
Glyceraldehyde as an Efficient Chemical Crosslinker Agent for the Formation of Chitosan Hydrogels



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Shear Viscosity Measurements and Entanglements
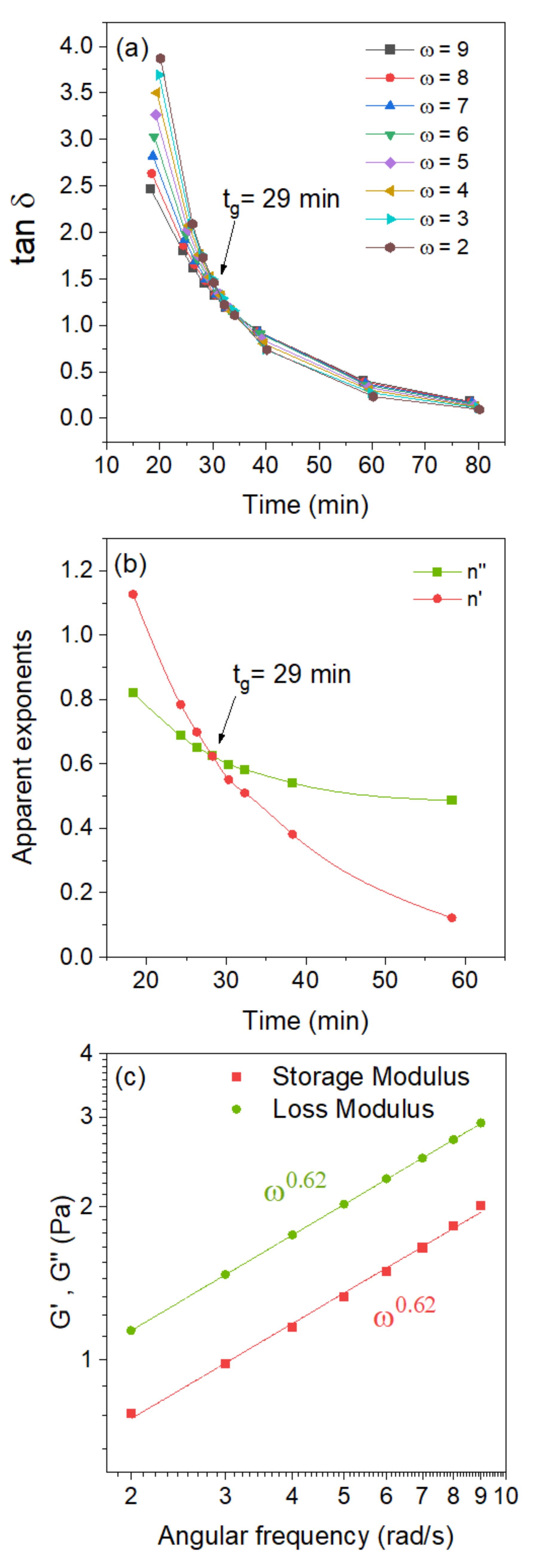
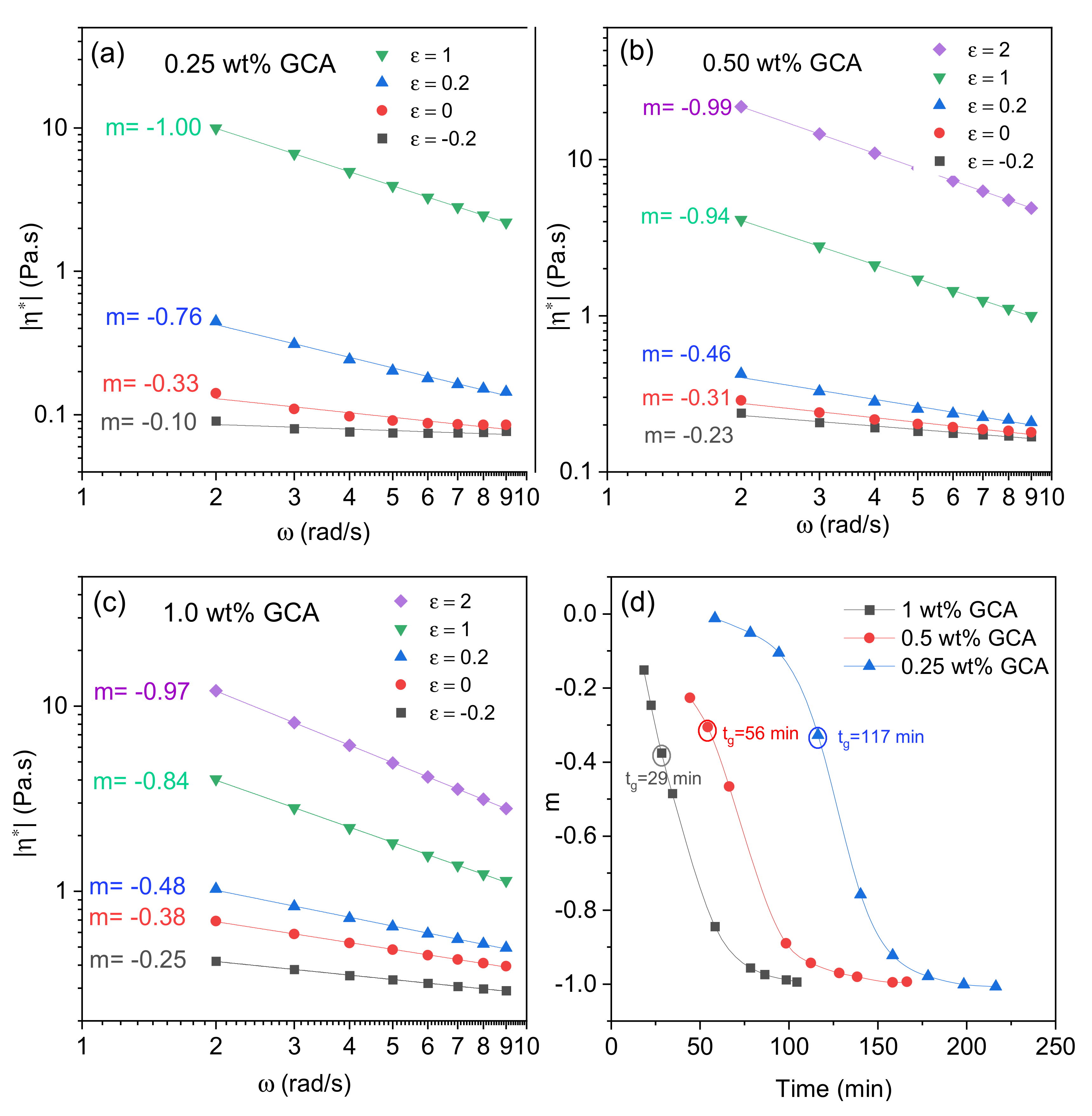
2.2. Gelation of Chitosan in the Presence of Glyceraldehyde
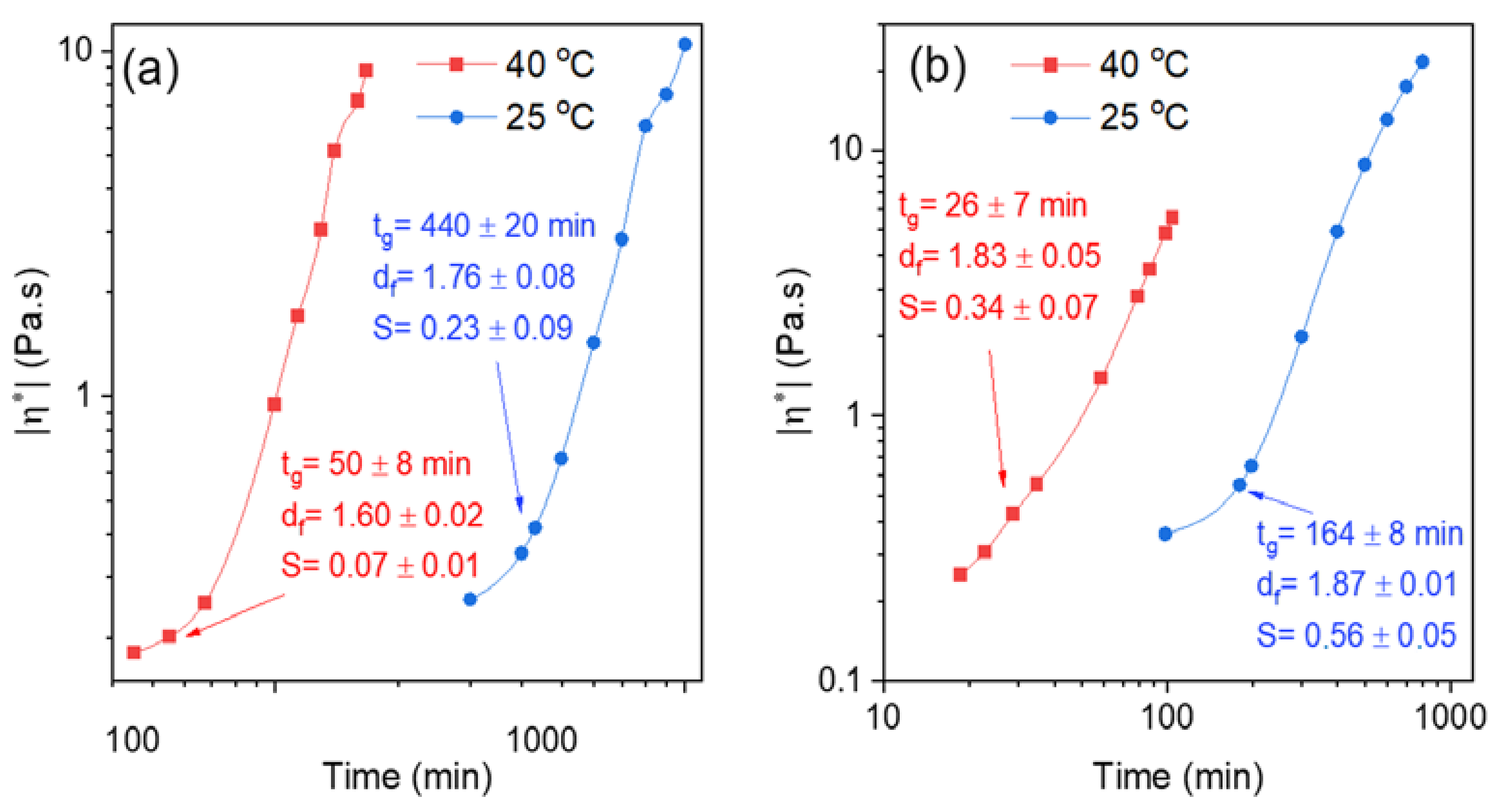
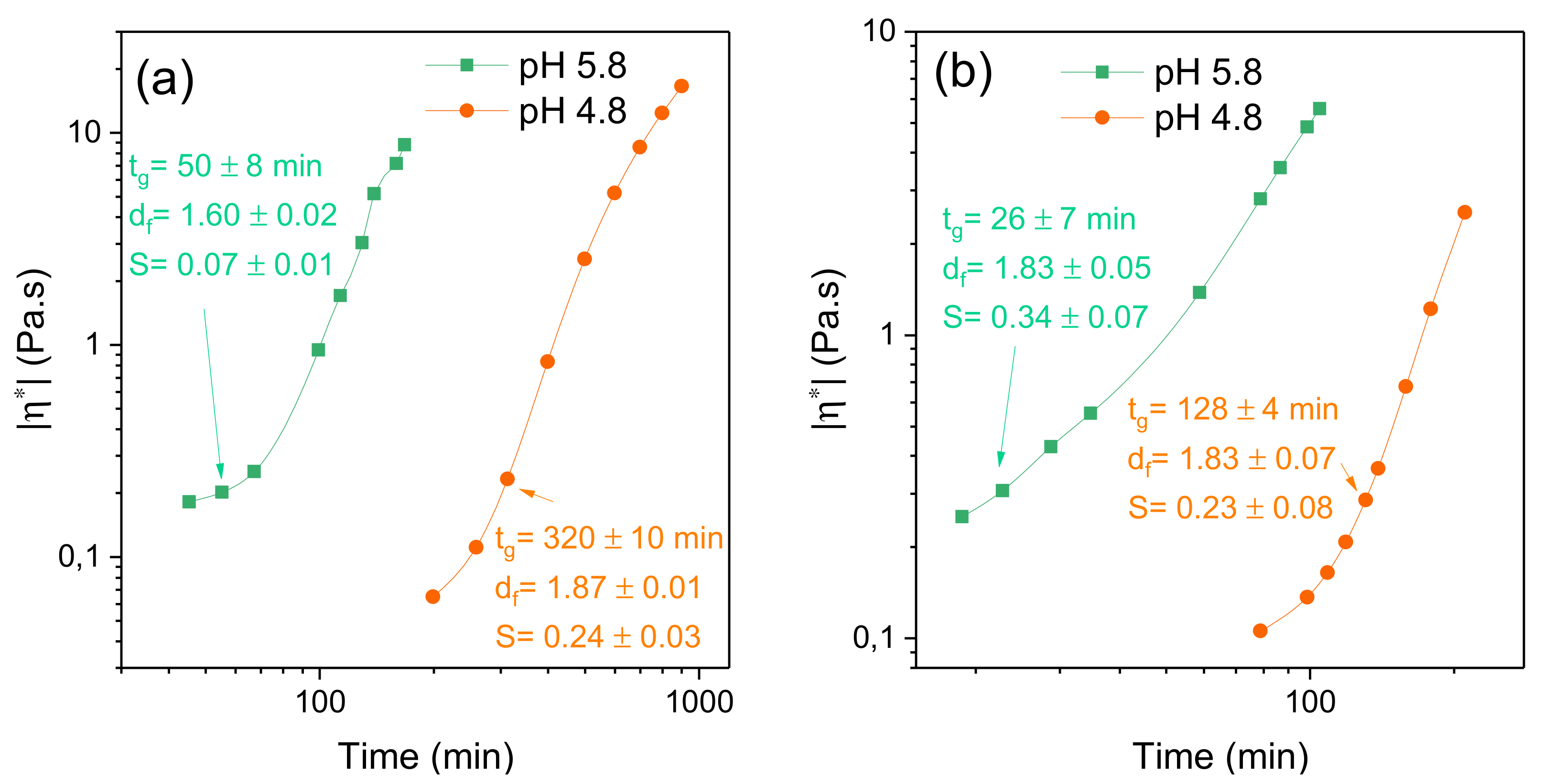
2.3. Effects of Temperature and pH on the Gelation Features
2.4. Effect of GCA on the Mesh Size of Mature Gels
2.5. Comparison of Gel Formation of Chitosan with Different Crosslinker Agents
3. Conclusions
4. Materials and Methods
4.1. Materials and Preparation of Gels
4.2. Rheology Experiments
4.3. Small Angle Neutron Scatering (SANS) Experiments
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Slaughter, B.V.; Khurshid, S.S.; Fisher, O.Z.; Khademhosseini, A.; Peppas, N.A. Hydrogels in regenerative medicine. Adv. Mater. 2009, 21, 3307–3329. [Google Scholar] [CrossRef] [Green Version]
- Kirschner, C.M.; Anseth, K.S. Hydrogels in healthcare: From static to dynamic material microenvironments. Acta Mater. 2013, 61, 931–944. [Google Scholar] [CrossRef] [Green Version]
- Muzzarelli, R.A.A.; Jeuniaux, C.; Gooday, G.W. Chitin in Nature and Technology; Plenum Press: New York, NY, USA, 1986. [Google Scholar]
- Hamedi, H.; Moradi, S.; Hudson, S.M.; Tonelli, A.E. Chitosan based hydrogels and their applications for drug delivery in wound dressings: A review. Carbohydr. Polym. 2018, 199, 445–460. [Google Scholar] [CrossRef] [PubMed]
- Struszczyk, H.; Wawro, D.; Niekraszewicz, A. Biodegradability of chitosan fibers. In Advances in Chitin and Chitosan; Elsevier: London, UK, 1991; pp. 580–585. [Google Scholar]
- Milosavljević, N.B.; Ristić, M.D.; Perić-Grujić, A.A.; Filipović, J.M.; Štrbac, S.B.; Rakočević, Z.L.; Krušić, M.T.K. Sorption of zinc by novel pH-sensitive hydrogels based on chitosan, itaconic acid and methacrylic acid. J. Hazard. Mater. 2011, 192, 846–854. [Google Scholar] [CrossRef]
- Gonçalves, J.O.; Santos, J.P.; Rios, E.C.; Crispim, M.M.; Dotto, G.L.; Pinto, L.A.A. Development of chitosan based hybrid hydrogels for dyes removal from aqueous binary system. J. Mol. Liq. 2017, 225, 265–270. [Google Scholar] [CrossRef]
