

Gelation Time of Network-Forming Polymer Solutions with Reversible Cross-Link Junctions of Variable Multiplicity


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
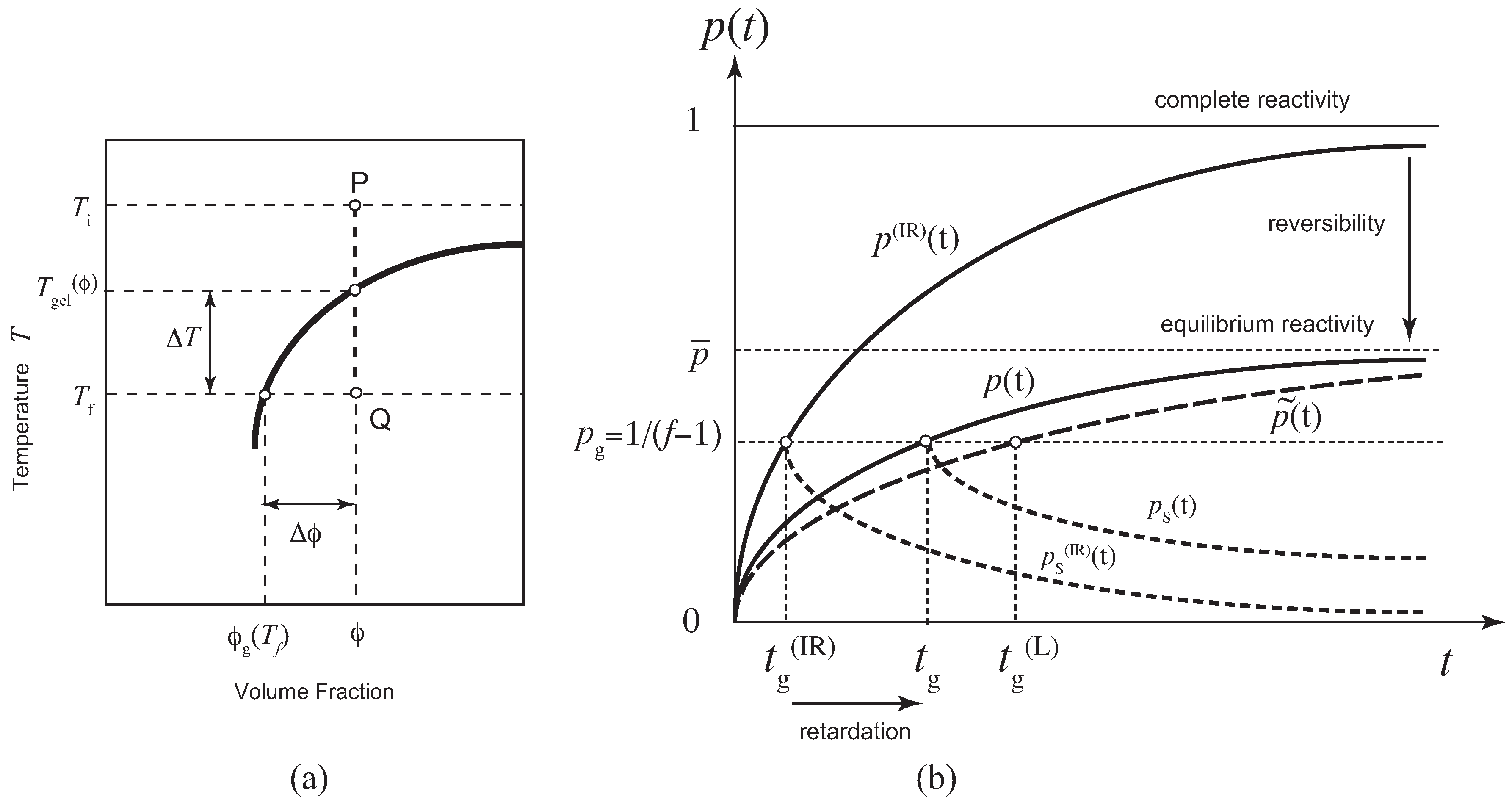
2. Theoretical Method
2.1. Pairwise Cross-Linking
2.2. Fixed-Multiplicity Model
