
“Pocket-sized RNA-Seq”: A Method to Capture New Mature microRNA Produced from a Genomic Region of Interest



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
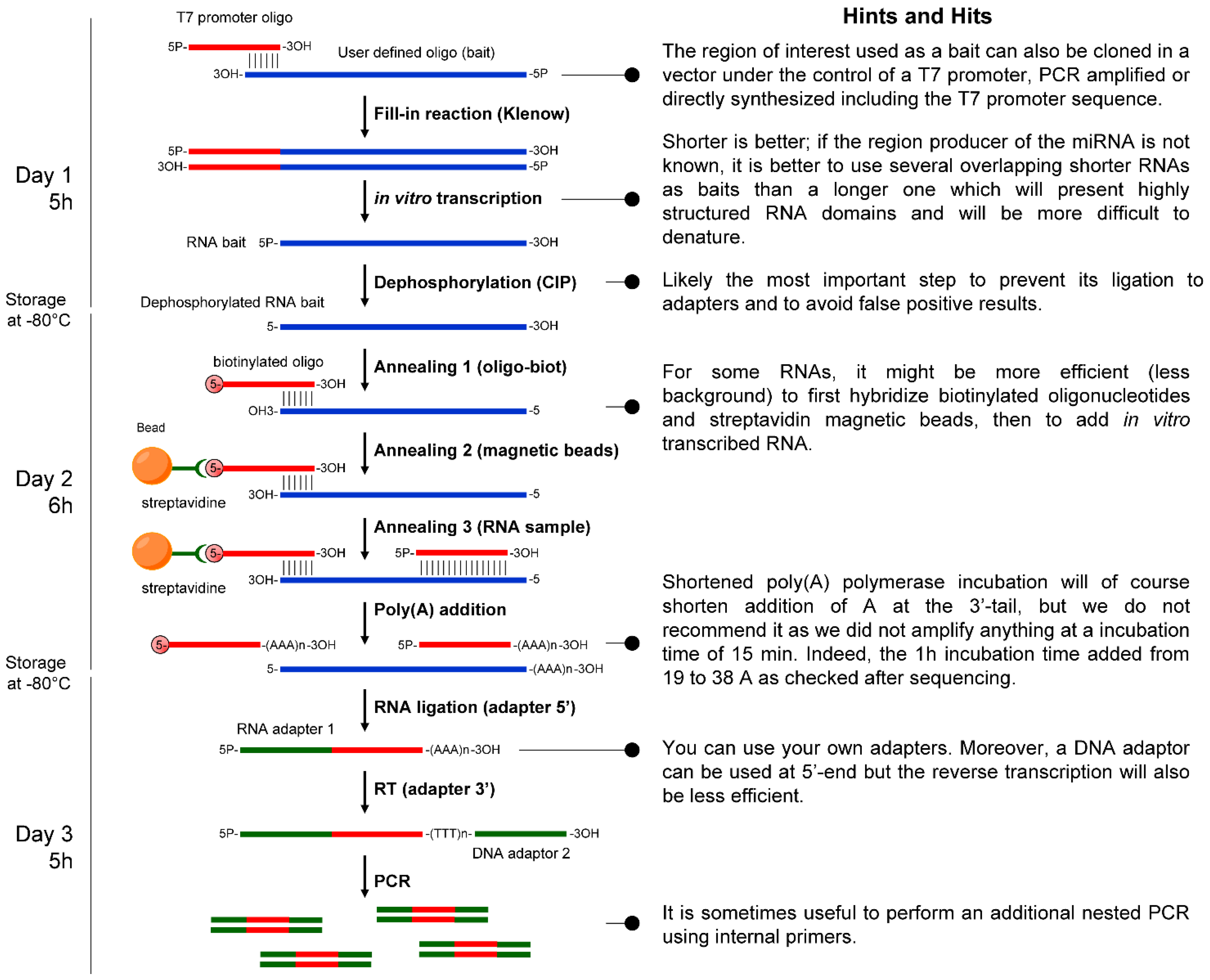
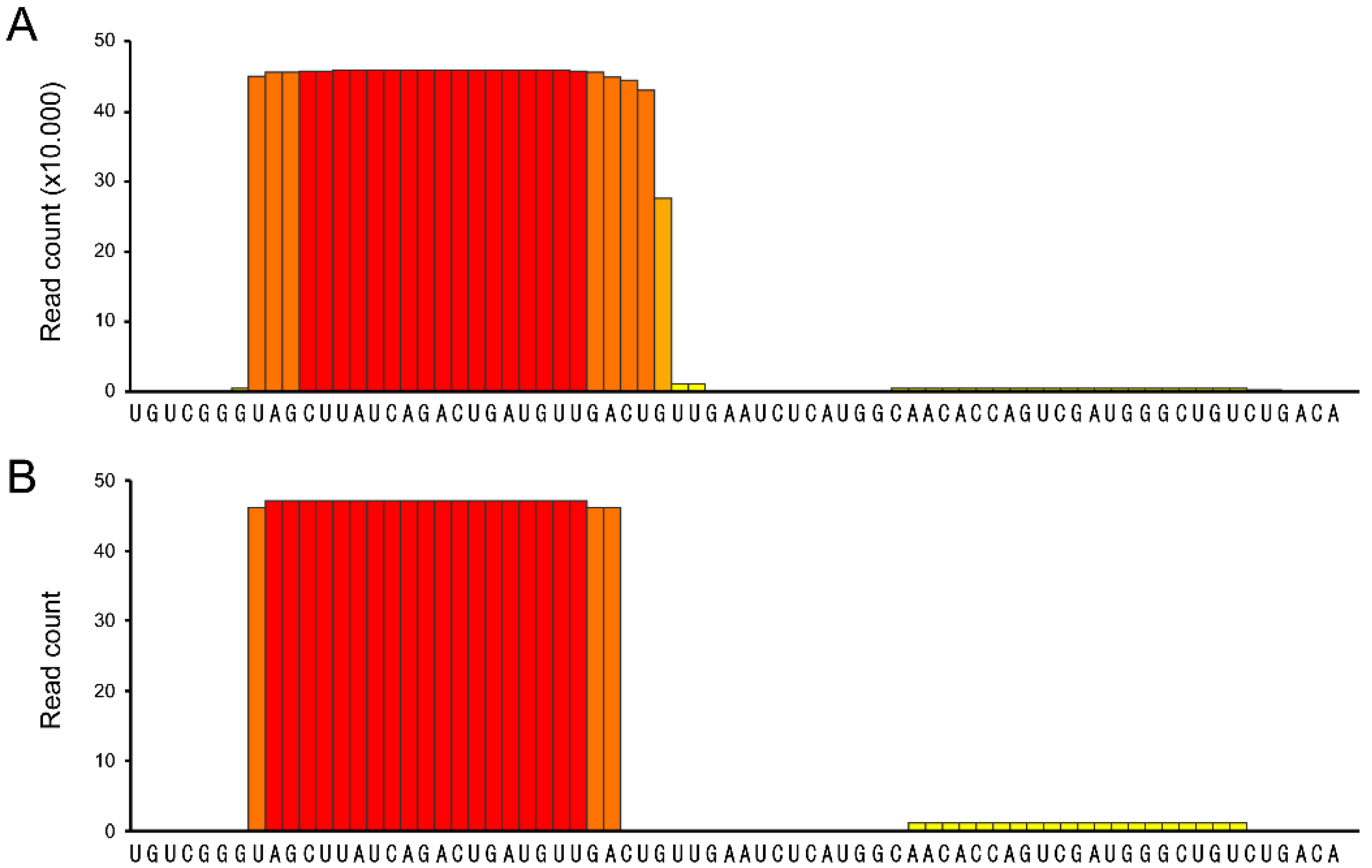
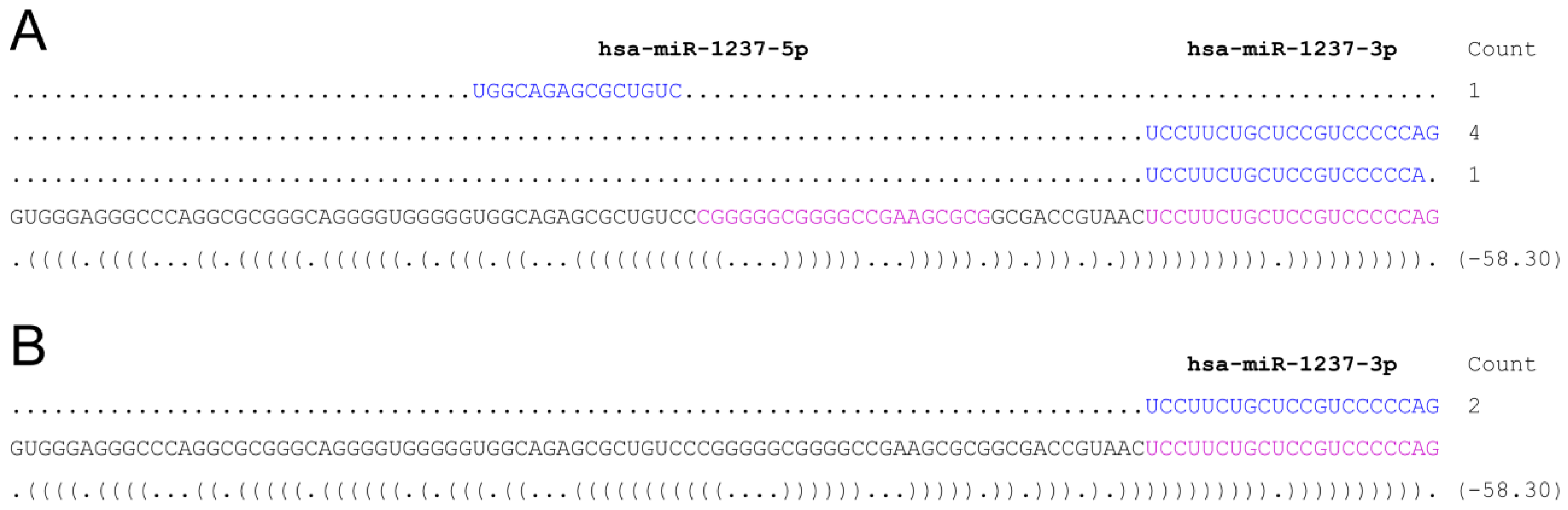
3. Discussion
- (1)
- psRNA-seq is a highly sensitive technique that can detect even low amounts of mature miRNAs (or any other short RNA), as exemplified by the amplification of the weakly expressed has-miR-21 star and hsa-miR-1237, that would not pass low-thresholding or elimination of isolated reads applied to eliminate high-throughput sequencing errors [16,17,18,19]. We can infer from our data that psRNA-seq is a method equally sensitive to high-throughput