- Delmar, K.; Bianco-Peled, H. Composite chitosan hydrogels for extended release of hydrophobic drugs. Carbohydr. Polym. 2016, 136, 570–580. [Google Scholar] [CrossRef] [PubMed]
- Sadeghi, M.; Hanifpour, F.; Taheri, R.; Javadian, H.; Ghasemi, M. Comparison of using formaldehyde and carboxy methylchitosan in preparation of Fe3O4 superparamagnetic nanoparticles-chitosan hydrogel network: Sorption behavior toward bovine serum albumin. Process Saf. Environ. Prot. 2016, 102, 119–128. [Google Scholar] [CrossRef]
- Jóźwiak, T.; Filipkowska, U.; Szymczyk, P.; Rodziewicz, J.; Mielcarek, A. Effect of ionic and covalent crosslinking agents on properties of chitosan beads and sorption effectiveness of reactive black 5 dye. React. Funct. Polym. 2017, 114, 58–74. [Google Scholar] [CrossRef]
- Alhwaige, A.A.; Ishida, H.; Qutubuddin, S. Poly(benzoxazine-f-chitosan) films: The role of aldehyde neighboring groups on chemical interaction of benzoxazine precursors with chitosan. Carbohydr. Polym. 2019, 209, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Mi, F.L.; Sung, H.W.; Shyu, S.S. Synthesis and characterization of a novel chitosan-based network prepared using naturally occurring crosslinker. J. Polym. Sci. A 2000, 38, 2804–2814. [Google Scholar] [CrossRef]
- Liang, H.-C.; Chang, Y.; Hsu, C.-K.; Lee, M.-H.; Sung, H.-W. Effects of crosslinking degree of an acellular biological tissue on its tissue regeneration pattern. Biomaterials 2004, 25, 3541–3552. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.F.; Ng, Y.-F.; Pudney, P.D.A. Mechanism and kinetics of the crosslinking reaction between biopolymers containing primary amine groups and genipin. J. Polym. Sci. A 2003, 41, 3941–3953. [Google Scholar] [CrossRef]
- Dimida, S.; Demitri, C.; De Benedictis, V.M.; Scalera, F.; Gervaso, F.; Sannino, A. Genipin-cross-linked chitosan-based hydrogels: Reaction kinetics and structure-related characteristics. J. Appl. Polym. Sci. 2015, 132, 42256/1–42256/8. [Google Scholar] [CrossRef]