2.3. Stepwise Association
3. Results
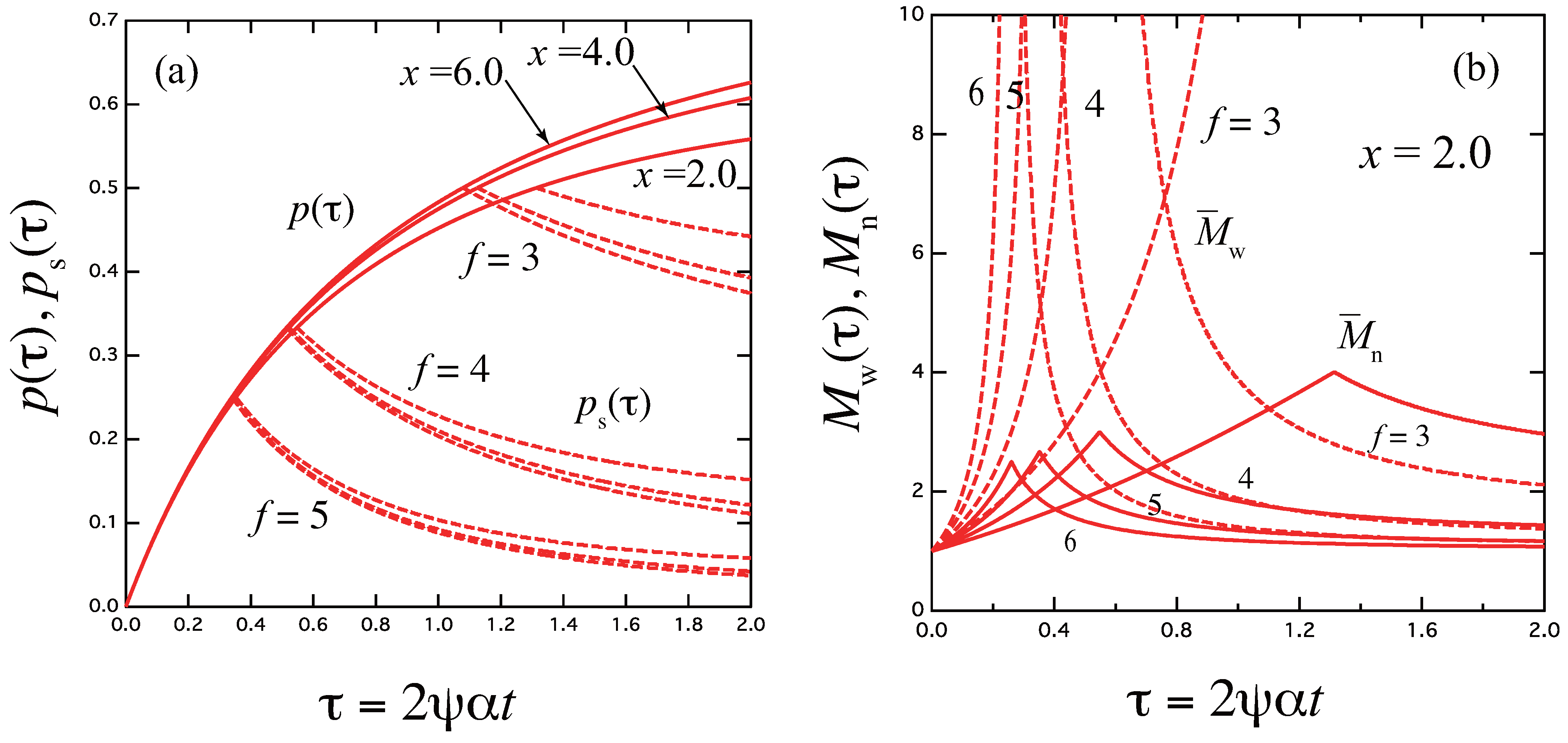
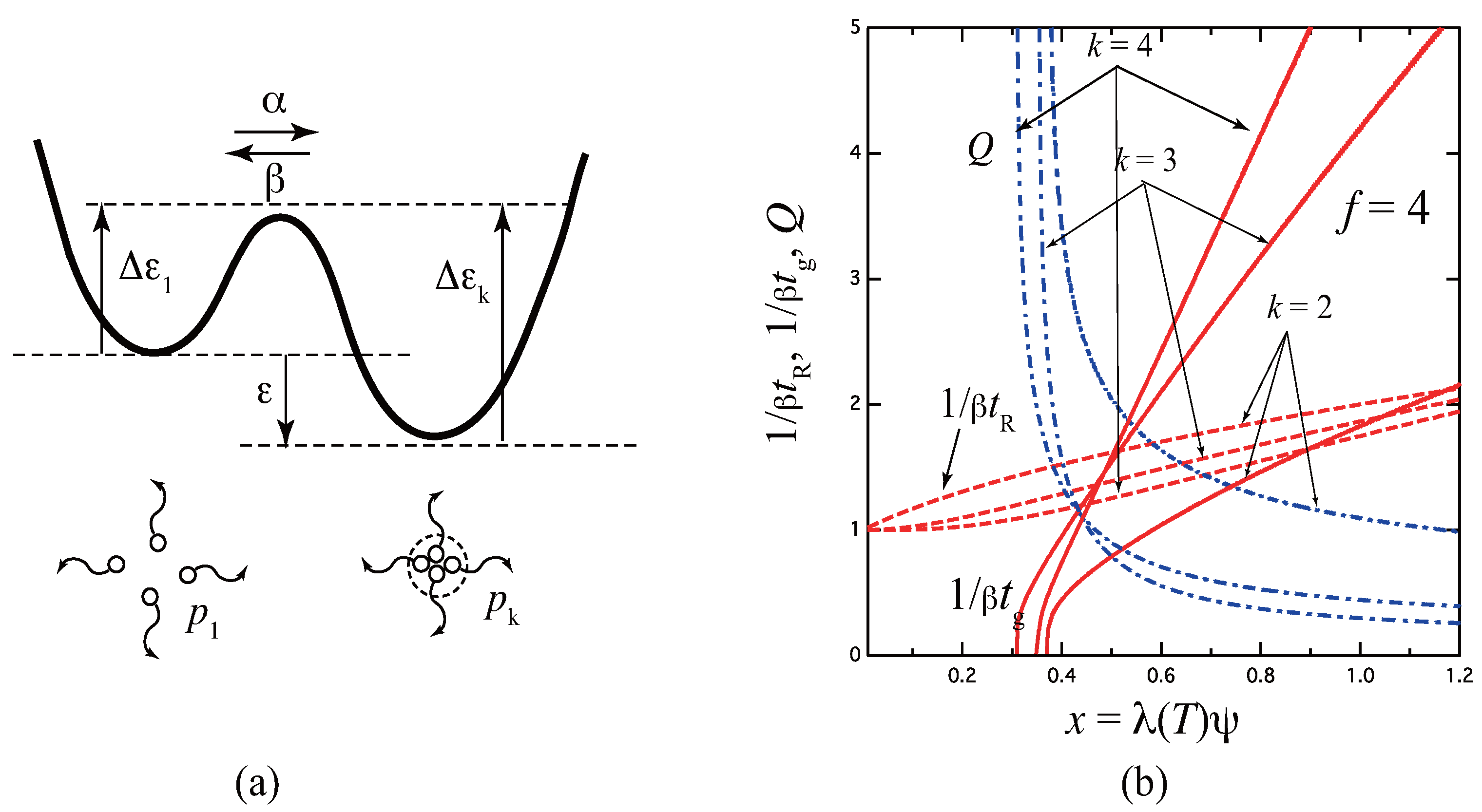
3.1. Three-State Model
3.1.1. Rate-Determining Step
3.1.2. Quasi-Stationary State Approximation
3.1.3. Slow-Mode Approximation
3.1.4. Some Numerical Results of the Relaxation Time
