
Stabilization of Urinary MicroRNAs by Association with Exosomes and Argonaute 2 Protein

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
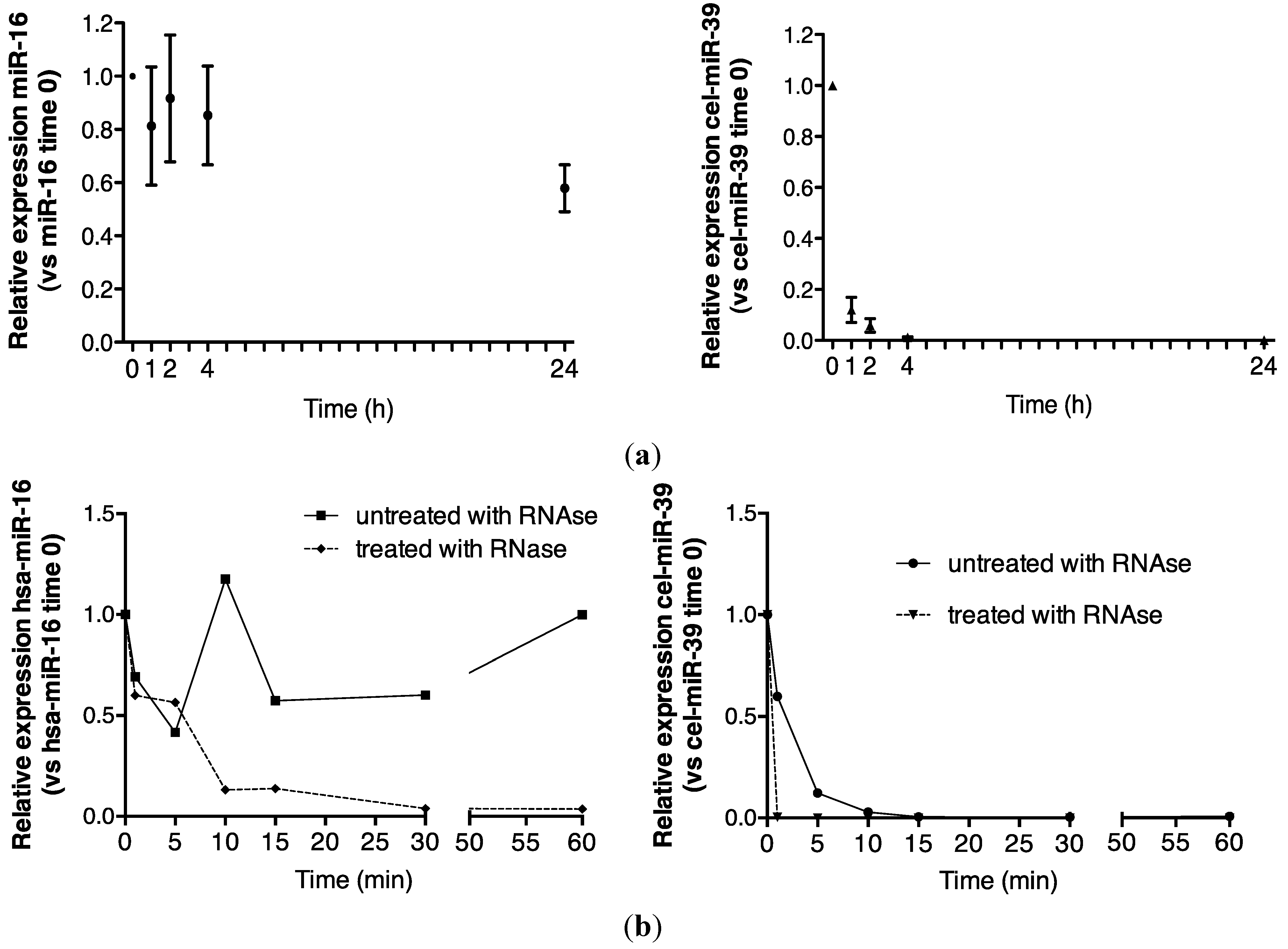
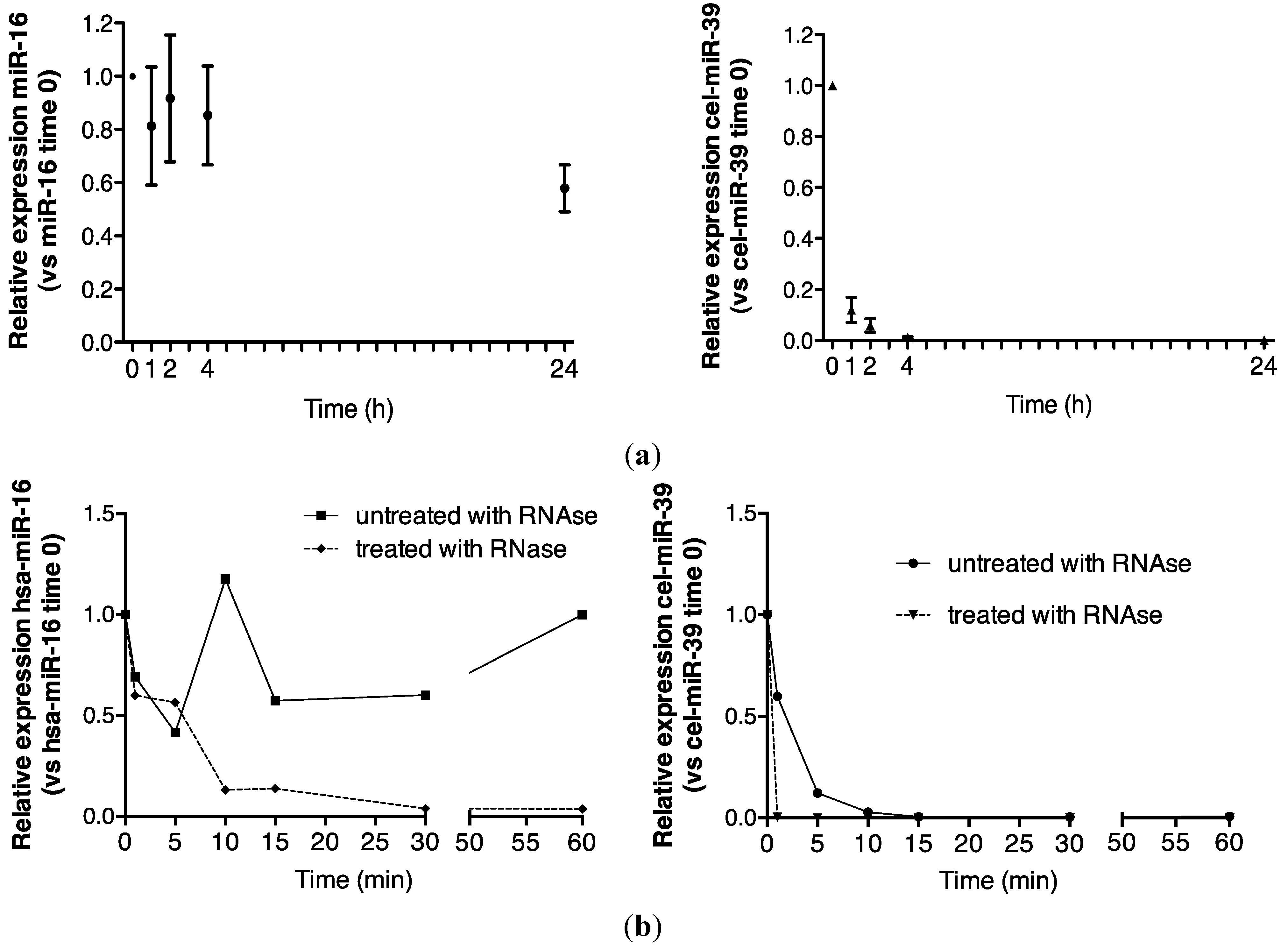
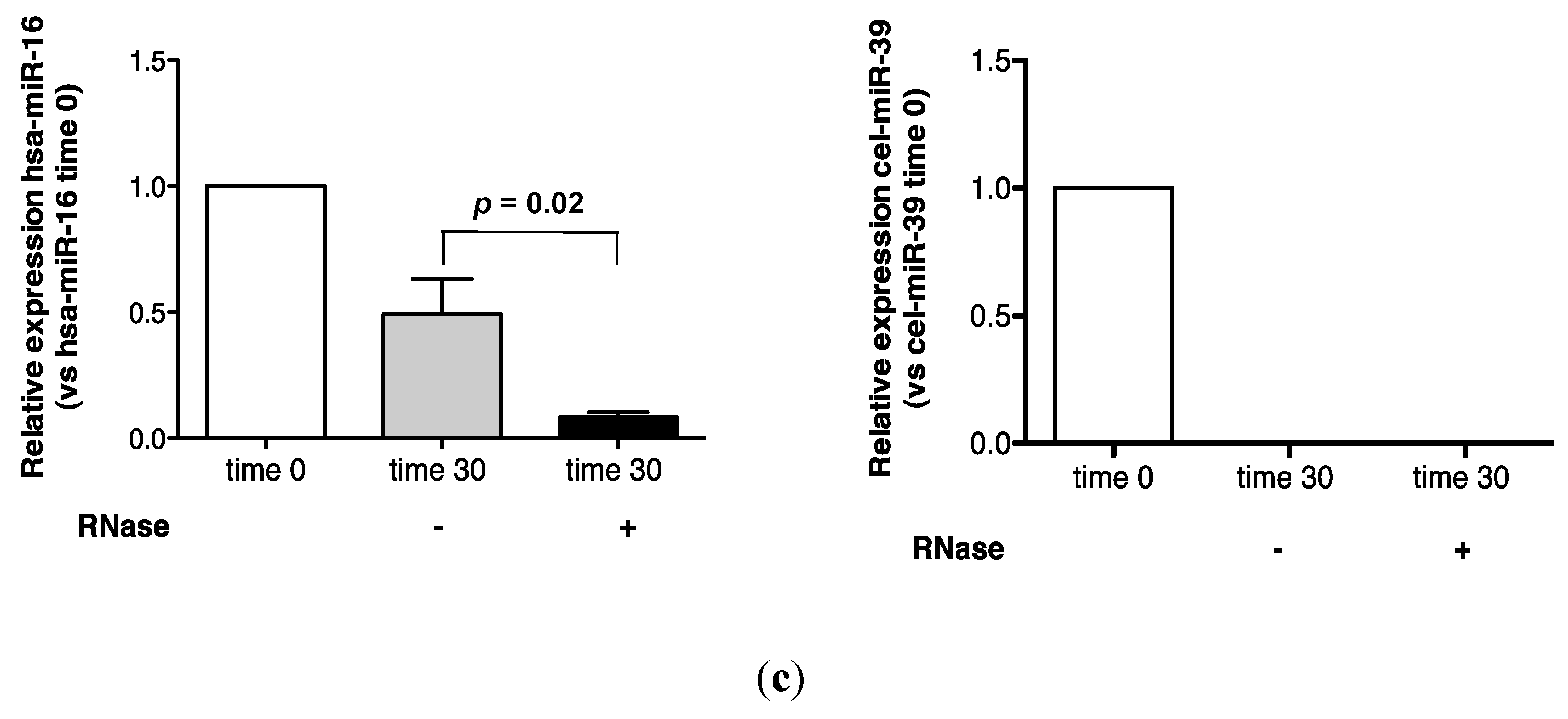
2.1. Endogenous Urinary MiR-16 Is Resistant to RNase Digestion
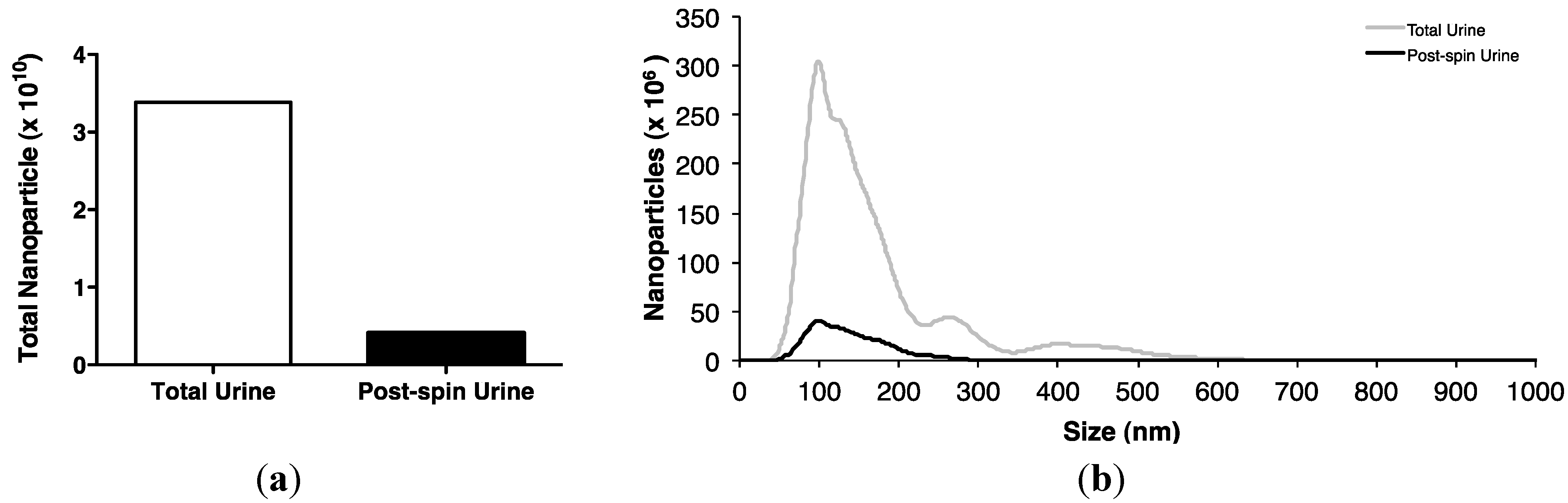
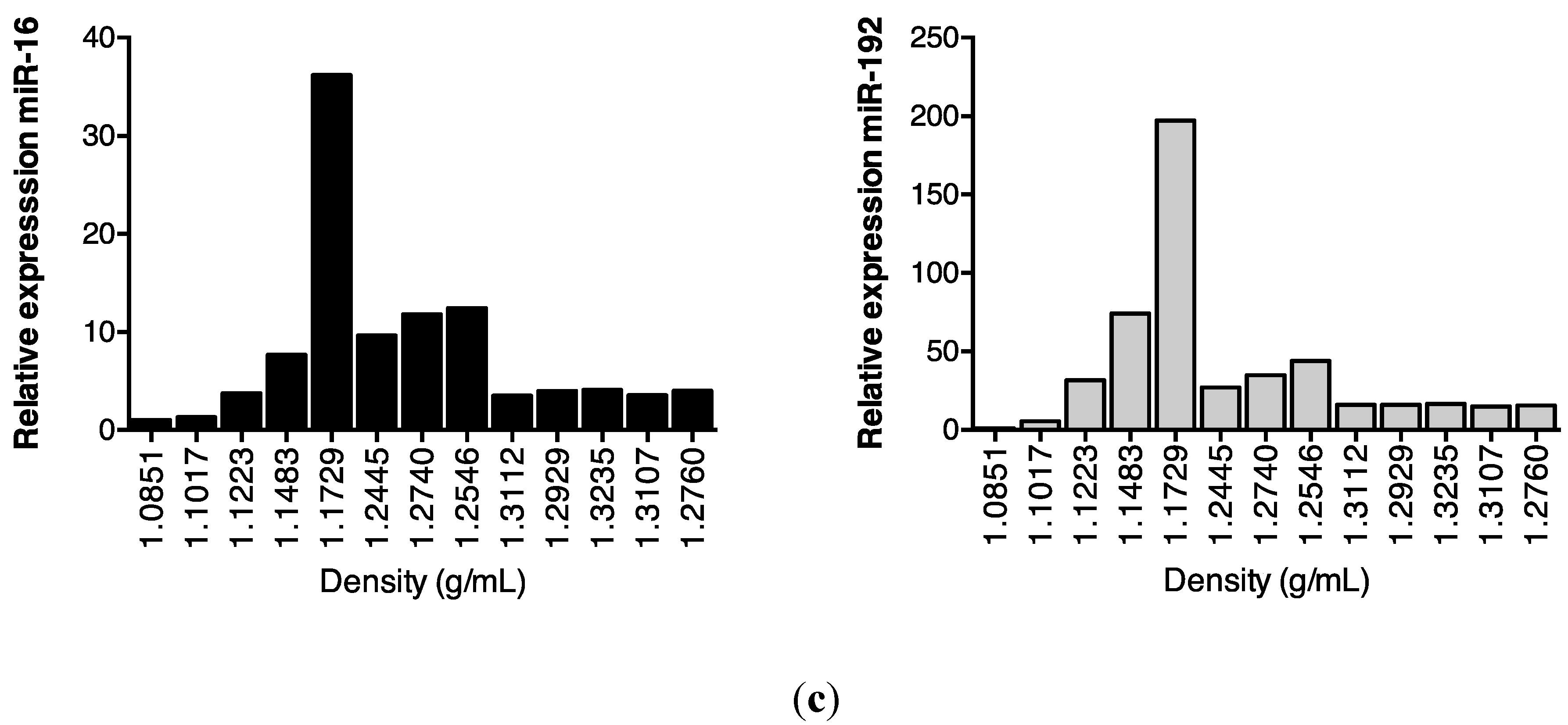
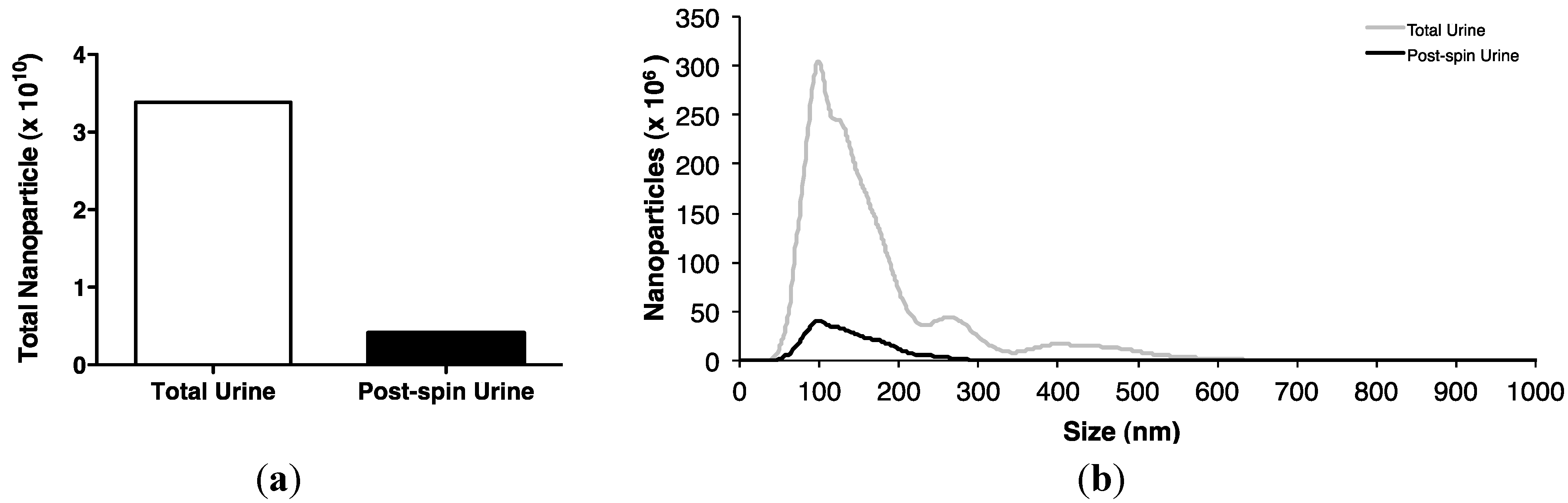
2.2. Association of Urinary MiR-16 and MiR-192 with Exosomes
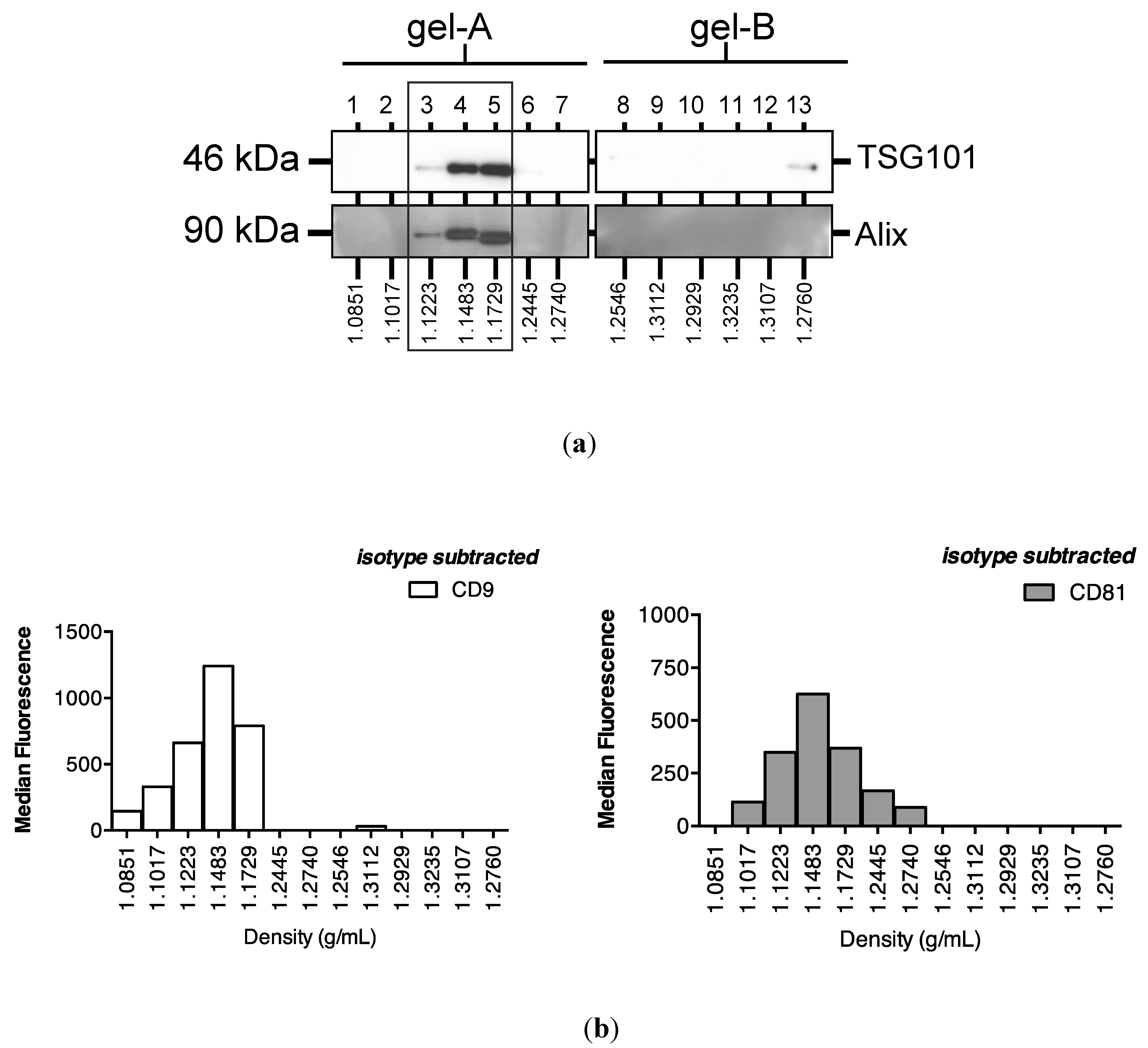
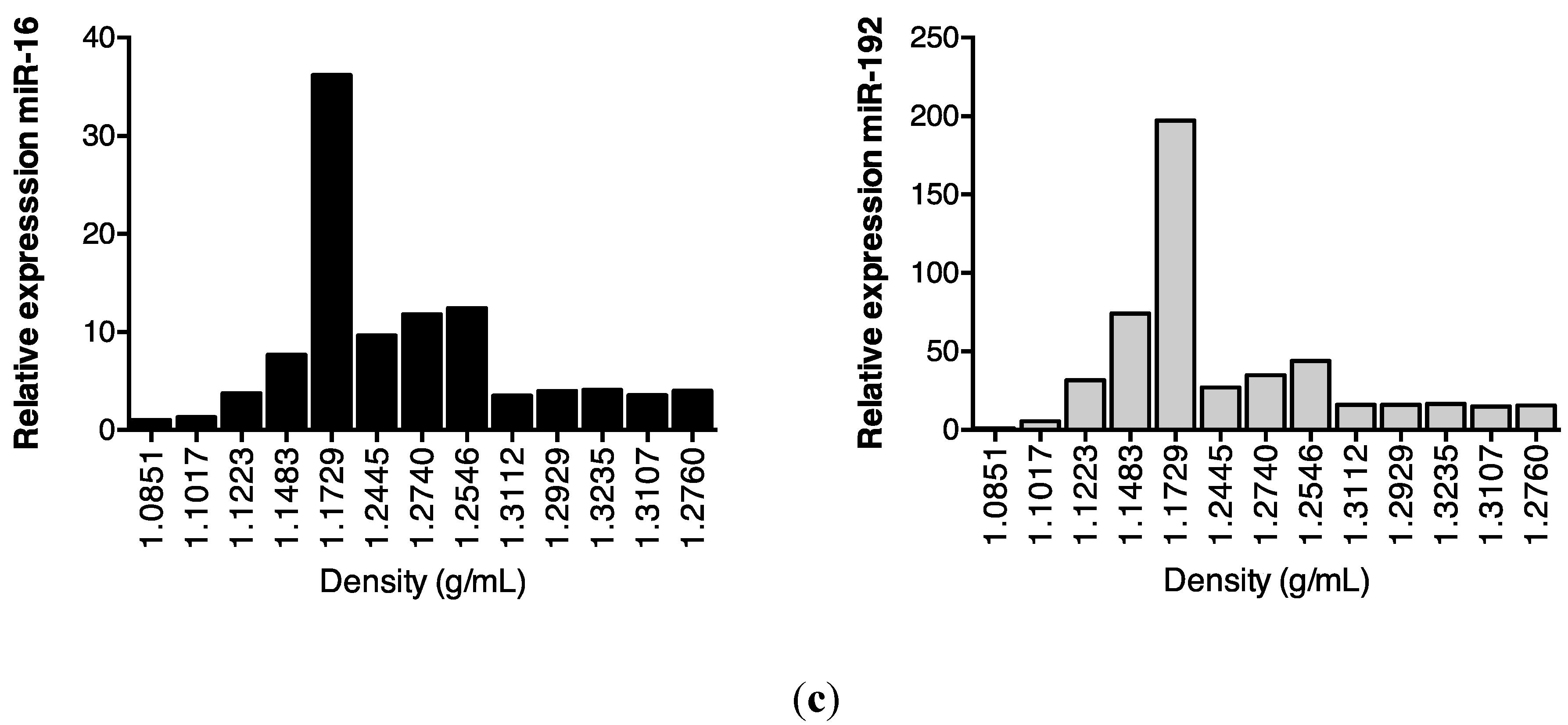
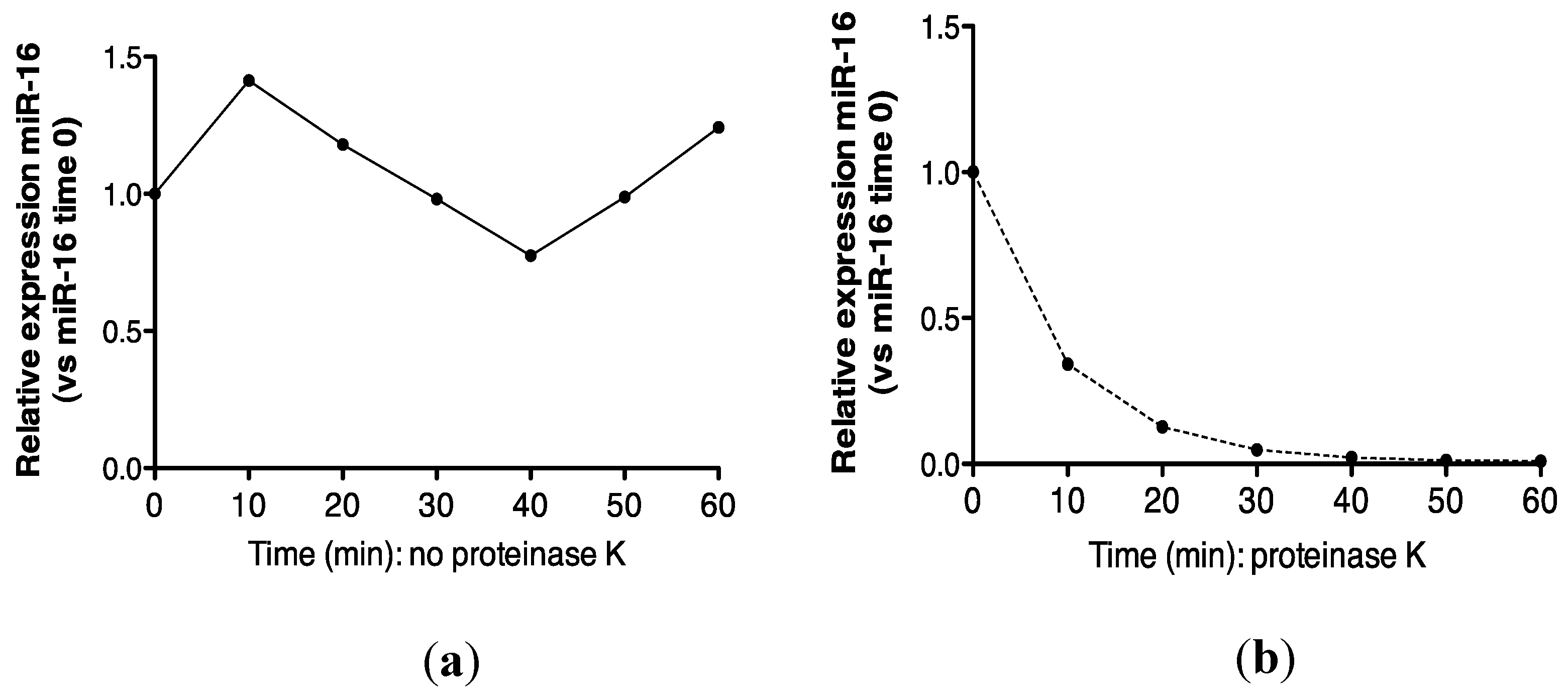
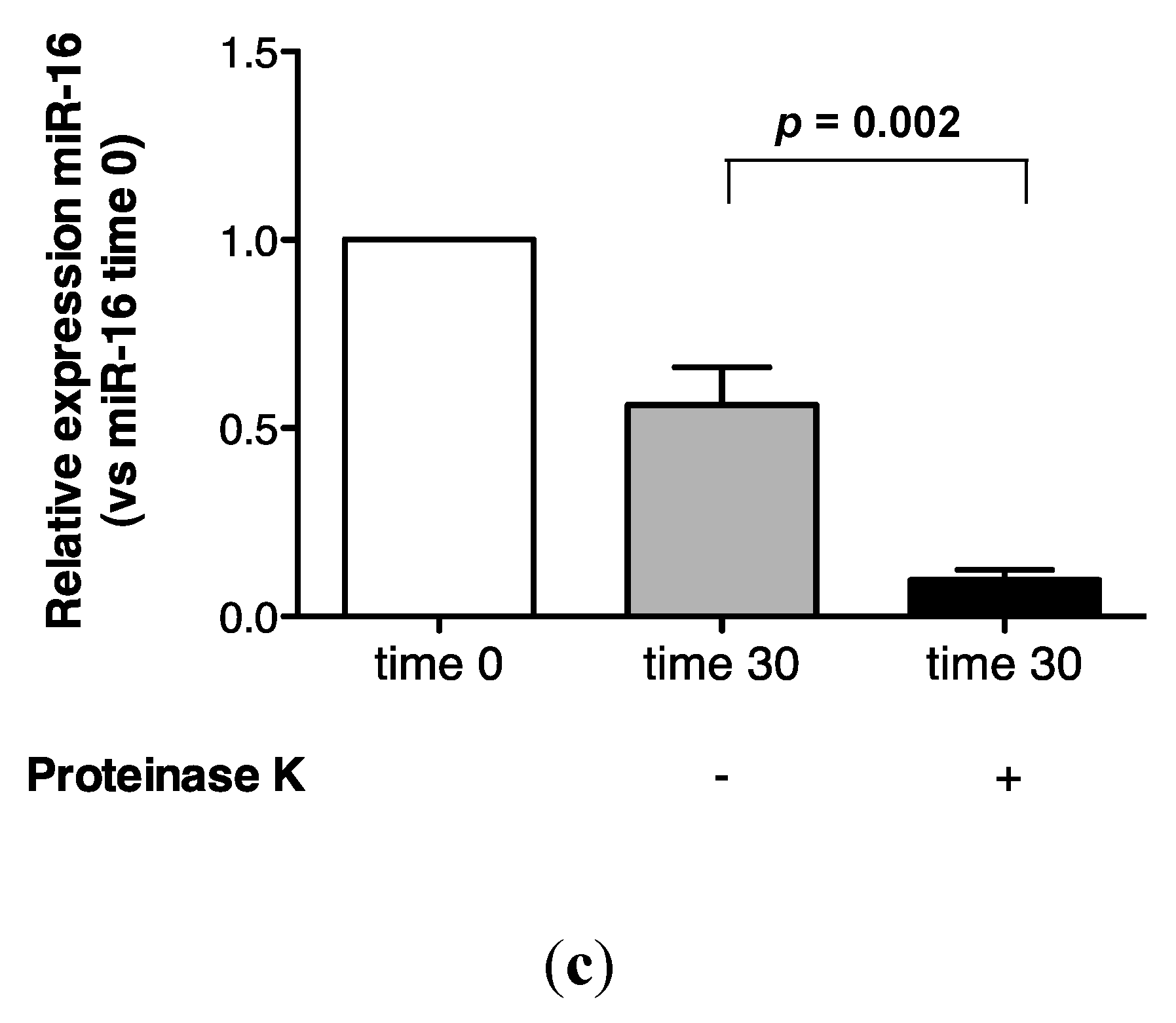
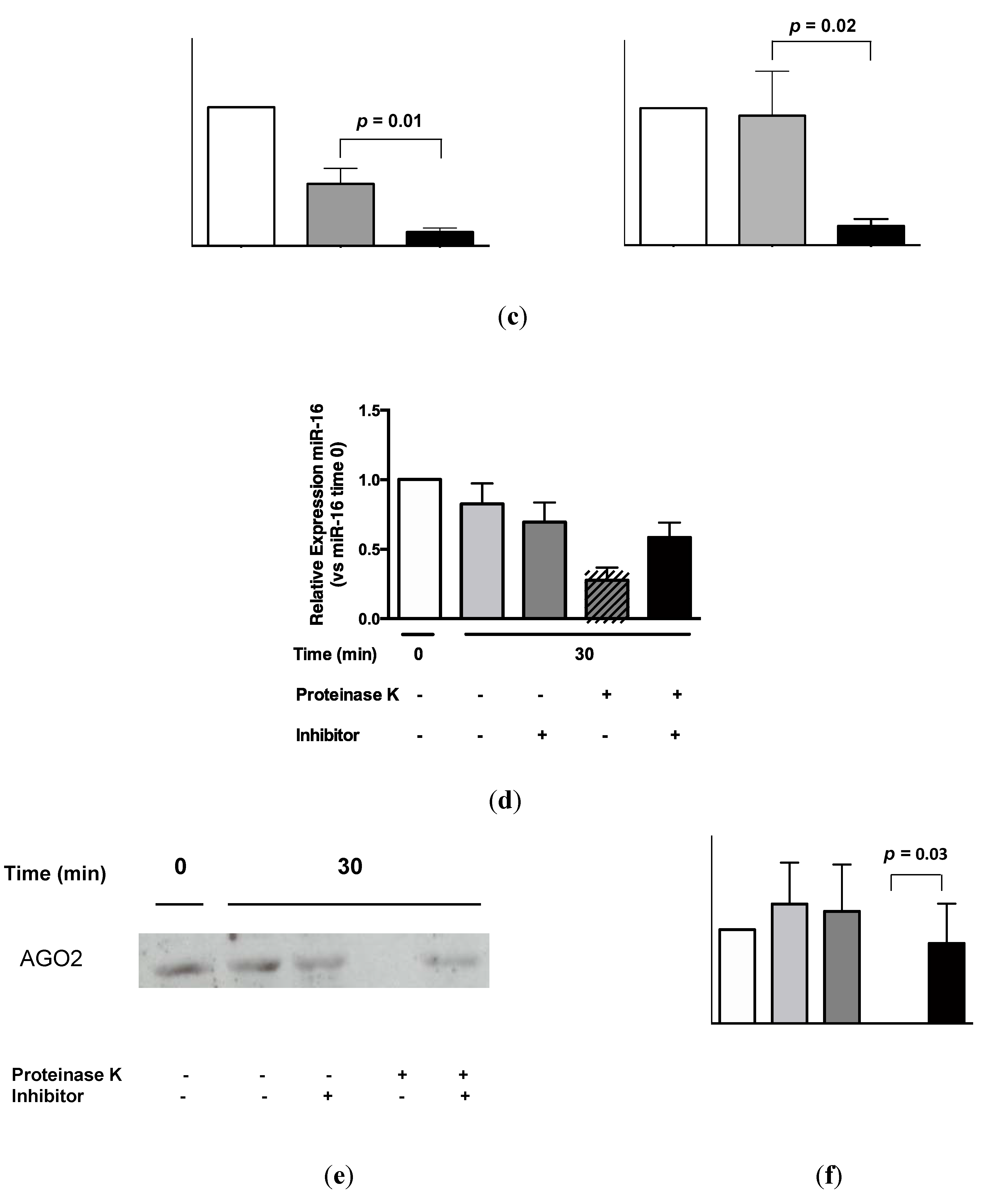
2.3. Protein Association Stabilizes Urinary MiR-16
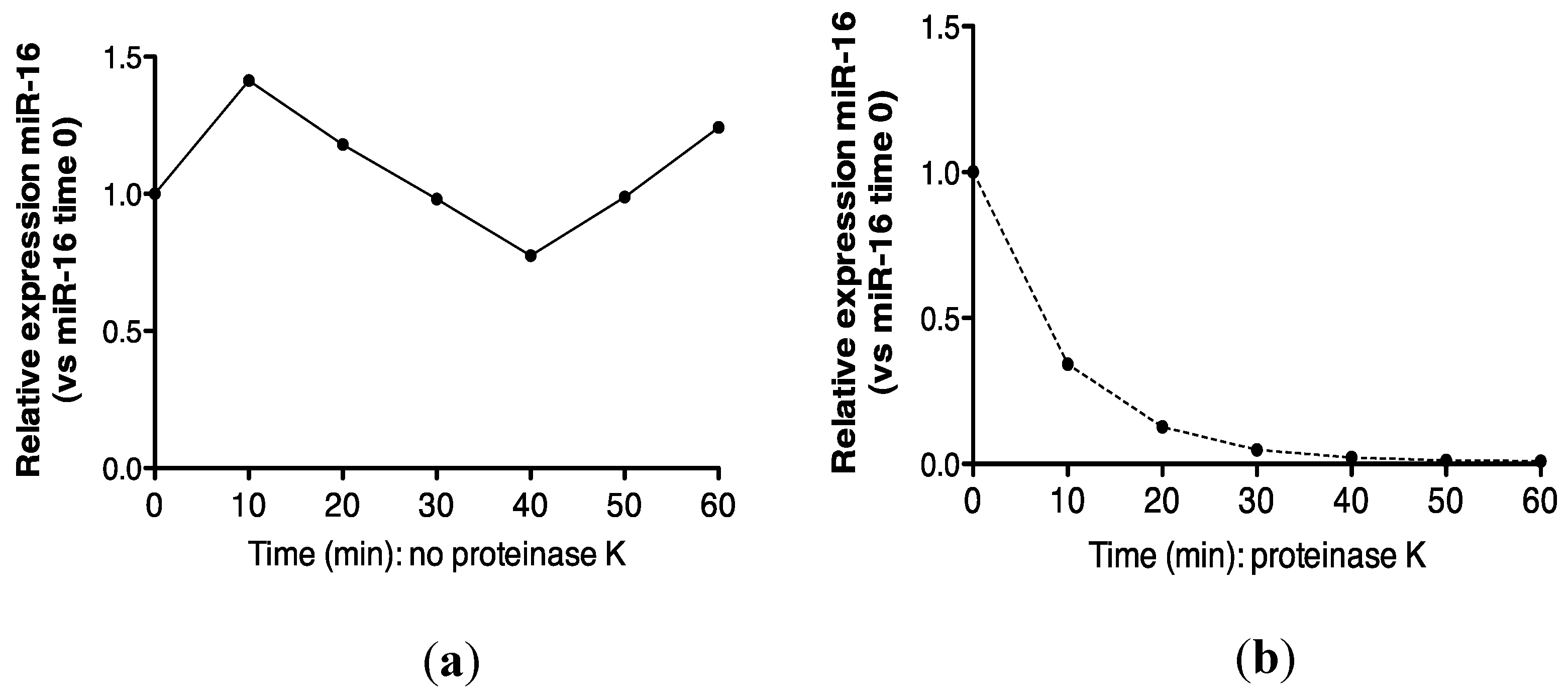
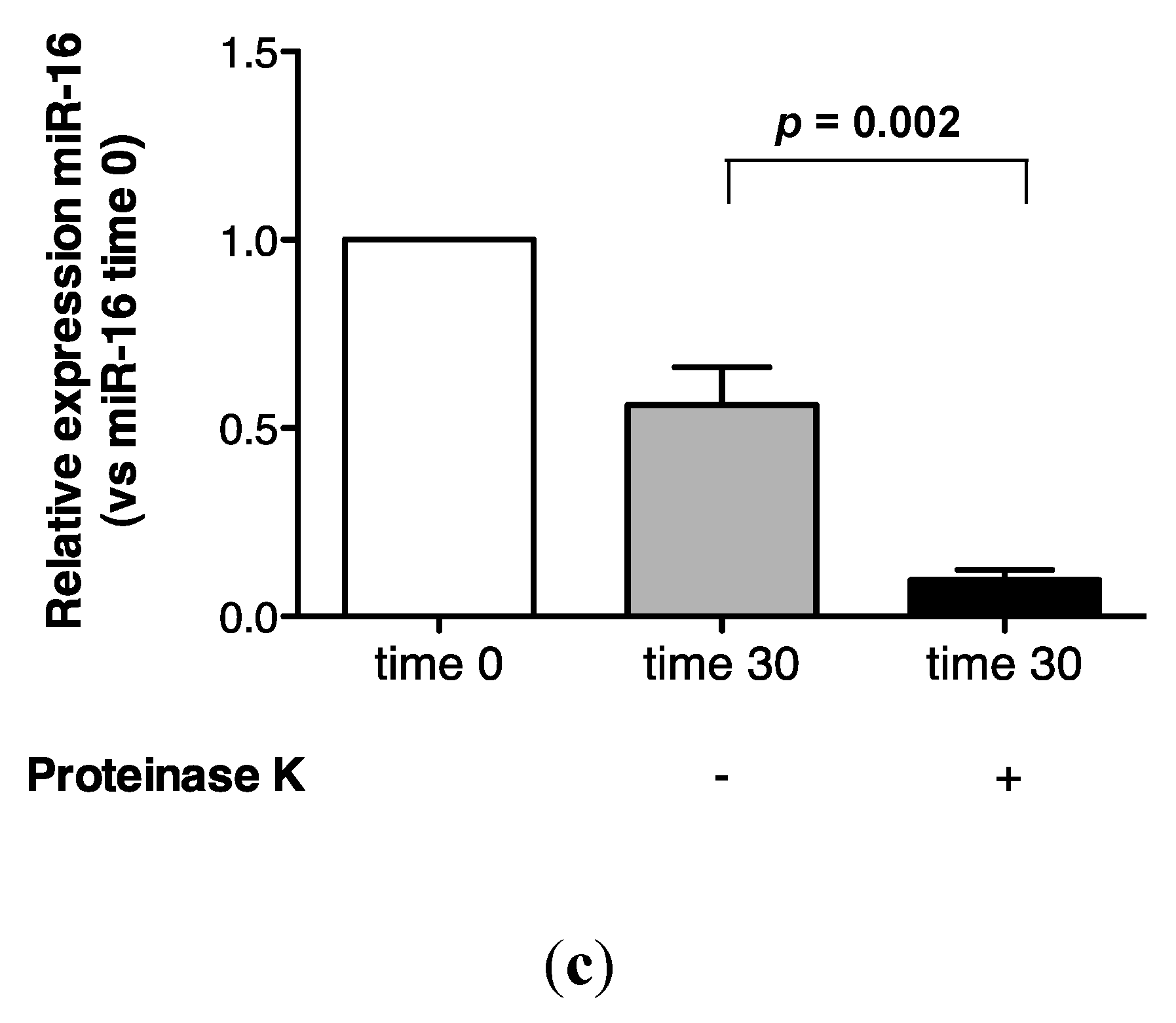
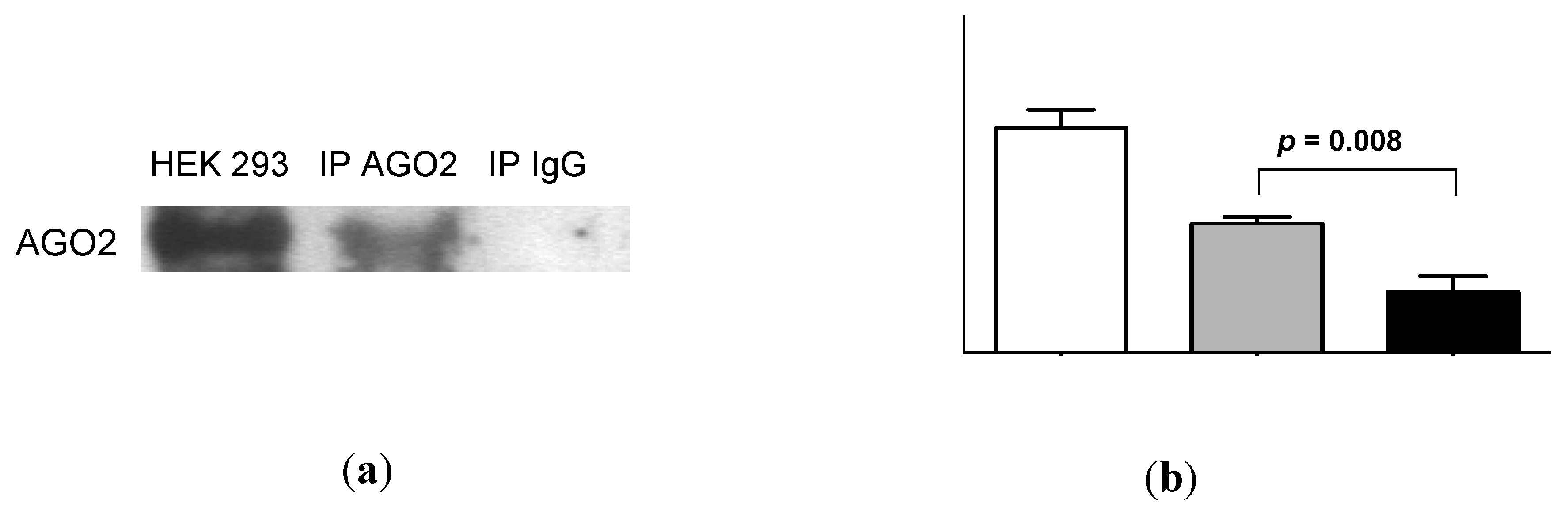
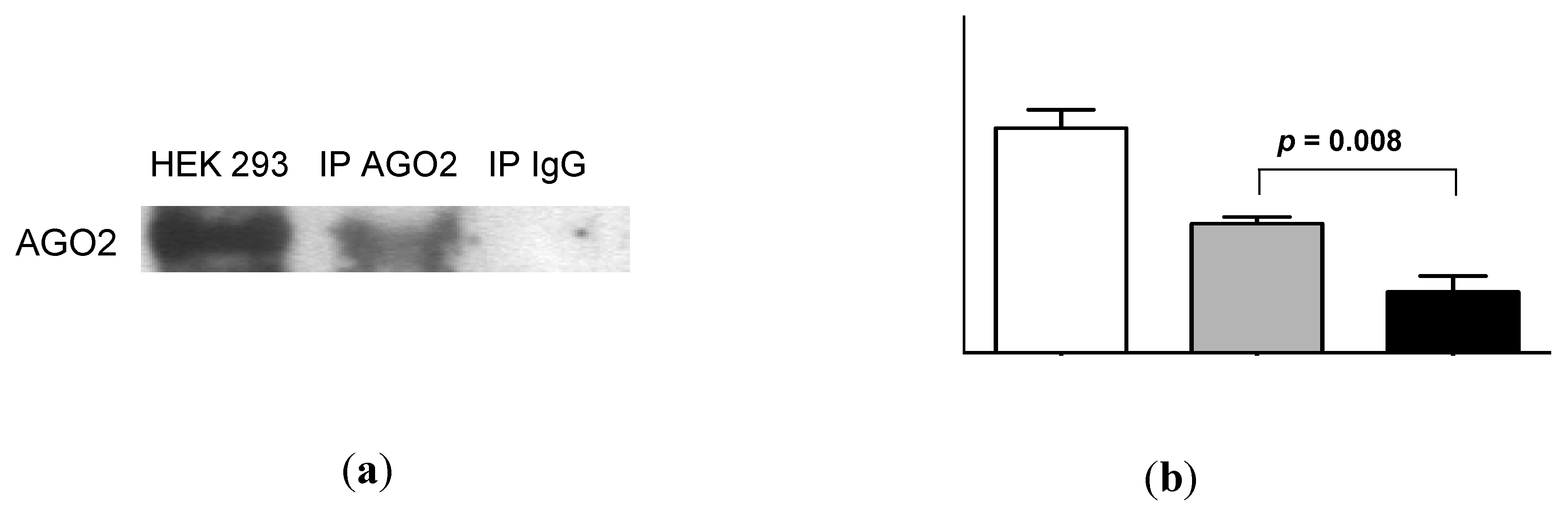
2.4. Urinary MiR-16 and MiR-192 Associate with AGO2
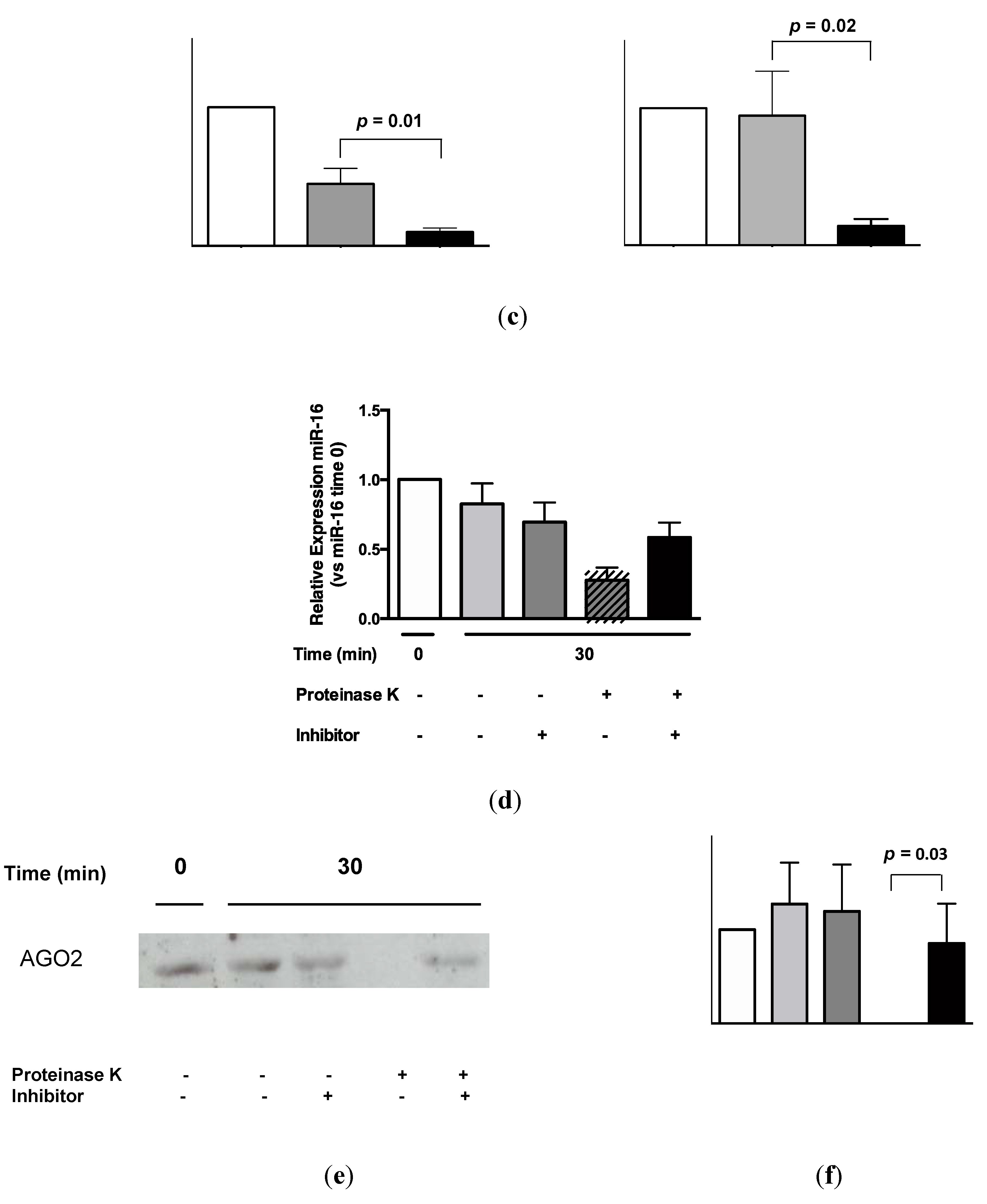
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Study Population
5.2. Urine Collection and RNA Isolation
5.3. Reverse Transcription-Quantitative PCR (RT-qPCR) Analysis
5.4. Room Temperature Urine Analysis over 24 h
5.5. RNase Treatment of Urine
5.6. Proteinase K Treatment of Urine
5.7. AGO2 Immunoprecipitation
5.8. Western Blotting
5.9. Isolation of Sedimented MiRNAs
5.10. Separation of Extracellular Vesicles by Density Gradient Centrifugation
