates NASH via Reducing mROS Output. Cell 2020, 183, 76–93. [Google Scholar] [CrossRef]
- Guo, X.Y.; Chen, J.N.; Sun, F.; Wang, Y.Q.; Pan, Q.; Fan, J.G. circRNA_0046367 Prevents Hepatoxicity of Lipid Peroxidation: An Inhibitory Role against Hepatic Steatosis. Oxid. Med. Cell. Longev. 2017, 2017, 3960197. [Google Scholar] [CrossRef]
- Li, J.; Qi, J.; Tang, Y.; Liu, H.; Zhou, K.; Dai, Z.; Yuan, L.; Sun, C. A nanodrug system overexpressed circRNA_0001805 alleviates nonalcoholic fatty liver disease via miR-106a-5p/miR-320a and ABCA1/CPT1 axis. J. Nanobiotechnology 2021, 19, 363. [Google Scholar] [CrossRef]
- Liu, C.H.; Jiang, W.; Zeng, Q.; Wu, D.; Li, H.; Zhou, L.; Bai, L.; Tang, H. CircRNA-PI4KB Induces Hepatic Lipid Deposition in Non-Alcoholic Fatty Liver Disease by Transporting miRNA-122 to Extra-Hepatocytes. Int. J. Mol. Sci. 2023, 24, 1297. [Google Scholar] [CrossRef]
- Ou, Q.; Zhao, Y.; Zhou, J.; Wu, X. Comprehensive circular RNA expression profiles in a mouse model of nonalcoholic steatohepatitis. Mol. Med. Rep. 2019, 19, 2636–2648. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yuan, B.; Wu, Z.; Dong, Y.; Zhang, L.; Zeng, Z. Microarray profiling of circular RNAs and the potential regulatory role of hsa_circ_0071410 in the activated human hepatic stellate cell induced by irradiation. Gene 2017, 629, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Chien, Y.; Tsai, P.H.; Lai, Y.H.; Lu, K.H.; Liu, C.Y.; Lin, H.F.; Huang, C.S.; Wu, W.W.; Wang, C.Y. CircularRNA as novel biomarkers in liver diseases. J. Chin. Med. Assoc. 2020, 83, 15–17. [Google Scholar] [CrossRef]
- Wang, P.; Huang, Z.; Peng, Y.; Li, H.; Lin, T.; Zhao, Y.; Hu, Z.; Zhou, Z.; Zhou, W.; Liu, Y.; et al. Circular RNA circBNC2 inhibits epithelial cell G2-M arrest to prevent fibrotic maladaptive repair. Nat. Commun. 2022, 13, 6502. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Wei, J.; Zeng, Y.; Liu, J.; Xiao, E.; Kang, Y.; Kang, Y. Mesenchymal stem cell-originated exosomal circDIDO1 suppresses hepatic stellate cell activation by miR-141-3p/PTEN/AKT pathway in human liver fibrosis. Drug Deliv. 2022, 29, 440–453. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Li, Y.; Chen, M.; Wang, W.; Zhao, Q.; He, B.; Zhang, M.; Jiang, Y. hMSCs-derived exosome circCDK13 inhibits liver fibrosis by regulating the expression of MFGE8 through miR-17-5p/KAT2B. Cell Biol. Toxicol. 2023, 39, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, Y.W.; Siomi, M.C.; Siomi, H. PIWI-Interacting RNA: Its Biogenesis and Functions. Annu. Rev. Biochem. 2015, 84, 405–433. [Google Scholar] [CrossRef] [PubMed]