3.2. Micellar Cross-Linking
4. Discussion
5. Conclusions
- (1)
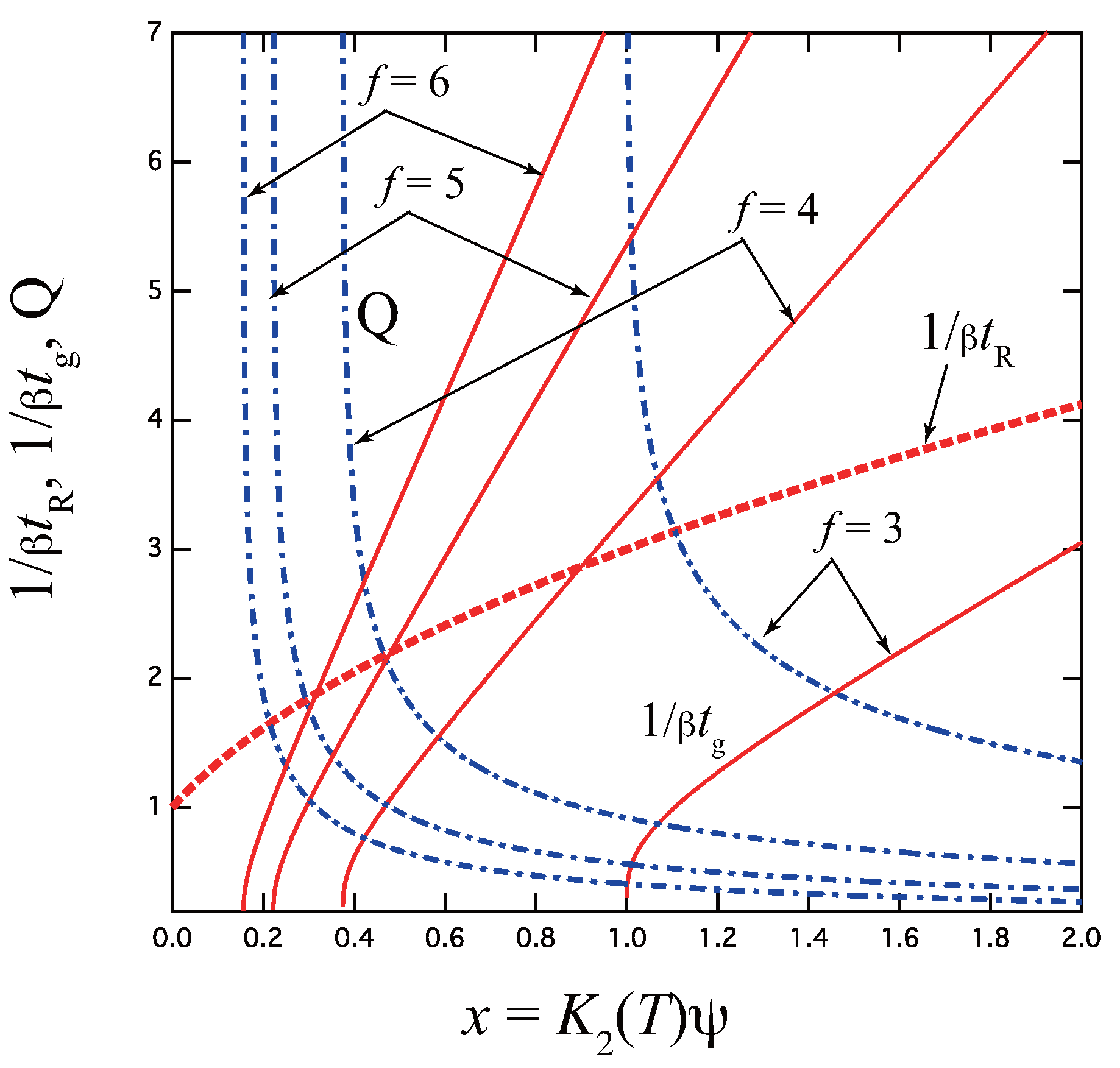
- The gelation time , the relaxation time , and the thermodynamic factor Q are all functions of a single variable (scaled concentration), where is the stepwise association constant at the final temperature T at which cross-linking reaction proceeds. Therefore, temperature and concentration are not separable, but give the same effect if they are properly scaled under a fixed value of x. Data for different concentrations can be superimposed onto a single curve by using an appropriate temperature shift factor.