- Tessier, F.J.; Monnier, V.M.; Sayre, L.M.; Kornfield, J.A. Triosidines: Novel Maillard reaction products and cross-links from the reaction of triose sugars with lysine and arginine residues. Biochem. J. 2003, 369, 705–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.; Takaoka, A.; Hoang, Q.V.; Trokel, S.L.; Paik, D.C. Pharmacologic alternatives to riboflavin photochemical cornealcross-linking: A comparison study of cell toxicity thresholds. Investig. Ophthalmol. Vis. Sci. 2014, 55, 3247–3257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerrard, J.A.; Brown, P.K.; Fayle, S.E. Crosslinking of food proteins. I: The reaction of glutaraldehyde, formaldehyde and glyceraldehyde with ribonuclease. Food Chem. 2002, 79, 343–349. [Google Scholar] [CrossRef]
- Caillard, R.; Remondetto, G.E.; Mateescu, M.A.; Subirade, M. Characterization of amino cross-linked soy protein hydrogels. J. Food Sci. 2008, 73, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Caillard, R.; Remondetto, G.E.; Subirade, M. Rheological investigation of soy protein hydrogels induced by Maillard-type reaction. Food Hydrocoll. 2010, 24, 81–87. [Google Scholar] [CrossRef]
- Conti, B.; Modena, T.; Genta, E.; Perugini, P.; Pavanetto, F.A. Proposed new method for the crosslinking of chitosan micro spheres. Drug Deliv. 1998, 5, 87–93. [Google Scholar] [CrossRef]
- De Gennes, P.-G. Scaling Concepts in Polymer Physics; Cornell University Press: Ithaca, NY, USA, 1979. [Google Scholar]
- Simha, R.; Zakin, J.L. Compression of flexible chain molecules in solution. J. Chem. Phys. 1960, 33, 1791–1793. [Google Scholar] [CrossRef]
- Larson, R.G. The Structure and Rheology of Complex Fluids; Oxford University Press: New York, NY, USA, 1999. [Google Scholar]
- Desbrieres, J. Viscosity of semiflexible chitosan solutions: Influence of concentration, temperature, and role of intermolecular interactions. Biomacromolecules 2002, 3, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Iversen, C.; Kjøniksen, A.-L.; Nyström, B.; Nakken, T.; Palmgren, O.; Tande, T. Linear and nonlinear rheological responses in aqueous systems of hydrophobically modified chitosan and its unmodified analogue. Polym. Bull. 1997, 39, 747–754. [Google Scholar] [CrossRef]
- Yan, H.; Jin, B. Influence of microstructural parameters on mechanical behavior of polymer gels. Int. J. Solids Struct. 2012, 49, 436–444. [Google Scholar] [CrossRef] [Green Version]
- Masubuchi, Y.; Uneyama, T. Retardation of reaction kinetics of polymers due to entanglement in post-gel stage in multi-chain slip-spring simulations. Soft Matter 2019, 15, 5109–5115. [Google Scholar] [CrossRef] [PubMed]