- Siomi, M.C.; Sato, K.; Pezic, D.; Aravin, A.A. PIWI-interacting small RNAs: The vanguard of genome defence. Nat. Rev. Mol. Cell Biol. 2011, 12, 246–258. [Google Scholar] [CrossRef]
- Ozata, D.M.; Gainetdinov, I.; Zoch, A.; O'Carroll, D.; Zamore, P.D. PIWI-interacting RNAs: Small RNAs with big functions. Nat. Rev. Genet. 2019, 20, 89–108. [Google Scholar] [CrossRef]
- Ma, X.; Huang, Y.; Ding, Y.; Shi, L.; Zhong, X.; Kang, M.; Li, C. Analysis of piRNA expression spectra in a non-alcoholic fatty liver disease mouse model induced by a methionine- and choline-deficient diet. Exp. Ther. Med. 2020, 19, 3829–3839. [Google Scholar] [CrossRef]
- Tang, X.; Xie, X.; Wang, X.; Wang, Y.; Jiang, X.; Jiang, H. The Combination of piR-823 and Eukaryotic Initiation Factor 3 B (EIF3B) Activates Hepatic Stellate Cells via Upregulating TGF-β1 in Liver Fibrogenesis. Med. Sci. Monit. 2018, 24, 9151–9165. [Google Scholar] [CrossRef] [PubMed]
- Pantazi, P.; Clements, T.; Venø, M.; Abrahams, V.M.; Holder, B. Distinct non-coding RNA cargo of extracellular vesicles from M1 and M2 human primary macrophages. J. Extracell. Vesicles 2022, 11, e12293. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Zhou, X.; Wang, Y.; Dai, L.; Yuan, J.; Peng, J.; Zhang, X.; Wang, C. Changes in the Small Noncoding RNAome During M1 and M2 Macrophage Polarization. Front. Immunol. 2022, 13, 799733. [Google Scholar] [CrossRef] [PubMed]
- Tosar, J.P.; Rovira, C.; Cayota, A. Non-coding RNA fragments account for the majority of annotated piRNAs expressed in somatic non-gonadal tissues. Commun. Biol. 2018, 1, 2. [Google Scholar] [CrossRef]
- Jeon, T.I.; Esquejo, R.M.; Roqueta-Rivera, M.; Phelan, P.E.; Moon, Y.A.; Govindarajan, S.S.; Esau, C.C.; Osborne, T.F. An SREBP-responsive microRNA operon contributes to a regulatory loop for intracellular lipid homeostasis. Cell Metab. 2013, 18, 51–61. [Google Scholar] [CrossRef]
- Liu, D.D.; Han, C.C.; Wan, H.F.; He, F.; Xu, H.Y.; Wei, S.H.; Du, X.H.; Xu, F. Effects of inhibiting PI3K-Akt-mTOR pathway on lipid metabolism homeostasis in goose primary hepatocytes. Animal 2016, 10, 1319–1327. [Google Scholar] [CrossRef]