- (2)
- These three factors obey a fundamental relation . The thermodynamic factor is logarithmically singular at the equilibrium gel point , while the relaxation time is continuous across the gel point. They are calculated for some important models of cross-link junctions, such as pairwise cross-linking, three-state model, cross-linking with fixed high multiplicity, and micellar cross-linking.
- (3)
- The gelation time of reversible cross-linking approaches the power law of the irreversible one in the asymptotic region of large x (either high concentration or high values of the association constant ). The power index of lies at somewhere between (simultaneous cross-linking) and (stepwise cross-linking). Hence, the reaction kinetics, simultaneous or stepwise, can be inferred by measuring the power.
- (4)
- For large micellar cross-link junctions, the gelation time is derived on the basis of the quasi-stationary approximation (Aniansson–Wall formula) for the relaxation time. Combination with the singular part of the thermodynamic factor estimated by our preceding equilibrium gelation theory provides an accurate estimation of the gelation time, and enables a comparison with experimental data.
Funding
Acknowledgments
Conflicts of Interest
Appendix A. Solution of the Kinetic Equation
Appendix B. Retardation Coefficient
Appendix C. Quasi-Stationary Approximation
References
- Guenet, J.M. Thermoreversible Gelation of Polymers and Biopolymers, 2nd ed.; Academic Press, Harcourt Brace Jovanovich Publishers: London, UK, 1992. [Google Scholar]
- te Nijenhuis, K. Thermoreversible Networks. Adv. Polym. Sci. 1997, 130, 1–252. [Google Scholar]
- Winter, H.H.; Mours, M. Rheology of Polymers near Liquid–Solid Transitions. Adv. Polym. Sci. 1997, 134, 165–234. [Google Scholar]
- Tanaka, F. Polymer Physics—Applications to Molecular Association and Thermoreversible Gelation; Cambridge University Press: Cambridge, UK, 2011. [Google Scholar]
- Zhang, J.; Hu, Y.; Li, Y. Gel Chemistry: Interactions, Structures and Properties; Springer: Singapore, 2018. [Google Scholar]
- Thakur, V.K.; Thakur, M.K. Polymer Gels: Science and Fundamentals, 1st ed.; Springer: Singapore, 2018. [Google Scholar]
- Thakur, V.K.; Thakur, M.K. Hydrogels: Recent Advances, 1st ed.; Springer: Singapore, 2018. [Google Scholar]
- Nakano, S.; Ogiso, T.; Kita, R.; Shinyashiki, N.; Yagihara, S.; Yoneyama, M.; Katsumoto, Y. Thermoreversible gelation of isotactic-rich poly(?it N-isopropylacrylamide) in water. J. Chem. Phys. 2011, 135, 114903. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Hashimoto, T.; Chuang, Y.-C.; Tanaka, T.; Chang, Y.-P.; Yang, T.-W.; Huang, M.-T. Physical Gelation of Aqueous Solutions of Atactic Poly(N-isopropylacrylamide). Macromolecules 2022, 55, 9152–9167. [Google Scholar] [CrossRef]
- Taylor, M.L.; Paul Tomlins, P.; Sahota, T.S. Thermoresponsive Gels. Gels 2017, 3, 4. [Google Scholar] [CrossRef]
- Zhang, K.; Xue, K.; Loh, X.J. Thermo-Responsive Hydrogels: From Recent Progress to Biomedical Applications. Gels 2021, 7, 77. [Google Scholar] [CrossRef]
- Patrickios, C.S. Amphiphilic Polymer Co-Networks: Synthesis, Properties, Modelling and Applications; Royal Society of Chemistry: London, UK, 2020. [Google Scholar]