- Nyström, B.; Kjøniksen, A.-L.; Beheshti, N.; Maleki, A.; Zhu, K.; Knudsen, K.D.; Pamies, R.; Cifre, J.G.H.; de la Torre, J.G. Characterization of polyelectrolyte features in polysaccharide systems and mucin. Adv. Colloid Interface Sci. 2010, 158, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Cui, W.; Eskin, N.A.M.; Goff, H.D. Stress relaxation in synergistically associated polysaccharides: Galactomananns and a non-pectic polysaccharide fraction from yellow mustard mucilage. Carbohydr. Polym. 2011, 84, 984–989. [Google Scholar] [CrossRef]
- Lopez, C.G. Entanglement properties of polyelectrolytes in salt-free and excess-salt solutions. ACS Macro Lett. 2019, 8, 979–983. [Google Scholar] [CrossRef]
- Lopez, C.G. Entanglement of semiflexible polyelectrolytes: Crossover concentrations and entanglement density of sodium carboxymethyl cellulose. J. Rheol. 2020, 64, 191–204. [Google Scholar] [CrossRef]
- Raspaud, E.; Lairez, D.; Adam, M. On the number of blobs per entanglement in semidilute and good solvent solution: Melt influence. Macromolecules 1995, 23, 927–933. [Google Scholar] [CrossRef]
- Brochard, F. Gel-like modes of polymer solutions in “θ” solvents. J. Phys. 1983, 44, 39–43. [Google Scholar] [CrossRef]
- Adam, M.; Delsanti, M. Viscosity and longest relaxation time of semi-dilute polymer solutions: II. Theta Solvent J. Phys. 1984, 45, 1513–1521. [Google Scholar]
- Colby, R.H.; Rubinstein, M. Two-parameter scaling for polymers in θ solvents. Macromolecules 1990, 23, 2753–2767. [Google Scholar] [CrossRef]
- Colby, R.H. Structure and linear viscoelasticity of flexible polymer solutions: Comparison of polyelectrolyte and neutral polymer solutions. Rheol. Acta 2010, 49, 425–442. [Google Scholar] [CrossRef]
- Rubinstein, M.; Colby, R.H.; Dobrynin, A.V. Dynamics of semidilute polyelectrolyte solutions. Phys. Rev. Lett. 1994, 73, 2776–2779. [Google Scholar] [CrossRef]
- Dobrynin, A.V.; Colby, R.H.; Rubinstein, M. Scaling theory of polyelectrolyte solutions. Macromolecules 1995, 28, 1859–1871. [Google Scholar] [CrossRef]
- Pinder, D.N.; Swanson, A.J.; Hebraud, P.; Hemar, Y. Micro-rheological investigations of dextran solutions using diffusing wave spectroscopy. Food Hydrocoll. 2006, 20, 240–244. [Google Scholar] [CrossRef]