- Wu, H.; Zhang, T.; Pan, F.; Steer, C.J.; Li, Z.; Chen, X.; Song, G. MicroRNA-206 prevents hepatosteatosis and hyperglycemia by facilitating insulin signaling and impairing lipogenesis. J. Hepatol. 2017, 66, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Li, P.; Yang, W.; Ruan, X.; Kiesewetter, K.; Zhu, J.; Cao, H. Integrative Transcriptome Analyses of Metabolic Responses in Mice Define Pivotal LncRNA Metabolic Regulators. Cell Metab. 2016, 24, 627–639. [Google Scholar] [CrossRef]
- Li, D.; Guo, L.; Deng, B.; Li, M.; Yang, T.; Yang, F.; Yang, Z. Long non-coding RNA HR1 participates in the expression of SREBP-1c through phosphorylation of the PDK1/AKT/FoxO1 pathway. Mol. Med. Rep. 2018, 18, 2850–2856. [Google Scholar] [CrossRef]
- Dattaroy, D.; Pourhoseini, S.; Das, S.; Alhasson, F.; Seth, R.K.; Nagarkatti, M.; Michelotti, G.A.; Diehl, A.M.; Chatterjee, S. Micro-RNA 21 inhibition of SMAD7 enhances fibrogenesis via leptin-mediated NADPH oxidase in experimental and human nonalcoholic steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G298–G312. [Google Scholar] [CrossRef]
- Sun, D.; Zhang, J.; Xie, J.; Wei, W.; Chen, M.; Zhao, X. MiR-26 controls LXR-dependent cholesterol efflux by targeting ABCA1 and ARL7. FEBS Lett. 2012, 586, 1472–1479. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.Y.; Sun, F.; Chen, J.N.; Wang, Y.Q.; Pan, Q.; Fan, J.G. circRNA_0046366 inhibits hepatocellular steatosis by normalization of PPAR signaling. World J. Gastroenterol. 2018, 24, 323–337. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, S.; Yang, M.; Zhao, Y.; Chen, X.; Zhang, F.; Li, N.; Yao, P.; Zhu, T.; Mei, H.; Wang, S.; et al. MicroRNA-451 Negatively Regulates Hepatic Glucose Production and Glucose Homeostasis by Targeting Glycerol Kinase-Mediated Gluconeogenesis. Diabetes 2016, 65, 3276–3288. [Google Scholar] [CrossRef]
- Trajkovski, M.; Hausser, J.; Soutschek, J.; Bhat, B.; Akin, A.; Zavolan, M.; Heim, M.H.; Stoffel, M. MicroRNAs 103 and 107 regulate insulin sensitivity. Nature 2011, 474, 649–653. [Google Scholar] [CrossRef]
- Huang, J.; Chen, S.; Cai, D.; Bian, D.; Wang, F. Long noncoding RNA lncARSR promotes hepatic cholesterol biosynthesis via modulating Akt/SREBP-2/HMGCR pathway. Life Sci. 2018, 203, 48–53. [Google Scholar] [CrossRef]
- Shen, M.; Pan, H.; Ke, J.; Zhao, F. NF-κB-upregulated miR-155-5p promotes hepatocyte mitochondrial dysfunction to accelerate the development of nonalcoholic fatty liver disease through downregulation of STC1. J. Biochem. Mol. Toxicol. 2022, 36, e23025. [Google Scholar] [CrossRef]