- Weiss, R.G.; Terech, P. Molecular Gels: Materials with Self-Assembled Fibrillar Networks; Springer: Dordrecht, The Netherlands, 2006; p. 978. [Google Scholar]
- Weiss, R.G. Controlling Variables in Molecular Gel Science: How Can We Improve the State of the Art? Gels 2018, 4, 25. [Google Scholar] [CrossRef]
- Morris, J.; Bietsch, J.; Bashaw, K.; Wang, G. Recently Developed Carbohydrate Based Gelators and Their Applications. Gels 2021, 7, 24. [Google Scholar] [CrossRef]
- Ohkura, M.; Kanaya, T.; Kaji, K. Gels of poly(vinyl alcohol) from dimethyl sulphoxide/water solutions. Polymer 1992, 33, 3686–3690. [Google Scholar] [CrossRef]
- Ohkura, M.; Kanaya, T.; Kaji, K. Gelation rates of poly(vinyl alcohol) solution. Polymer 1992, 33, 5044–5048. [Google Scholar] [CrossRef]
- Mal, S.; Maiti, P.; Nandi, A.K. On the Gelation Rates of Thermoreversible Poly(vinylidene fluoride) gels. Macromolecules 1995, 28, 2371–2376. [Google Scholar] [CrossRef]
- Hong, P.-D.; Chou, C.-M. Phase separation and gelation behaviors in poly(vinylidene fluoride)/tetra(ethylene glycol) dimethyl ether solutions. Polymer 2000, 41, 8311–8320. [Google Scholar] [CrossRef]
- Tobitani, A.; Ross-Murphy, S.B. Heat-Induced Gelation of Globular Proteins. 1. Model for the Effects of Time and Temperature on the Gelation Time of BSA Gels. Macromolecules 1997, 30, 4845–4854. [Google Scholar] [CrossRef]
- Tobitani, A.; Ross-Murphy, S.B. Heat-Induced Gelation of Globular Proteins. 2. Effect of Environmental Factors on Single-Component and Mixed-Protein Gels. Macromolecules 1997, 30, 4855–4862. [Google Scholar] [CrossRef]
- Fukui, K.; Yamabe, T. A General Theory of Gel Formation with Multifunctional Interunit Junctions. Bull. Chem. Soc. Jpn 1967, 40, 2052–2063. [Google Scholar] [CrossRef]
- Tanaka, F.; Stockmayer, W.H. Thermoreversible Gelation with Junctions of Variable Multiplicity. Macromolecules 1994, 27, 3943–3954. [Google Scholar] [CrossRef]
- Tanaka, F. Thermoreversible Gelation Interfering with Phase Separation in Multicomponent Mixtures of Associating Polymers. Macromolecules 2022, 55, 5233–5248. [Google Scholar] [CrossRef]
- Madbouly, S.A.; Otaigbe, J.U. Kinetic Analysis of Fractal Gel Formation in Waterborne Polyurethane Dispersions Undergoing High Deformation Flows. Macromolecules 2006, 39, 4144–4151. [Google Scholar] [CrossRef]
- Ponton, A.; Griesmar, P.; Barboux-Doeuff, S.; Sanchez, C. Rheological investigation of the sol–gel transition: Effect of hydrolysis variation in silicon oxide and titanium oxide based on matrices. J. Mater. Chem. 2001, 11, 3125–3129. [Google Scholar] [CrossRef]
- Ponton, A.; Warlus, S.; Griesmar, P. Rheological Study of Sol-Gel Transition in Silica Alkoxides. J. Colloid Interface Sci. 2002, 249, 209–216. [Google Scholar] [CrossRef]
- Zhou, J.-F.; Johari, G.P. Gelation Time during Polymerization by Ultrasonic Shear Waves Propagation. Macromolecules 1997, 30, 8085–8087. [Google Scholar] [CrossRef]
- Normand, V.; Muller, S.; Ravey, J.C.; Parker, A. Gelation Kinetics of Gelatin: A Master Curve and Network Modeling. Macromolecules 2000, 33, 1063–1071. [Google Scholar] [CrossRef]
- Sperinde, J.J.; Griffith, L.G. Control and Prediction of Gelation Kinetics in Enzymatically Cross-Linked Poly(ethylene glycol) Hydrogels. Macromolecules 2000, 33, 5476–5480. [Google Scholar] [CrossRef]