5.11. Flow Cytometric Analyses of Exosome-Coated Beads
5.12. Nanoparticle Tracking Analysis
5.13. Statistical Data
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Levey, A.S.; Atkins, R.; Coresh, J.; Cohen, E.P.; Collins, A.J.; Eckardt, K.U.; Nahas, M.E.; Jaber, B.L.; Jadoul, M.; Levin, A.; et al. Chronic kidney disease as a global public health problem: Approaches and initiatives—A position statement from kidney disease improving global outcomes. Kidney Int. 2007, 72, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, J.; Remuzzi, G.; Saran, R.; Williams, D.E.; Rios-Burrows, N.; Powe, N.; Team, C.-C.S.; Bruck, K.; Wanner, C.; Stel, V.S.; et al. Taming the chronic kidney disease epidemic: A global view of surveillance efforts. Kidney Int. 2014, 86, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.A.; Baxter, D.H.; Zhang, S.; Huang, D.Y.; Huang, K.H.; Lee, M.J.; Galas, D.J.; Wang, K. The microRNA spectrum in 12 body fluids. Clin. Chem. 2010, 56, 1733–1741. [Google Scholar] [CrossRef] [PubMed]
- Lorenzen, J.M.; Thum, T. Circulating and urinary microRNAs in kidney disease. Clin. J. Am. Soc. Nephrol. 2012, 7, 1528–1533. [Google Scholar] [CrossRef] [PubMed]
- Bowen, T.; Jenkins, R.H.; Fraser, D.J. MicroRNAs, transforming growth factor beta-1, and tissue fibrosis. J. Pathol. 2013, 229, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Lorenzen, J.M.; Haller, H.; Thum, T. MicroRNAs as mediators and therapeutic targets in chronic kidney disease. Nat. Rev. Nephrol. 