sequencing for an already known miRNA, highly expressed in the relevant cellular system. Obviously, no conclusion can be drawn regarding yet unidentified small RNAs.
- (2)
- psRNA-seq does not require knowing the biogenesis pathway of the small RNA of interest. Therefore, not only miRNA, but also mirtrons, simtrons, snoRNAs, snRNAs, and others small ncRNAs can potentially be detected and cloned using psRNA-seq provided the genomic location, even approximate, of the sequence containing the small RNA is known. It is indeed simple to refine the position using overlapping sequences as we have done with hsa-miR-21. We used 60 to 102 nt long baits to minimize secondary structures that may form, although we have experience of RNA pull down assays using much longer bait RNAs, which efficiency has to be tested individually [29,30].
- (3)
- psRNA-seq can be performed on rare samples like adult stem cells or patient biopsies since it does not require large amounts of material. While microarrays and RNA deep sequencing needs are in the range of several micrograms of sample RNA, we used as less as 10 ng of fractionated RNA, an amount that can be further reduced. Indeed, from all the RNA samples that we prepared, we used 1/100th by PCR reactions and obtained thousands of colonies after bacterial transformation, clearly suggesting that 0.1 ng of fractionated RNA should be enough to obtain similar results (0.1 ng of small fractionated RNA represents about 1 ng of total RNA and as little as 100 human cells).