- Tili, E.; Michaille, J.J.; Cimino, A.; Costinean, S.; Dumitru, C.D.; Adair, B.; Fabbri, M.; Alder, H.; Liu, C.G.; Calin, G.A.; et al. Modulation of miR-155 and miR-125b levels following lipopolysaccharide/TNF-alpha stimulation and their possible roles in regulating the response to endotoxin shock. J. Immunol. 2007, 179, 5082–5089. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Wang, S.; Gao, L.; Zhou, Z.; Yang, Z.; Lin, J.; Ren, S.; Xing, H.; Wu, B. Oscillating lncRNA Platr4 regulates NLRP3 inflammasome to ameliorate nonalcoholic steatohepatitis in mice. Theranostics 2021, 11, 426–444. [Google Scholar] [CrossRef]
- Shi, N.; Sun, K.; Tang, H.; Mao, J. The impact and role of identified long noncoding RNAs in nonalcoholic fatty liver disease: A narrative review. J. Clin. Lab. Anal. 2023, 37, e24943. [Google Scholar] [CrossRef]
- Zhang, Q.; Yu, K.; Cao, Y.; Luo, Y.; Liu, Y.; Zhao, C. miR-125b promotes the NF-κB-mediated inflammatory response in NAFLD via directly targeting TNFAIP3. Life Sci. 2021, 270, 119071. [Google Scholar] [CrossRef]
- Jiang, X.; Ning, Q. The mechanism of lncRNA H19 in fibrosis and its potential as novel therapeutic target. Mech. Ageing Dev. 2020, 188, 111243. [Google Scholar] [CrossRef]
- Fu, X.; Dong, B.; Tian, Y.; Lefebvre, P.; Meng, Z.; Wang, X.; Pattou, F.; Han, W.; Wang, X.; Lou, F.; et al. MicroRNA-26a regulates insulin sensitivity and metabolism of glucose and lipids. J. Clin. Investig. 2015, 125, 2497–2509. [Google Scholar] [CrossRef]
- Xihua, L.; Shengjie, T.; Weiwei, G.; Matro, E.; Tingting, T.; Lin, L.; Fang, W.; Jiaqiang, Z.; Fenping, Z.; Hong, L. Circulating miR-143-3p inhibition protects against insulin resistance in Metabolic Syndrome via targeting of the insulin-like growth factor 2 receptor. Transl. Res. 2019, 205, 33–43. [Google Scholar] [CrossRef]
- Jordan, S.D.; Krüger, M.; Willmes, D.M.; Redemann, N.; Wunderlich, F.T.; Brönneke, H.S.; Merkwirth, C.; Kashkar, H.; Olkkonen, V.M.; Böttger, T.; et al. Obesity-induced overexpression of miRNA-143 inhibits insulin-stimulated AKT activation and impairs glucose metabolism. Nat. Cell Biol. 2011, 13, 434–446. [Google Scholar] [CrossRef]