2011, 7, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Beltrami, C.; Clayton, A.; Phillips, A.O.; Fraser, D.J.; Bowen, T. Analysis of urinary microRNAs in chronic kidney disease. Biochem. Soc. Trans. 2012, 40, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, M.L.; DiStefano, J.K. Towards microRNA-based therapeutics for diabetic nephropathy. Diabetologia 2013, 56, 444–456. [Google Scholar] [CrossRef] [PubMed]
- Macconi, D.; Tomasoni, S.; Romagnani, P.; Trionfini, P.; Sangalli, F.; Mazzinghi, B.; Rizzo, P.; Lazzeri, E.; Abbate, M.; Remuzzi, G.; et al. MicroRNA-324-3p promotes renal fibrosis and is a target of ACE inhibition. J. Am. Soc. Nephrol. 2012, 23, 1496–1505. [Google Scholar] [CrossRef] [PubMed]
- Laterza, O.F.; Lim, L.; Garrett-Engele, P.W.; Vlasakova, K.; Muniappa, N.; Tanaka, W.K.; Johnson, J.M.; Sina, J.F.; Fare, T.L.; Sistare, F.D.; et al. Plasma microRNAs as sensitive and specific biomarkers of tissue injury. Clin. Chem. 2009, 55, 1977–1983. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Wang, X.; Yang, J.; Duan, X.; Yao, Y.; Shi, X.; Chen, Z.; Fan, Z.; Liu, X.; Qin, S.; et al. A translational study of urine miRNAs in acute myocardial infarction. J. Mol. Cell. Cardiol. 2012, 53, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhao, J.; Evan, G.; Xiao, C.; Cheng, Y.; Xiao, J. Circulating microRNAs: Novel biomarkers for cardiovascular diseases. J. Mol. Med. 2012, 90, 865–875. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P.S.; Parkin, R.K.; Kroh, E.M.; Fritz, B.R.; Wyman, S.K.; Pogosova-Agadjanyan, E.L.; Peterson, A.; Noteboom, J.; O'Briant, K.C.; Allen, A.; et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. USA 2008, 105, 10513–10518. [Google Scholar] [CrossRef] [PubMed]
- Schwarzenbach, H.; Hoon, D.S.; Pantel, K. Cell-free nucleic acids as biomarkers in cancer patients. Nat. Rev. Cancer 2011, 11, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Arroyo, J.D.; Chevillet, J.R.; Kroh, E.M.; Ruf, I.K.; Pritchard, C.C.; Gibson, D.F.; Mitchell, P.S.; Bennett, C.F.; Pogosova-Agadjanyan, E.L.; Stirewalt, D.L.; et al. Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc. Natl. Acad. Sci. USA 2011, 108, 5003–5008. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Sun, X.; Scicluna, B.J.; Coleman, B.M.; Hill, A.F. Characterization and deep sequencing analysis of exosomal and non-exosomal miRNA in human urine. Kidney Int. 2014, 86, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Chevillet, J.R.; Kang, Q.; Ruf, I.K.; Briggs, H.A.; Vojtech, L.N.; Hughes, S.M.; Cheng, H.H.; Arroyo, J.D.; Meredith, E.K.; Gallichotte, E.N.; et al. Quantitative and stoichiometric analysis of the microRNA content of exosomes. Proc. Natl. Acad. Sci. USA 2014, 111, 14888–14893. [Google Scholar] [CrossRef] [PubMed]
- Turchinovich, A.; Burwinkel, B. Distinct AGO1 and AGO2 associated miRNA profiles in human cells and blood plasma. RNA Biol. 2012, 9, 1066–1075. [Google Scholar] [CrossRef] [PubMed]