- Ganesan, M.; Knier, S.; Younger, J.G.; Solomon, M.J. Associative and entanglement contributions to the solution rheology of a bacterial polysaccharide. Macromolecules 2016, 49, 8313–8321. [Google Scholar] [CrossRef]
- Del Giudice, F.; Tassieri, M.; Oelschlaeger, C.; Shen, A.Q. When microrheology, bulk rheology, and microfluidics meet: Broadband rheology of hydroxyethyl cellulose water solutions. Macromolecules 2017, 50, 2951–2963. [Google Scholar] [CrossRef] [Green Version]
- Lopez, C.G.; Voleske, L.; Richtering, W. Scaling laws of entangled polysaccharides. Carbohydr. Polym. 2020, 234, 115886. [Google Scholar] [CrossRef]
- Xu, X.; Liu, W.; Zhang, L. Rheological behavior of aeromonas gum in aqueous solutions. Food Hydrocoll. 2006, 20, 723–729. [Google Scholar] [CrossRef]
- Chambon, F.; Winter, H.H. Linear viscoelasticity at the gel point of a crosslinking PDMS with imbalanced stoichiometry. J. Rheol. 1987, 31, 683–697. [Google Scholar] [CrossRef]
- Winter, H.H. Can the gel point of a crosslinking polymer be detected by the G’-G" crossover? Polym. Eng. Sci. 1987, 27, 1698–1702. [Google Scholar] [CrossRef]
- Scanlan, J.C.; Winter, H.H. Composition dependence of the viscoelasticity of end-linked poly(dimethylsiloxane) at the gel point. Macromolecules 1991, 24, 47–54. [Google Scholar] [CrossRef]
- Hodgson, D.F.; Amis, E.J. Dynamic viscoelasticity during sol-gel reactions. J. Non-Cryst. Solids 1991, 131, 913–920. [Google Scholar] [CrossRef]
- Muthukumar, M. Screening effect on viscoelasticity near the gel point. Macromolecules 1989, 22, 4656–4658. [Google Scholar] [CrossRef]
- Kenari, H.S.; Imani, M.; Dashtimoghadam, E.; Maleki, A.; Nyström, B.; Nodehi, A. Oscillatory rheometric tracing of dextran crosslinking reaction in aqueous semidilute solutions–effects of formulation on the gelation properties. Polymer 2013, 54, 2999–3007. [Google Scholar] [CrossRef]
- Kenari, H.S.; Mollaie, F.; Dashtimoghadam, E.; Imani, M.; Nyström, B. Effects of chain length of the cross-linking agent on rheological and swelling characteristics of dextran hydrogels. Carbohydr. Polym. 2018, 181, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Winter, H.H. Transient networks –evolution of rheology during chemical gelation. Prog. Colloid Polym. Sci. 1987, 75, 104–110. [Google Scholar] [CrossRef]