- Lin, X.; Du, Y.; Lu, W.; Gui, W.; Sun, S.; Zhu, Y.; Wang, G.; Eserberg, D.T.; Zheng, F.; Zhou, J.; et al. CircRNF111 Protects Against Insulin Resistance and Lipid Deposition via Regulating miR-143-3p/IGF2R Axis in Metabolic Syndrome. Front. Cell Dev. Biol. 2021, 9, 663148. [Google Scholar] [CrossRef] [PubMed]
- Cockcroft, S. Mammalian lipids: Structure, synthesis and function. Essays Biochem. 2021, 65, 813–845. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.W.; Ha, J.; Yoon, K.S.; Kang, I.; Choi, T.G.; Kim, S.S. Innate Immune System in the Pathogenesis of Non-Alcoholic Fatty Liver Disease. Nutrients 2023, 15, 2068. [Google Scholar] [CrossRef] [PubMed]
- Arrese, M.; Cabrera, D.; Kalergis, A.M.; Feldstein, A.E. Innate Immunity and Inflammation in NAFLD/NASH. Dig. Dis. Sci. 2016, 61, 1294–1303. [Google Scholar] [CrossRef]
- Saponaro, C.; Gaggini, M.; Carli, F.; Gastaldelli, A. The Subtle Balance between Lipolysis and Lipogenesis: A Critical Point in Metabolic Homeostasis. Nutrients 2015, 7, 9453–9474. [Google Scholar] [CrossRef] [PubMed]
- Bertolio, R.; Napoletano, F.; Mano, M.; Maurer-Stroh, S.; Fantuz, M.; Zannini, A.; Bicciato, S.; Sorrentino, G.; Del Sal, G. Sterol regulatory element binding protein 1 couples mechanical cues and lipid metabolism. Nat. Commun. 2019, 10, 1326. [Google Scholar] [CrossRef]
- Eberlé, D.; Hegarty, B.; Bossard, P.; Ferré, P.; Foufelle, F. SREBP transcription factors: Master regulators of lipid homeostasis. Biochimie 2004, 86, 839–848. [Google Scholar] [CrossRef]
- Liu, X.; Zhao, J.; Liu, Q.; Xiong, X.; Zhang, Z.; Jiao, Y.; Li, X.; Liu, B.; Li, Y.; Lu, Y. MicroRNA-124 promotes hepatic triglyceride accumulation through targeting tribbles homolog 3. Sci. Rep. 2016, 6, 37170. [Google Scholar] [CrossRef] [PubMed]
- Wójcicka, G.; Jamroz-Wiśniewska, A.; Horoszewicz, K.; Bełtowski, J. Liver X receptors (LXRs). Part I: Structure, function, regulation of activity, and role in lipid metabolism. Adv. Hyg. Exp. Med. 2007, 61, 736–759. [Google Scholar]
- Todisco, S.; Santarsiero, A.; Convertini, P.; De Stefano, G.; Gilio, M.; Iacobazzi, V.; Infantino, V. PPAR Alpha as a Metabolic Modulator of the Liver: Role in the Pathogenesis of Nonalcoholic Steatohepatitis (NASH). Biology 2022, 11, 792. [Google Scholar] [CrossRef]
- Prasun, P.; Ginevic, I.; Oishi, K. Mitochondrial dysfunction in nonalcoholic fatty liver disease and alcohol related liver disease. Transl. Gastroenterol. Hepatol. 2021, 6, 4. [Google Scholar] [CrossRef]