- Landgraf, P.; Rusu, M.; Sheridan, R.; Sewer, A.; Iovino, N.; Aravin, A.; Pfeffer, S.; Rice, A.; Kamphorst, A.O.; Landthaler, M.; et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell 2007, 129, 1401–1414. [Google Scholar] [CrossRef] [PubMed]
- Krupa, A.; Jenkins, R.; Luo, D.D.; Lewis, A.; Phillips, A.; Fraser, D. Loss of microRNA-192 promotes fibrogenesis in diabetic nephropathy. J. Am. Soc. Nephrol. 2010, 21, 438–447. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, R.H.; Martin, J.; Phillips, A.O.; Bowen, T.; Fraser, D.J. Transforming growth factor beta1 represses proximal tubular cell microRNA-192 expression through decreased hepatocyte nuclear factor DNA binding. Biochem. J. 2012, 443, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, R.H.; Davies, L.C.; Taylor, P.R.; Akiyama, H.; Cumbes, B.; Beltrami, C.; Carrington, C.P.; Phillips, A.O.; Bowen, T.; Fraser, D.J. MiR-192 induces G2/M growth arrest in aristolochic acid nephropathy. Am. J. Pathol. 2014, 184, 996–1009. [Google Scholar] [CrossRef] [PubMed]
- Thery, C.; Amigorena, S.; Raposo, G.; Clayton, A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. 2006. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(t)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Clayton, A. Cancer cells use exosomes as tools to manipulate immunity and the microenvironment. Oncoimmunology 2012, 1, 78–80. [Google Scholar] [CrossRef] [PubMed]