- Saalwachter, K.; Gottlieb, M.; Liu, R.; Oppermann, W. Gelation as Studied by Proton Multiple-Quantum NMR. Macromolecules 2007, 40, 1555–1561. [Google Scholar] [CrossRef]
- Kurakazu, M.; Takuya Katashima, T.; Chijiishi, M.; Nishi, K.; Akagi, Y.; Matsunaga, T.; Shibayama, M.; Chung, U.; Sakai, T. Evaluation of Gelation Kinetics of Tetra-PEG Gel. Macromolecules 2010, 43, 3935–3940. [Google Scholar] [CrossRef]
- Stockmayer, W.H. Theory of Molecular Size Distribution and Gel Formation in Branched-Chain Polymers. J. Chem. Phys. 1943, 11, 45–55. [Google Scholar] [CrossRef]
- Stockmayer, W.H. Theory of Molecular Size Distribution and Gel Formation in Branched Polymers II. General Cross Linking. J. Chem. Phys. 1944, 12, 125–131. [Google Scholar] [CrossRef]
- Ziff, R.M.; Stell, G. Kinetics of Polymer Gelation. J. Chem. Phys. 1980, 73, 3492–3499. [Google Scholar] [CrossRef]
- Ziff, R.M. Kinetics of Polymerization. J. Stat. Phys. 1980, 23, 241–263. [Google Scholar] [CrossRef]
- Ziff, R.M.; Hendriks, E.M.; Ernst, M.H. Critical Properties for Gelation: A Kinetic Approarch. Phys. Rev. Lett. 1982, 49, 593–595. [Google Scholar] [CrossRef]
- Ziff, R.M.; Ernst, M.H.; Hendriks, E.M. Kinetics of Gelation and Universality. J. Phys. A: Math. Gen. 1983, 16, 2293–2320. [Google Scholar] [CrossRef]
- van Dongen, P.G.J.; Ernst, M.H. Kinetics of Reversible Polymerization. J. Stat. Phys. 1984, 37, 301–324. [Google Scholar] [CrossRef]
- Flory, P.J. Principles of Polymer Chemistry; Cornell University Press: Ithaca, NY, USA, 1953. [Google Scholar]
- Flory, P.J. Molecular Size Distribution in Three Dimensional Polymers I. Gelation. J. Am. Chem. Soc. 1941, 63, 3083–3090. [Google Scholar] [CrossRef]
- Flory, P.J. Molecular Size Distribution in Three Dimensional Polymers II. Trifunctional Branching Units. J. Am. Chem. Soc. 1941, 63, 3091–3096. [Google Scholar] [CrossRef]
- Flory, P.J. Molecular Size Distribution in Three Dimensional Polymers III. Tetrafunctional Branching Units. J. Am. Chem. Soc. 1941, 63, 3096–3100. [Google Scholar] [CrossRef]
- Kresheck, G.C.; Hamori, E.; Davenport, G.; Scheraga, H.A. Determination of the Dissociation Rate of Dodecylpyridinium Iodide Micelles by a Temperature-Jump Technique18. J. Am. Chem. Soc. 1966, 88, 246–253. [Google Scholar] [CrossRef]
- Muller, N. Kinetics of Micelle Dissociation by Temperature-Jump Techniques. A Reinterpretation. J. Phys. Chem. 1972, 70, 3017–3020. [Google Scholar] [CrossRef]
- Aniansson, E.A.; Wall, S.N. On the Kinetics of Step-Wise Micelle Association. J. Phys. Chem. 1974, 78, 1024–1030. [Google Scholar] [CrossRef]
- Aniansson, E.A.; Wall, S.N.; Almgren, M. Theory of the Kinetics of Micellar Equilibria and Quantitative Interpretation of Chemical Relaxation Studies of Micellar Solutions of Ionic Surfactants. J. Phys. Chem. 1976, 80, 905–922. [Google Scholar] [CrossRef]
- Patist, A.; Oh, S.G.; Leung, R.; Shah, D.O. Kinetics of micellization: Its significance to technological processes. Colloids Surf. A Physicochem. Eng. Asp. 2001, 176, 3–16. [Google Scholar] [CrossRef]
- Clark, A.H.; Ross-Murphy, S.B. Structural and Mechanical Properties of Biopolymer Gels. Adv. Polym. Sci. 1987, 83, 57–192. [Google Scholar]
- Djabourov, M.; Nishinari, K.; Ross-Murphy, S.B. Physical Gels from Biological and Synthetic Polymers; Cambridge University Press: New York, NY, USA, 2013. [Google Scholar]