- Descorbeth, M.; Figueroa, K.; Serrano-Illán, M.; De León, M. Protective effect of docosahexaenoic acid on lipotoxicity-mediated cell death in Schwann cells: Implication of PI3K/AKT and mTORC2 pathways. Brain Behav. 2018, 8, e01123. [Google Scholar] [CrossRef]
- Römer, A.; Linn, T.; Petry, S.F. Lipotoxic Impairment of Mitochondrial Function in β-Cells: A Review. Antioxidants 2021, 10, 293. [Google Scholar] [CrossRef]
- Hoxhaj, G.; Manning, B.D. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef]
- Knight, B.L.; Hebbachi, A.; Hauton, D.; Brown, A.M.; Wiggins, D.; Patel, D.D.; Gibbons, G.F. A role for PPARalpha in the control of SREBP activity and lipid synthesis in the liver. Biochem. J. 2005, 389, 413–421. [Google Scholar] [CrossRef]
- Dávalos, A.; Goedeke, L.; Smibert, P.; Ramírez, C.M.; Warrier, N.P.; Andreo, U.; Cirera-Salinas, D.; Rayner, K.; Suresh, U.; Pastor-Pareja, J.C.; et al. miR-33a/b contribute to the regulation of fatty acid metabolism and insulin signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 9232–9237. [Google Scholar] [CrossRef]
- Rayner, K.J.; Suárez, Y.; Dávalos, A.; Parathath, S.; Fitzgerald, M.L.; Tamehiro, N.; Fisher, E.A.; Moore, K.J.; Fernández-Hernando, C. MiR-33 contributes to the regulation of cholesterol homeostasis. Science 2010, 328, 1570–1573. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, H.; Umar, M.I.; Imran, S.; Javaid, F.; Syed, S.K.; Riaz, R.; Hassan, W. TGF-β1 signaling can worsen NAFLD with liver fibrosis backdrop. Exp. Mol. Pathol. 2022, 124, 104733. [Google Scholar] [CrossRef] [PubMed]
- Tomita, K.; Teratani, T.; Suzuki, T.; Shimizu, M.; Sato, H.; Narimatsu, K.; Okada, Y.; Kurihara, C.; Irie, R.; Yokoyama, H.; et al. Free cholesterol accumulation in hepatic stellate cells: Mechanism of liver fibrosis aggravation in nonalcoholic steatohepatitis in mice. Hepatology 2014, 59, 154–169. [Google Scholar] [CrossRef] [PubMed]
- Negishi, M.; Wongpalee, S.P.; Sarkar, S.; Park, J.; Lee, K.Y.; Shibata, Y.; Reon, B.J.; Abounader, R.; Suzuki, Y.; Sugano, S.; et al. A new lncRNA, APTR, associates with and represses the CDKN1A/p21 promoter by recruiting polycomb proteins. PLoS ONE 2014, 9, e95216. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Zheng, J.; Mao, Y.; Dong, P.; Li, G.; Lu, Z.; Guo, C.; Liu, Z.; Fan, X. Long non-coding RNA APTR promotes the activation of hepatic stellate cells and the progression of liver fibrosis. Biochem. Biophys. Res. Commun. 2015, 463, 679–685. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Han, X.; Zhang, Z.; Zheng, L.; Hu, Z.; Yao, Q.; Cui, H.; Shu, G.; Si, M.; Li, C.; et al. The liver-enriched lnc-LFAR1 promotes liver fibrosis by activating TGFβ and Notch pathways. Nat. Commun. 2017, 8, 144. [Google Scholar] [CrossRef]