- Martin, J.E.; Adolf, D.; Wilcoxon, J.P. Viscoelasticity of near-critical gels. Phys. Rev. Lett. 1988, 61, 2620–2623. [Google Scholar] [CrossRef]
- Durand, D.; Delsanti, M.; Adam, M.; Luck, J.M. Frequency dependence of viscoelastic properties of branched polymers near gelation threshold. Europhys. Lett. 1987, 3, 297–301. [Google Scholar] [CrossRef]
- Rubinstein, M.; Colby, R.H.; Gillmor, J.R. Dynamic scaling for polymer gelation. Am. Chem. Soc. Polym. Prepr. 1989, 30, 81–82. [Google Scholar]
- Alexander, S. Is the elastic energy of amorphous materials rotationally invariant? J. Phys. 1984, 45, 1939–1945. [Google Scholar] [CrossRef]
- Izuka, A.; Winter, H.H.; Hashimoto, T. Molecular weight dependence of viscoelasticity of polycaprolactone critical gels. Macromolecules 1992, 25, 2422–2428. [Google Scholar] [CrossRef]
- Izuka, A.; Winter, H.H.; Hashimoto, T. Temperature dependence of viscoelasticity of polycaprolactone critical gels. Macromolecules 1994, 27, 6883–6888. [Google Scholar] [CrossRef] [Green Version]
- Higham, A.K.; Garber, L.A.; Latshaw, D.C.; Hall, C.K.; Pojman, J.A.; Khan, S.A. Gelation and cross-linking in multifunctional thiol and multifunctional acrylate systems involving an in situ comonomer catalyst. Macromolecules 2014, 47, 821–829. [Google Scholar] [CrossRef]
- Nyström, B.; Kjøniksen, A.-L.; Iversen, C. Characterization of association phenomena in aqueous systems of chitosan of different hydrophobicity. Adv. Colloid Interface Sci. 1999, 79, 81–103. [Google Scholar] [CrossRef]
- Hamdine, M.; Heuzey, M.-C.; Bégin, A. Viscoelastic properties of phosphoric and oxalic acid-based chitosan hydrogels. Rheol. Acta 2006, 45, 659–675. [Google Scholar] [CrossRef]
- Kjøniksen, A.-L.; Nyström, B. Effects of polymer concentration and cross-linking density on rheology of chemically cross-linked poly(vinyl alcohol) near gelation threshold. Macromolecules 1996, 29, 5215–5222. [Google Scholar] [CrossRef]
- Maleki, A.; Kjøniksen, A.-L.; Knudsen, K.D.; Nyström, B. Dynamical and structural behaviors of hydroxyethylcellulose hydrogels obtained by chemical gelation. Polym. Int. 2006, 55, 365–374. [Google Scholar] [CrossRef]