- (4)
- psRNA-seq is a simple, rapid, and inexpensive method to set up. It does not require special skills in molecular biology or bioinformatics. Indeed, it does not require long and tricky procedures to prepare libraries to be sequenced by RNA-seq technologies or challenging bioinformatical analysis of large datasets.
- (5)
- psRNA-seq is highly flexible. It is not specific to miRNAs (canonical and from intron origin) and can also be used to capture plenty of other ncRNAs such as snRNAs, snoRNAs and many other still-unknown small RNAs. The method is also easily transposable to other species, with some restrictions (see below).
4. Material and Methods
4.1. Bait Preparation
4.2. RNA Sample Preparation
4.3. Hybridization of the Bait with Size-fractionated Short RNAs
4.4. Polyadenylation of the Captured RNAs
4.5. Adapted RACE-PCR Amplification of the Captured Products
4.6. Stem-Loop RT-qPCR
4.7. RNA-seq Datasets Used
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Griffiths-Jones, S. Annotating noncoding RNA genes. Annu. Rev. Genomics Hum. Genet. 2007, 8, 279–298. [Google Scholar] [CrossRef]
- Holley, C.L.; Topkara, V.K. An introduction to small non-coding RNAs: miRNA and snoRNA. Cardiovasc. Drugs Ther. 2011, 25, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Shin, C. MicroRNA-target interactions: New insights from genome-wide approaches. Ann. N. Y. Acad. Sci. 2012, 1271, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Mo, Y.Y. MicroRNA regulatory networks and human disease. Cell Mol. Life Sci. 2012, 69, 3529–3531. [Google Scholar] [CrossRef] [PubMed]
- Ruegger, S.; Grosshans, H. MicroRNA turnover: When, how, and why. Trends Biochem. Sci. 2012, 37, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Liu, X.; Li, D.; Wang, P.; Li, N.; Lu, L.; Cao, X. MicroRNA-466l upregulates IL-10 expression in TLR-triggered macrophages by antagonizing RNA-binding protein tristetraprolin-mediated IL-10 mRNA degradation. J. Immunol. 2010, 184, 6053–6059. [Google Scholar] [CrossRef] [PubMed]
- Place, R.F.; Li, L.C.; Pookot, D.; Noonan, E.J.; Dahiya, R. MicroRNA-373 induces expression of genes with complementary promoter sequences. Proc. Natl. Acad. Sci. USA 2008, 105, 1608–1613. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Kim, M.; Han, J.; Yeom, K.H.; Lee, S.; Baek, S.H.; Kim, V.N. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004, 23, 4051–4060. [Google Scholar] [CrossRef] [PubMed]
- Chureau, C.; Prissette, M.; Bourdet, A.; Barbe, V.; Cattolico, L.; Jones, L.; Eggen, A.; Avner, P.; Duret, L. Comparative sequence analysis of the X-inactivation center region in mouse, human, and bovine. Genome Res. 2002, 12, 894–908. [Google Scholar] [PubMed]
- Curtis, H.J.; Sibley, C.R.; Wood, M.J. Mirtrons, an emerging class of atypical miRNA. Wiley Interdiscip. Rev. RNA 2012, 3, 617–632. [Google Scholar] [CrossRef] [PubMed]
- Eis, P.S.; Tam, W.; Sun, L.; Chadburn, A.; Li, Z.; Gomez, M.F.; Lund, E.; Dahlberg, J.E. Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proc. Natl. Acad. Sci. USA 2005, 102, 3627–3632. [Google Scholar] [CrossRef] [PubMed]
- Ulveling, D.; Francastel, C.; Hube, F. When one is better than two: RNA with dual functions. Biochimie 2011, 93, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Uva, P.; Da, S.L.; Del, C.M.; Baldassarre, A.; Sestili, P.; Orsini, M.; Palma, A.; Gessani, S.; Masotti, A. Rat mir-155 generated from the lncRNA Bic is ‘hidden’ in the alternate genomic assembly and reveals the existence of novel mammalian miRNAs and clusters. RNA 2013, 19, 365–379. [Google Scholar] [CrossRef] [PubMed]