- Turchinovich, A.; Weiz, L.; Langheinz, A.; Burwinkel, B. Characterization of extracellular circulating microRNA. Nucleic Acids Res. 2011, 39, 7223–7233. [Google Scholar] [CrossRef] [PubMed]
- Gallo, A.; Tandon, M.; Alevizos, I.; Illei, G.G. The majority of microRNAs detectable in serum and saliva is concentrated in exosomes. PloS ONE 2012, 7, e30679. [Google Scholar] [CrossRef] [PubMed]
- Raposo, G.; Nijman, H.W.; Stoorvogel, W.; Liejendekker, R.; Harding, C.V.; Melief, C.J.; Geuze, H.J. B lymphocytes secrete antigen-presenting vesicles. J. Exp. Med. 1996, 183, 1161–1172. [Google Scholar] [CrossRef] [PubMed]
- Shelke, G.V.; Lasser, C.; Gho, Y.S.; Lötvall, J. Importance of exosome depletion protocols to eliminate functional and RNA-containing extracellular vesicles from fetal bovine serum. J. Extracell. Vesicles 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Olejniczak, S.H.; La Rocca, G.; Gruber, J.J.; Thompson, C.B. Long-lived microRNA-argonaute complexes in quiescent cells can be activated to regulate mitogenic responses. Proc. Natl. Acad. Sci. USA 2013, 110, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, R.H.; Blank, A.; Dekker, C.A. Multiple ribonucleases of human urine. Biochemistry 1981, 20, 2268–2274. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Ba, Y.; Ma, L.; Cai, X.; Yin, Y.; Wang, K.; Guo, J.; Zhang, Y.; Chen, J.; Guo, X.; et al. Characterization of microRNAs in serum: A novel class of biomarkers for diagnosis of cancer and other diseases. Cell Res. 2008, 18, 997–1006. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zou, J.; Wang, G.K.; Zhang, J.T.; Huang, S.; Qin, Y.W.; Jing, Q. Uracils at nucleotide position 9–11 are required for the rapid turnover of miR-29 family. Nucleic Acids Res. 2011, 39, 4387–4395. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Qin, Y.W.; Brewer, G.; Jing, Q. MicroRNA degradation and turnover: Regulating the regulators. Wiley Interdiscip. Rev. RNA 2012, 3, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Valadi, H.; Ekstrom, K.; Bossios, A.; Sjostrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Montecalvo, A.; Larregina, A.T.; Shufesky, W.J.; Stolz, D.B.; Sullivan, M.L.; Karlsson, J.M.; Baty, C.J.; Gibson, G.A.; Erdos, G.; Wang, Z.; et al. Mechanism of transfer of functional microRNAs between mouse dendritic cells via exosomes. Blood 2012, 119, 756–766. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, R.H.; Martin, J.; Phillips, A.O.; Bowen, T.; Fraser, D.J. Pleiotropy of microRNA-192 in the kidney. Biochem. Soc. Trans. 2012, 40, 762–767. [Google Scholar] [CrossRef] [PubMed]
- Van Deun, J.; Mestdagh, P.; Sormunen, R.; Cocquyt, V.; Vermaelen, K.; Vandesompele, J.; Bracke, M.; De Wever, O.; Hendrix, A. The impact of disparate isolation methods for extracellular vesicles on downstream RNA profiling. J. Extracell. Vesicles 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Diederichs, S.; Haber, D.A. Dual role for argonautes in microRNA processing and posttranscriptional regulation of microRNA expression. Cell 2007, 131, 1097–1108. [Google Scholar] [CrossRef] [PubMed]