- Raghavan, S.R.; Chen, L.A.; McDowell, C.; Khan, S.A.; Hwang, R.; White, S. Rheological study of cross-linking and gelation in chlorobutyl elastomer systems. Polymer 1996, 37, 5869–5875. [Google Scholar] [CrossRef]
- Sandolo, C.; Matricardi, P.; Alhaique, F.; Coviello, T. Effect of temperature and cross-linking density on rheology of chemical cross-linked guar gum at the gel point. Food Hydrocoll. 2009, 23, 210–220. [Google Scholar] [CrossRef]
- Kalaee, M.R.; Famili, M.H.N.; Mahdavi, H. Cure kinetic of poly (alkyltetrasulfide) using a rheological method. Polym. Plast. Technol. Eng. 2009, 48, 627–632. [Google Scholar] [CrossRef]
- Park, J.W.; Choi, K.-H.; Park, K.K. Acid-base equilibria and related properties of chitosan. Bull. Korean Chem. Soc. 1983, 4, 68–72. [Google Scholar]
- Rinaudo, M. Chitin and chitosan: Properties and applications. Prog. Polym. Sci. 2006, 31, 603–632. [Google Scholar] [CrossRef]
- Pita-Lopez, M.L.; Fletes-Vargas, G.; Espinosa-Andrews, H.; Rodriguez-Rodriguez, R. Physically cross-linked chitosan-based hydrogels for tissue engineering applications: A state-of-the-art review. Eur. Polym. J. 2021, 145, 110176. [Google Scholar] [CrossRef]
- Domard, A. pH and C.D. measurements on a fully deacetylated chitosan: Applications to CuII-polymer interactions. Int. J. Biol. Macromol. 1987, 9, 98–104. [Google Scholar] [CrossRef]
- Haggerty, L.; Sugarman, J.H.; Prud’homme, R.K. Diffusion of polymers through polyacrylamide gels. Polymer 1988, 29, 1058–1063. [Google Scholar] [CrossRef]
- Schurz, J. Rheology of polymer solutions of the network type. Prog. Polym. Sci. 1991, 16, 1–53. [Google Scholar] [CrossRef]
- Flory, P.J. Principles of Polymer Chemistry; Cornell University Press: Ithaca, NY, USA, 1953. [Google Scholar]
- Treloar, L.R.G. Physics of Rubber Elasticity; Oxford University Press: Oxford, UK, 1975. [Google Scholar]
- Seidel, C.; Kulicke, W.-M.; Hess, C.; Hartmann, B.; Lechner, M.D.; Lazik, W. Influence of the cross-linking agent on the gel structure of starch derivatives. Starch/Stärke 2001, 53, 305–310. [Google Scholar] [CrossRef]
- Wang, J.; Ugaz, V.M. Using in situ rheology to characterize the microstructure in photopolymerized polyacrylamide gels for DNA electrophoresis. Electrophoresis 2006, 27, 3349–3358. [Google Scholar] [CrossRef]