- Westholm, J.O.; Lai, E.C. Mirtrons: microRNA biogenesis via splicing. Biochimie 2011, 93, 1897–1904. [Google Scholar] [CrossRef] [PubMed]
- Havens, M.A.; Reich, A.A.; Duelli, D.M.; Hastings, M.L. Biogenesis of mammalian microRNAs by a non-canonical processing pathway. Nucleic Acids Res. 2012, 40, 4626–4640. [Google Scholar] [CrossRef] [PubMed]
- Bullard, J.H.; Purdom, E.; Hansen, K.D.; Dudoit, S. Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinform. 2010, 11, 94. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Lin, L.; Jiang, P.; Wang, D.; Xing, Y. A comparison of RNA-Seq and high-density exon array for detecting differential gene expression between closely related species. Nucleic Acids Res. 2011, 39, 578–588. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [PubMed]
- Tarazona, S.; Garcia-Alcalde, F.; Dopazo, J.; Ferrer, A.; Conesa, A. Differential expression in RNA-seq: A matter of depth. Genome Res. 2011, 21, 2213–2223. [Google Scholar] [CrossRef] [PubMed]
- Bar, M.; Wyman, S.K.; Fritz, B.R.; Qi, J.; Garg, K.S.; Parkin, R.K.; Kroh, E.M.; Bendoraite, A.; Mitchell, P.S.; Nelson, A.M.; et al. MicroRNA discovery and profiling in human embryonic stem cells by deep sequencing of small RNA libraries. Stem Cells 2008, 26, 2496–2505. [Google Scholar] [CrossRef] [PubMed]
- Stark, M.S.; Tyagi, S.; Nancarrow, D.J.; Boyle, G.M.; Cook, A.L.; Whiteman, D.C.; Parsons, P.G.; Schmidt, C.; Sturm, R.A.; Hayward, N.K. Characterization of the Melanoma miRNAome by Deep Sequencing. PLoS ONE 2010, 5, e9685. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.Y.; Pfuhl, T.; Motsch, N.; Barth, S.; Nicholls, J.; Grasser, F.; Meister, G. Identification of novel Epstein-Barr virus microRNA genes from nasopharyngeal carcinomas. J. Virol. 2009, 83, 3333–3341. [Google Scholar] [CrossRef] [PubMed]
- Varkonyi-Gasic, E.; Hellens, R.P. Quantitative stem-loop RT-PCR for detection of microRNAs. Methods Mol. Biol. 2011, 744, 145–157. [Google Scholar] [PubMed]
- Berezikov, E.; Chung, W.J.; Willis, J.; Cuppen, E.; Lai, E.C. Mammalian mirtron genes. Mol. Cell 2007, 28, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Farazi, T.A.; Horlings, H.M.; Ten Hoeve, J.J.; Mihailovic, A.; Halfwerk, H.; Morozov, P.; Brown, M.; Hafner, M.; Reyal, F.; van, K.M.; et al. MicroRNA sequence and expression analysis in breast tumors by deep sequencing. Cancer Res. 2011, 71, 4443–4453. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; Baek, M.; Gusev, Y.; Brackett, D.J.; Nuovo, G.J.; Schmittgen, T.D. Systematic evaluation of microRNA processing patterns in tissues, cell lines, and tumors. RNA 2008, 14, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Neilson, J.R.; Zheng, G.X.; Burge, C.B.; Sharp, P.A. Dynamic regulation of miRNA expression in ordered stages of cellular development. Genes Dev. 2007, 21, 578–589. [Google Scholar] [CrossRef] [PubMed]
- Van Rooij, E. The art of microRNA research. Circ. Res. 2011, 108, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Ferri, F.; Bouzinba-Segard, H.; Velasco, G.; Hube, F.; Francastel, C. Non-coding murine centromeric transcripts associate with and potentiate Aurora B kinase. Nucleic Acids Res. 2009, 37, 5071–5080. [Google Scholar] [CrossRef] [PubMed]