- Welton, J.L.; Khanna, S.; Giles, P.J.; Brennan, P.; Brewis, I.A.; Staffurth, J.; Mason, M.D.; Clayton, A. Proteomics analysis of bladder cancer exosomes. Mol. Cell. Proteomics 2010, 9, 1324–1338. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beltrami, C.; Clayton, A.; Newbury, L.J.; Corish, P.; Jenkins, R.H.; Phillips, A.O.; Fraser, D.J.; Bowen, T. Stabilization of Urinary MicroRNAs by Association with Exosomes and Argonaute 2 Protein. Non-Coding RNA 2015, 1, 151-166. https://doi.org/10.3390/ncrna1020151
Beltrami C, Clayton A, Newbury LJ, Corish P, Jenkins RH, Phillips AO, Fraser DJ, Bowen T. Stabilization of Urinary MicroRNAs by Association with Exosomes and Argonaute 2 Protein. Non-Coding RNA. 2015; 1(2):151-166. https://doi.org/10.3390/ncrna1020151
Chicago/Turabian StyleBeltrami, Cristina, Aled Clayton, Lucy J. Newbury, Peter Corish, Robert H. Jenkins, Aled O. Phillips, Donald J. Fraser, and Timothy Bowen. 2015. "Stabilization of Urinary MicroRNAs by Association with Exosomes and Argonaute 2 Protein" Non-Coding RNA 1, no. 2: 151-166. https://doi.org/10.3390/ncrna1020151
APA StyleBeltrami, C., Clayton, A., Newbury, L. J., Corish, P., Jenkins, R. H., Phillips, A. O., Fraser, D. J., & Bowen, T. (2015). Stabilization of Urinary MicroRNAs by Association with Exosomes and Argonaute 2 Protein. Non-Coding RNA, 1(2), 151-166. https://doi.org/10.3390/ncrna1020151