- Annable, T.; Buscall, R.; Ettelaie, R.; Whittlestone, D. The Rheology of Solutions of Associating Polymers: Comparison of Experimental Behavior with Transient Network Theory. J. Rheol. 1993, 37, 695–726. [Google Scholar] [CrossRef]
- Annable, T.; Buscall, R.; Ettelaie, R.; Shepherd, P.; Whittlestone, D. Influence of Surfactants on the Rheology of Associating Polymers in Solution. Langmuir 1994, 10, 1060–1070. [Google Scholar] [CrossRef]
- Yekta, A.; Xu, B.; Duhamel, J.; Adiwidjaja, H.; Winnik, M.A. Fluorescence Studies of Associating Polymers in Water: Determination of the Chain End Aggregation Number and a Model for the Association Process. Macromolecules 1995, 28, 956–966. [Google Scholar] [CrossRef]
- Kujawa, P.; Watanabe, H.; Tanaka, F.; Winnik, F.M. Amphiphhilic Telechelic Poly(?it N-isopropylacrylamide) in Water: From Micelles to Gels. Eur. Phys. J. E 2005, 17, 129–137. [Google Scholar] [CrossRef]
- Kujawa, P.; Segui, F.; Shaban, S.; Diab, C.; Okada, Y.; Tanaka, F.; Winnik, F.M. Impact of End-Group Association and Main-Chain Hydration on the Thermosensitive Properties of Hydrophobically Modified Telechelic Poly(?it N-isopropylacrylamide) in Water. Macromolecules 2006, 39, 341–348. [Google Scholar] [CrossRef]
- Kujawa, P.; Tanaka, F.; Winnik, F.M. Temperature-Dependent Properties of Telechelic Hydrophobically Modified Poly(?it N-isopropylacrylamides) in Water: Evidence from Light Scattering and Fluorescence Spectroscopy for the Formation of Stable Mesoglobules at Elevated Temperatures. Macromolecules 2006, 39, 3048–3055. [Google Scholar] [CrossRef]
- Kujawa, P.; Aseyev, V.; Tenhu, H.; Winnik, F.M. Temperature-Sensitive Properties of Poly(?it N-isopropylacrylamides) Mesoglobules Formed in Dilute Aqueous Solutions Heated above Their Demixing Point in Water: Evidence from Light Scattering and Fluorescence Spectroscopy for the Formation of Stable Mesoglobules at Elevated Temperatures. Macromolecules 2006, 39, 7686–7693. [Google Scholar]
- Quellet, C.; Eicke, H.-F.; Xu, G.; Hauger, Y. Transient Networks in ABA Block Copolymer-Microemulsion Systems. Macromolecules 1990, 23, 3347–3352. [Google Scholar] [CrossRef]
- Mortensen, K.; Brown, W.; Norden, B. Inverse Melting Transition and Evidence of Three-Dimentional Cubatic Structure in a Block-Copolymer Miceller System. Phys. Rev. Lett. 1992, 68, 2340–2343. [Google Scholar] [CrossRef]
- Odenwald, M.; Eicke, H.-F.; Meier, W. Transient Networks in ABA Block Copolymers and Microemulsions: A Rheological Study. Macromolecules 1995, 28, 5069–5074. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tanaka, F. Gelation Time of Network-Forming Polymer Solutions with Reversible Cross-Link Junctions of Variable Multiplicity. Gels 2023, 9, 379. https://doi.org/10.3390/gels9050379
Tanaka F. Gelation Time of Network-Forming Polymer Solutions with Reversible Cross-Link Junctions of Variable Multiplicity. Gels. 2023; 9(5):379. https://doi.org/10.3390/gels9050379
Chicago/Turabian StyleTanaka, Fumihiko. 2023. "Gelation Time of Network-Forming Polymer Solutions with Reversible Cross-Link Junctions of Variable Multiplicity" Gels 9, no. 5: 379. https://doi.org/10.3390/gels9050379
APA StyleTanaka, F. (2023). Gelation Time of Network-Forming Polymer Solutions with Reversible Cross-Link Junctions of Variable Multiplicity. Gels, 9(5), 379. https://doi.org/10.3390/gels9050379