- Ali, W.; Gebert, B.; Hennecke, T.; Graf, K.; Ulbricht, M.; Gutman, J.G. Design of thermally responsive polymeric hydrogels for brackish water desalination: Effect of architecture on swelling, deswelling, and salt rejection. ACS Appl. Mater. Interfaces 2015, 7, 15696–15706. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, M.; Krause, P.; Miruchna, V.; Von Klitzing, R. Tailoring PNIPAM hydrogels for large temperature-triggered changes in mechanical properties. Colloid Polym. Sci. 2019, 297, 633–640. [Google Scholar] [CrossRef]
- Pescosolido, L.; Feruglio, L.; Farra, R.; Fiorentino, S.; Colombo, I.; Coviello, T.; Matricardi, P.; Hennink, W.E.; Vermonden, T.; Grassi, M. Mesh size distribution determination of interpenetrating polymer network hydrogels. Soft Matter 2012, 8, 7708–7715. [Google Scholar] [CrossRef]
- Sacco, P.; Cok, M.; Asaro, F.; Paoletti, P.; Donati, I. The role played by the molecular weight and acetylation degree in modulating the stiffness and elasticity of chitosan gels. Carbohydr. Polym. 2018, 196, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Kopac, T.; Rucigaj, A.; Krajnc, M. The mutual effect of the crosslinker and biopolymer concentration on the desired hydrogel properties. Int. J. Biol. Macromol. 2020, 159, 557–569. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carmona, P.; Tasici, A.M.; Sande, S.A.; Knudsen, K.D.; Nyström, B. Glyceraldehyde as an Efficient Chemical Crosslinker Agent for the Formation of Chitosan Hydrogels. Gels 2021, 7, 186. https://doi.org/10.3390/gels7040186
Carmona P, Tasici AM, Sande SA, Knudsen KD, Nyström B. Glyceraldehyde as an Efficient Chemical Crosslinker Agent for the Formation of Chitosan Hydrogels. Gels. 2021; 7(4):186. https://doi.org/10.3390/gels7040186
Chicago/Turabian StyleCarmona, Pierre, Anca M. Tasici, Sverre A. Sande, Kenneth D. Knudsen, and Bo Nyström. 2021. "Glyceraldehyde as an Efficient Chemical Crosslinker Agent for the Formation of Chitosan Hydrogels" Gels 7, no. 4: 186. https://doi.org/10.3390/gels7040186
APA StyleCarmona, P., Tasici, A. M., Sande, S. A., Knudsen, K. D., & Nyström, B. (2021). Glyceraldehyde as an Efficient Chemical Crosslinker Agent for the Formation of Chitosan Hydrogels. Gels, 7(4), 186. https://doi.org/10.3390/gels7040186