- Yu, F.; Jiang, Z.; Chen, B.; Dong, P.; Zheng, J. NEAT1 accelerates the progression of liver fibrosis via regulation of microRNA-122 and Kruppel-like factor 6. J. Mol. Med. 2017, 95, 1191–1202. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Sánchez, N.; Díaz-Orozco, L.E. Editorial: International Consensus Recommendations to Replace the Terminology of Non-Alcoholic Fatty Liver Disease (NAFLD) with Metabolic-Associated Fatty Liver Disease (MAFLD). Med. Sci. Monit. 2021, 27, e933860. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, P.; Hellerbrand, C. Non-alcoholic fatty liver disease, obesity and the metabolic syndrome. Best. Pract. Res. Clin. Gastroenterol. 2014, 28, 637–653. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef]
- Godoy-Matos, A.F.; Silva Júnior, W.S.; Valerio, C.M. NAFLD as a continuum: From obesity to metabolic syndrome and diabetes. Diabetol. Metab. Syndr. 2020, 12, 60. [Google Scholar] [CrossRef]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef]
- Czech, M.P. Insulin action and resistance in obesity and type 2 diabetes. Nat. Med. 2017, 23, 804–814. [Google Scholar] [CrossRef]
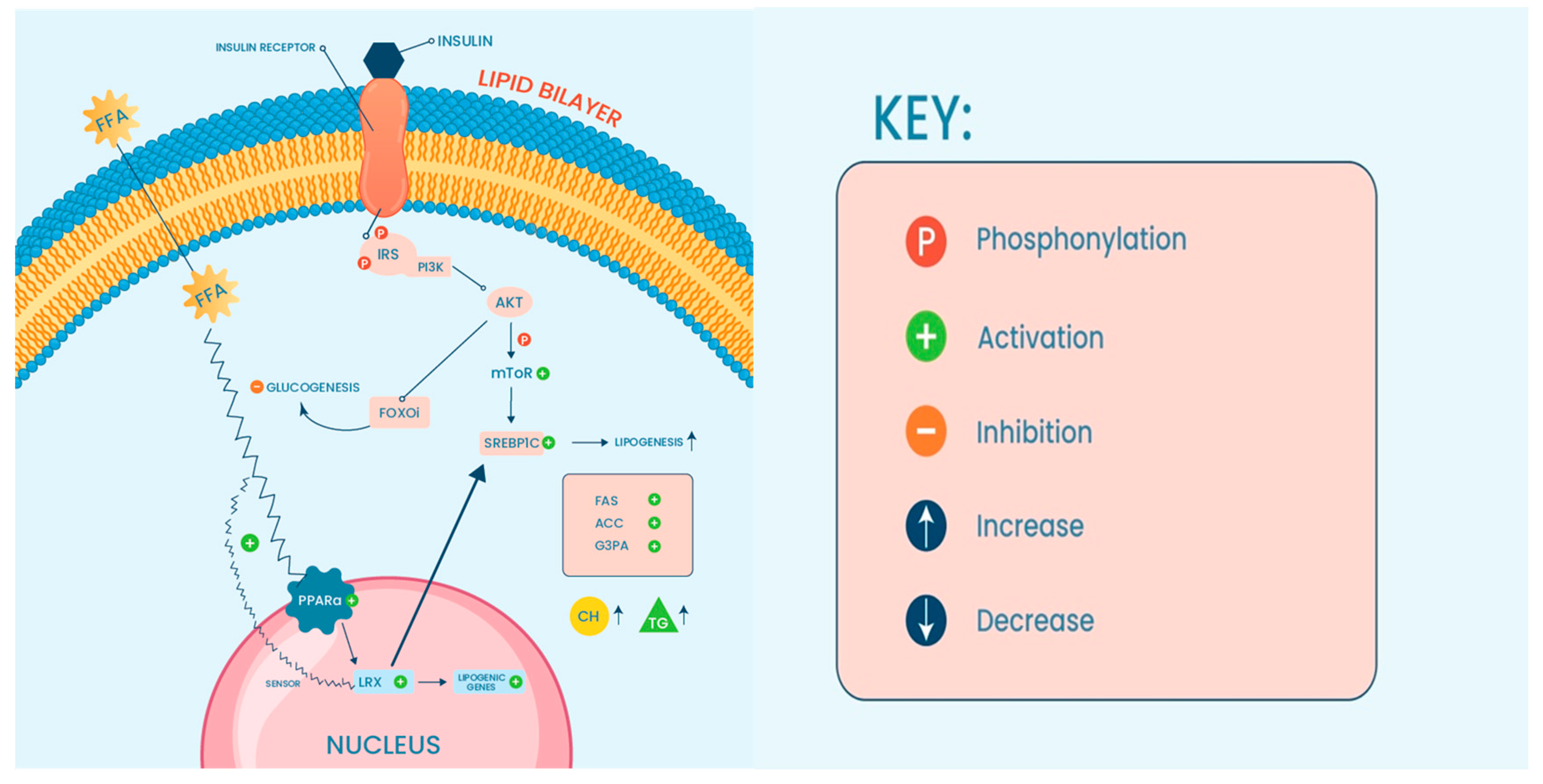




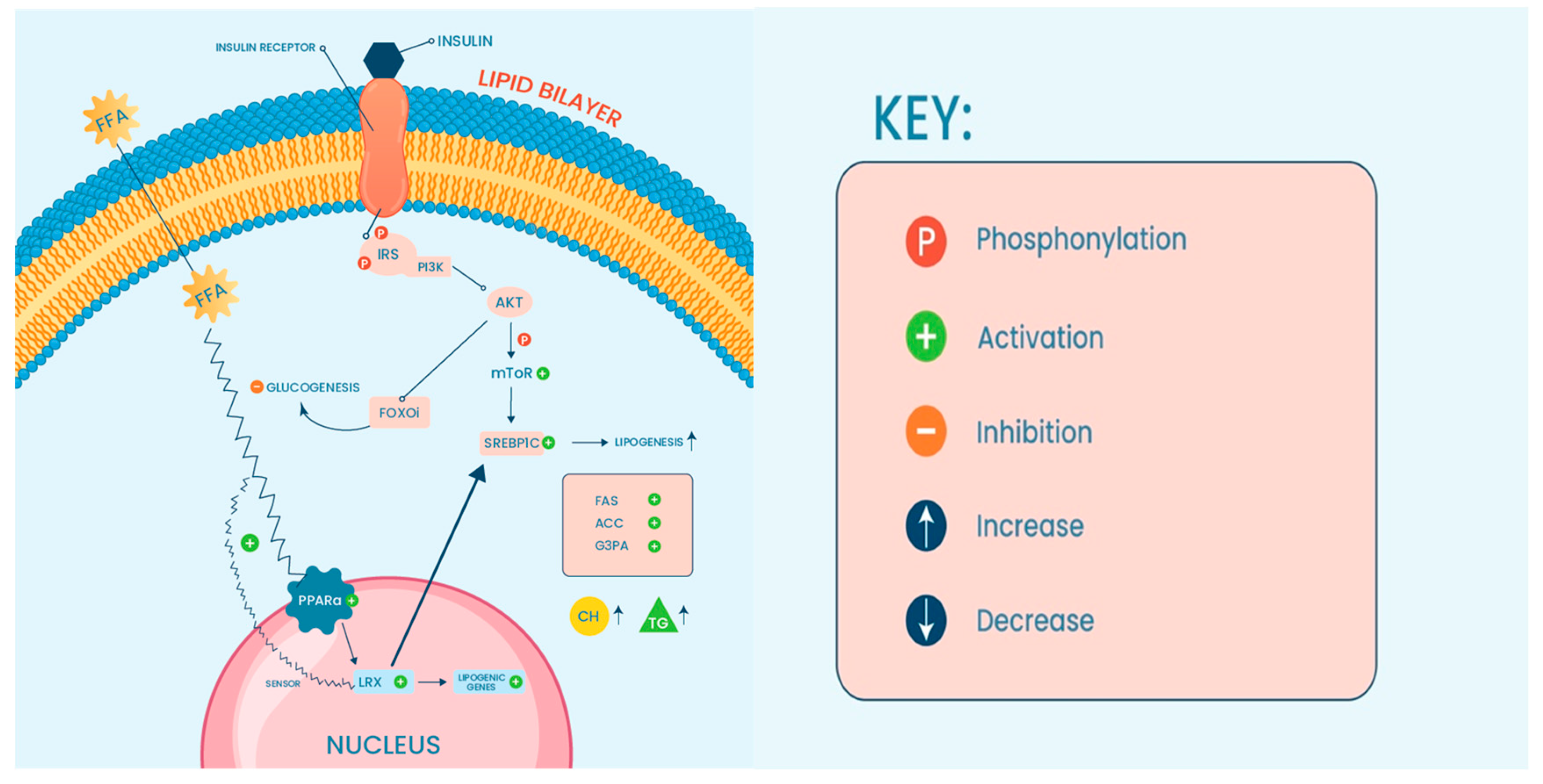{kind=link}
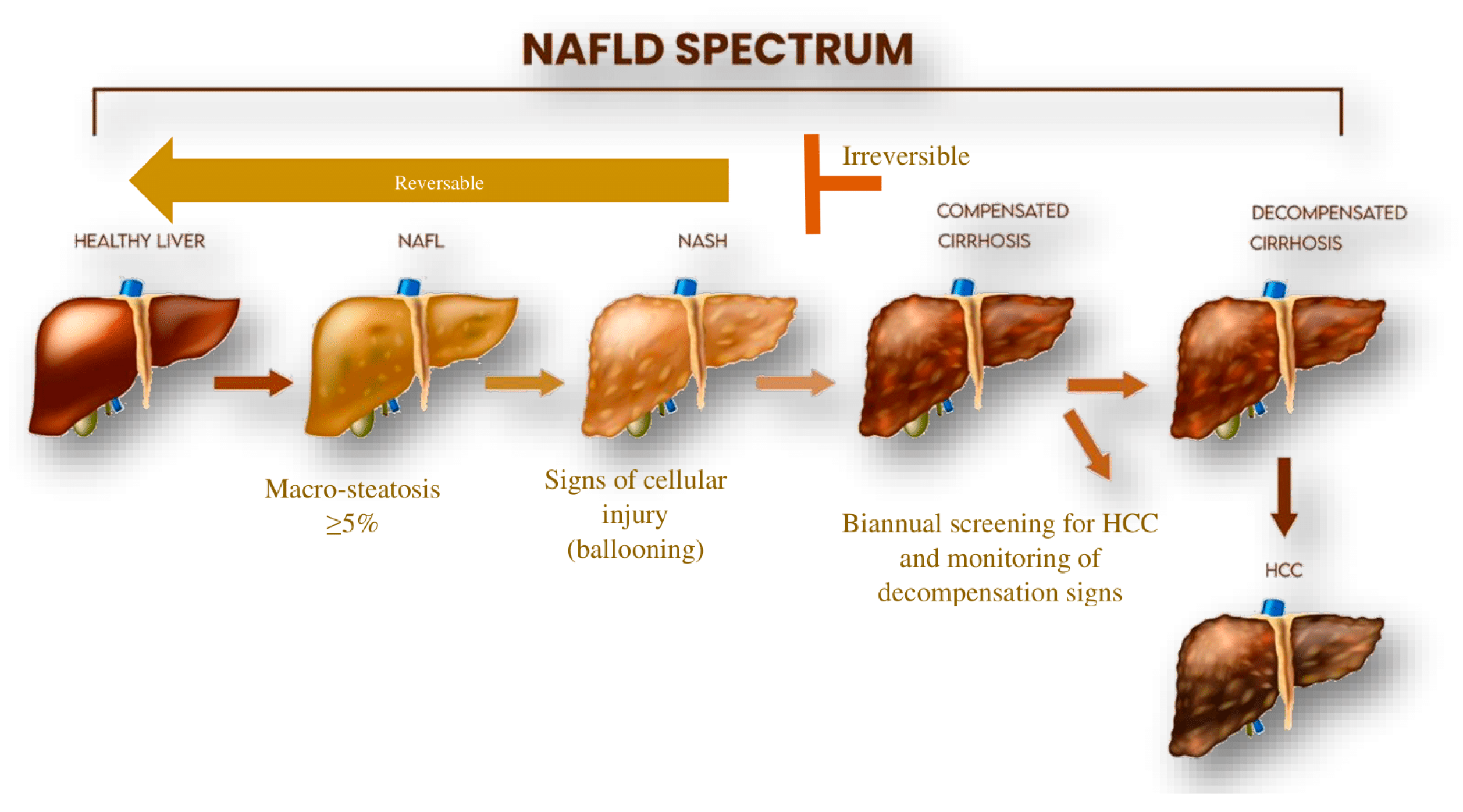{kind=link}
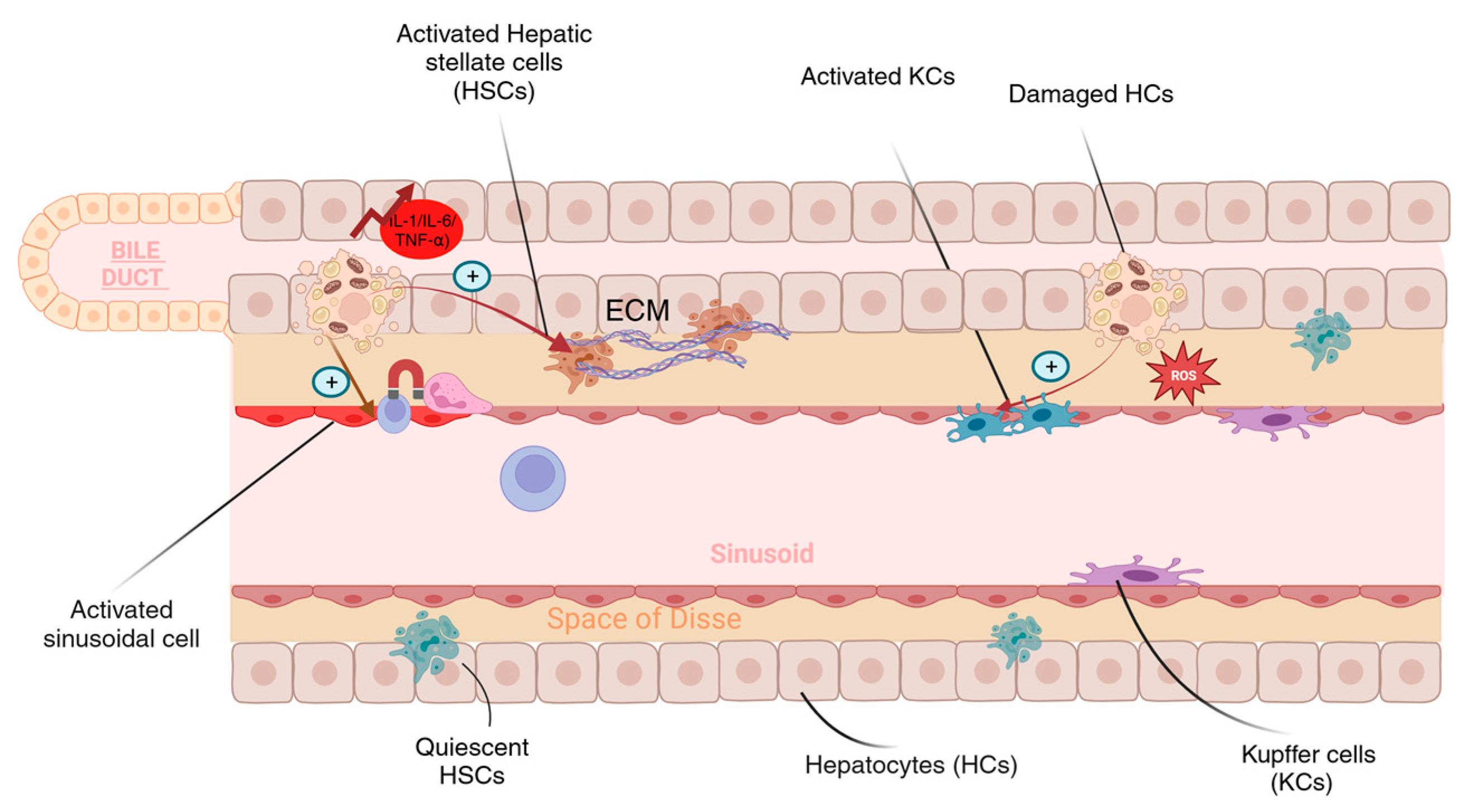{kind=link}
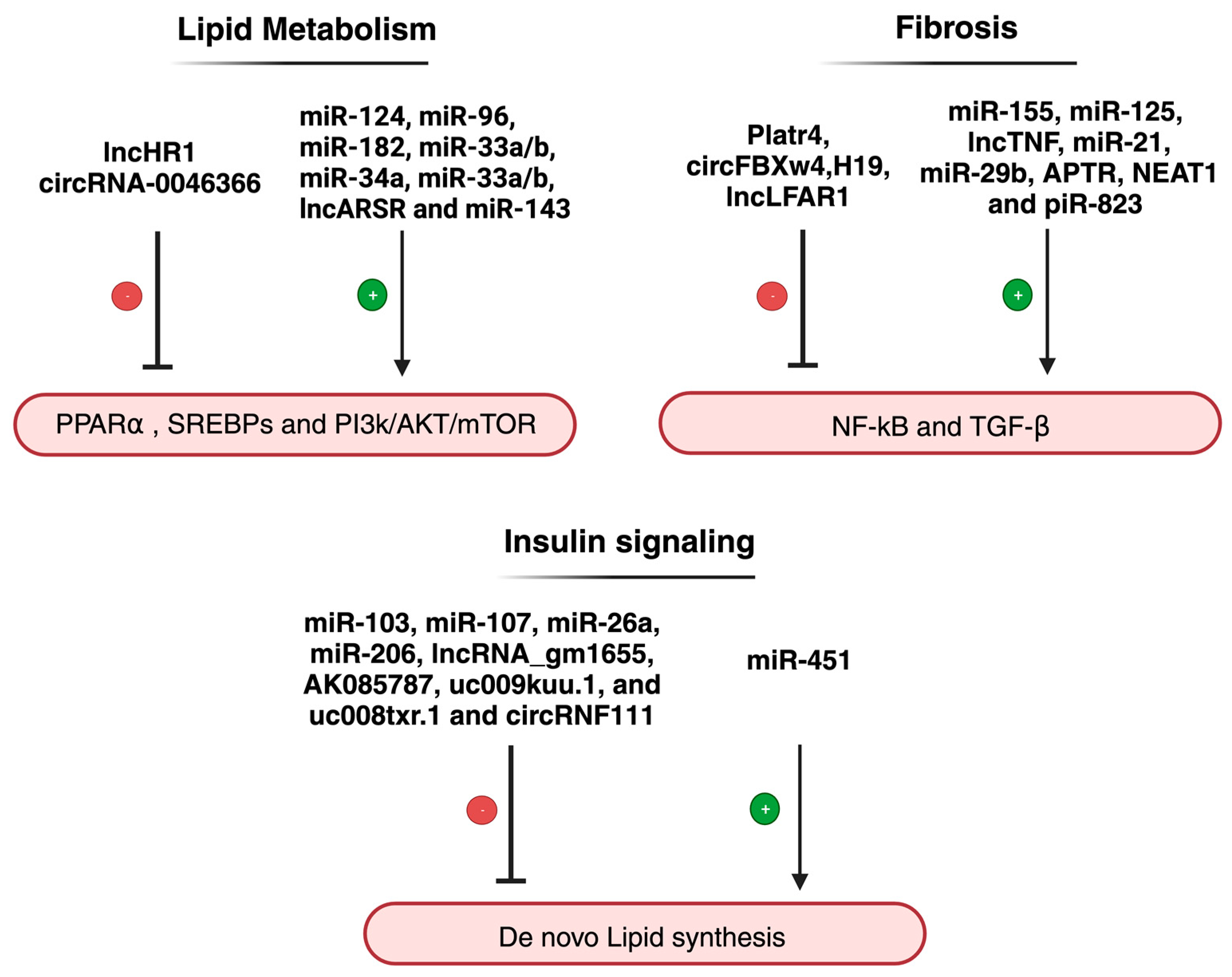{kind=link}
| Non-Coding RNA | Expression | Primary Function | Reference |
|---|---|---|---|
| miR-96-124-182-183 | Upregulated | These ncRNAs modulate lipid synthesis through SREBP-1c | [89,90] |
| miR-206 | Downregulated | [91] | |
| Gm16551 | Downregulated | [92] | |
| lncHR1 | Downregulated | [93] | |
| miR-122-3075-21-192-5p | Upregulated | These ncRNAs are involved in the stimulation or inhibition of HCs and HC-related injuries | [46,49,54,56,57,58,74,77,94] |
| piR-823 | Upregulated | [85] | |
| circBNC2 | Upregulated | [78] | |
| circ_0071410 | [77] | ||
| circPI4KB | [74] | ||
| circRNA_0046367 | Downregulated | [72] | |
| miR-26 | Downregulated | miR-26 expression depends on LXRs and has a protective role | [95] |