- Hube, F.; Velasco, G.; Rollin, J.; Furling, D.; Francastel, C. Steroid receptor RNA activator protein binds to and counteracts SRA RNA-mediated activation of MyoD and muscle differentiation. Nucleic Acids Res. 2011, 39, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, Z.; Yu, B.; Liu, J.; Chen, X. Methylation protects miRNAs and siRNAs from a 3′-end uridylation activity in Arabidopsis. Curr. Biol. 2005, 15, 1501–1507. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Ebright, Y.W.; Yu, B.; Chen, X. HEN1 recognizes 21-24 nt small RNA duplexes and deposits a methyl group onto the 2′ OH of the 3′ terminal nucleotide. Nucleic Acids Res. 2006, 34, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Sakaguchi, Y.; Suzuki, T.; Suzuki, T.; Siomi, H.; Siomi, M.C. Pimet, the Drosophila homolog of HEN1, mediates 2′-O-methylation of Piwi- interacting RNAs at their 3′ ends. Genes Dev. 2007, 21, 1603–1608. [Google Scholar] [CrossRef] [PubMed]
- Krichevsky, A.M.; Gabriely, G. miR-21: A small multi-faceted RNA. J. Cell Mol. Med. 2009, 13, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Kumarswamy, R.; Volkmann, I.; Thum, T. Regulation and function of miRNA-21 in health and disease. RNA Biol. 2011, 8, 706–713. [Google Scholar] [CrossRef] [PubMed]
- Selcuklu, S.D.; Donoghue, M.T.; Spillane, C. miR-21 as a key regulator of oncogenic processes. Biochem. Soc. Trans. 2009, 37, 918–925. [Google Scholar] [CrossRef] [PubMed]
- Velasco, G.; Hube, F.; Rollin, J.; Neuillet, D.; Philippe, C.; Bouzinba-Segard, H.; Galvani, A.; Viegas-Pequignot, E.; Francastel, C. Dnmt3b recruitment through E2F6 transcriptional repressor mediates germ-line gene silencing in murine somatic tissues. Proc. Natl. Acad. Sci. USA 2010, 107, 9281–9286. [Google Scholar] [CrossRef] [PubMed]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef] [PubMed]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Tilgner, H.; Knowles, D.G.; Johnson, R.; Davis, C.A.; Chakrabortty, S.; Djebali, S.; Curado, J.; Snyder, M.; Gingeras, T.R.; Guigo, R. Deep sequencing of subcellular RNA fractions shows splicing to be predominantly co-transcriptional in the human genome but inefficient for lncRNAs. Genome Res. 2012, 22, 1616–1625. [Google Scholar] [CrossRef] [PubMed]
- Hube, F.; Guo, J.; Chooniedass-Kothari, S.; Cooper, C.; Hamedani, M.K.; Dibrov, A.A.; Blanchard, A.A.; Wang, X.; Deng, G.; Myal, Y.; et al. Alternative splicing of the first intron of the steroid receptor RNA activator (SRA) participates in the generation of coding and noncoding RNA isoforms in breast cancer cell lines. DNA Cell Biol. 2006, 25, 418–428. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hubé, F.; Francastel, C. “Pocket-sized RNA-Seq”: A Method to Capture New Mature microRNA Produced from a Genomic Region of Interest. Non-Coding RNA 2015, 1, 127-138. https://doi.org/10.3390/ncrna1020127
Hubé F, Francastel C. “Pocket-sized RNA-Seq”: A Method to Capture New Mature microRNA Produced from a Genomic Region of Interest. Non-Coding RNA. 2015; 1(2):127-138. https://doi.org/10.3390/ncrna1020127
Chicago/Turabian StyleHubé, Florent, and Claire Francastel. 2015. "“Pocket-sized RNA-Seq”: A Method to Capture New Mature microRNA Produced from a Genomic Region of Interest" Non-Coding RNA 1, no. 2: 127-138. https://doi.org/10.3390/ncrna1020127
APA StyleHubé, F., & Francastel, C. (2015). “Pocket-sized RNA-Seq”: A Method to Capture New Mature microRNA Produced from a Genomic Region of Interest. Non-Coding RNA, 1(2), 127-138. https://doi.org/10.3390/ncrna1020127