| miR-34a | Upregulated | Both ncRNAs were found to be associated with the regulation of hepatic fat content through PPARα | [96] |
| circRNA_0046366 | Downregulated | [96] | |
| miR-451 | Downregulated | Involved regulation of glucose homeostasis | [97] |
| miR-103/miR-107 | Upregulated | [98] | |
| LncARSR | Upregulated | The expression of LncARSR correlates with the activated state of PI3K/AKT/mTOR pathway | [99] |
| miR-155 | Upregulated | These ncRNASs have an influence on the induction of inflammation via NF-κB pathway | [100,101] |
| Platr4 | Upregulated | [102] | |
| LncTNF | Upregulated | [103] | |
| miR-125 | Upregulated | The expression of these ncRNAs contributes to the activation of pro-inflammatory mediators | [104] |
| circFBXW4 | Downregulated | [14] | |
| miR-29b | Downregulated | These ncRNAs are involved in fibrosis mediation through TGF-β1 | [94] |
| H19 | Downregulated | [65,105] | |
| piR-823 | Upregulated | [85] | |
| miR-26a-143-145 | Upregulated | These ncRNAs have been found to be modulators in insulin sensitivity | [106,107,108] |
| AK085787-uc009kuu.1-uc008txr.1 | Upregulated | [92] | |
| CircRNF111-circRNA_0001805 | Downregulated | [73,109] | |
| CircRNA SCAR | Upregulated | This ncRNA may play a role in activating liver fibroblasts | [71] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zailaie, S.A.; Khoja, B.B.; Siddiqui, J.J.; Mawardi, M.H.; Heaphy, E.; Aljagthmi, A.; Sergi, C.M. Investigating the Role of Non-Coding RNA in Non-Alcoholic Fatty Liver Disease. Non-Coding RNA 2024, 10, 10. https://doi.org/10.3390/ncrna10010010
Zailaie SA, Khoja BB, Siddiqui JJ, Mawardi MH, Heaphy E, Aljagthmi A, Sergi CM. Investigating the Role of Non-Coding RNA in Non-Alcoholic Fatty Liver Disease. Non-Coding RNA. 2024; 10(1):10. https://doi.org/10.3390/ncrna10010010
Chicago/Turabian StyleZailaie, Samar A., Basmah B. Khoja, Jumana J. Siddiqui, Mohammad H. Mawardi, Emily Heaphy, Amjad Aljagthmi, and Consolato M. Sergi. 2024. "Investigating the Role of Non-Coding RNA in Non-Alcoholic Fatty Liver Disease" Non-Coding RNA 10, no. 1: 10. https://doi.org/10.3390/ncrna10010010
APA StyleZailaie, S. A., Khoja, B. B., Siddiqui, J. J., Mawardi, M. H., Heaphy, E., Aljagthmi, A., & Sergi, C. M. (2024). Investigating the Role of Non-Coding RNA in Non-Alcoholic Fatty Liver Disease. Non-Coding RNA, 10(1), 10. https://doi.org/10.3390/ncrna10010010