
Instantaneous Degelling Thermoresponsive Hydrogel



Abstract
:1. Introduction
2. Results
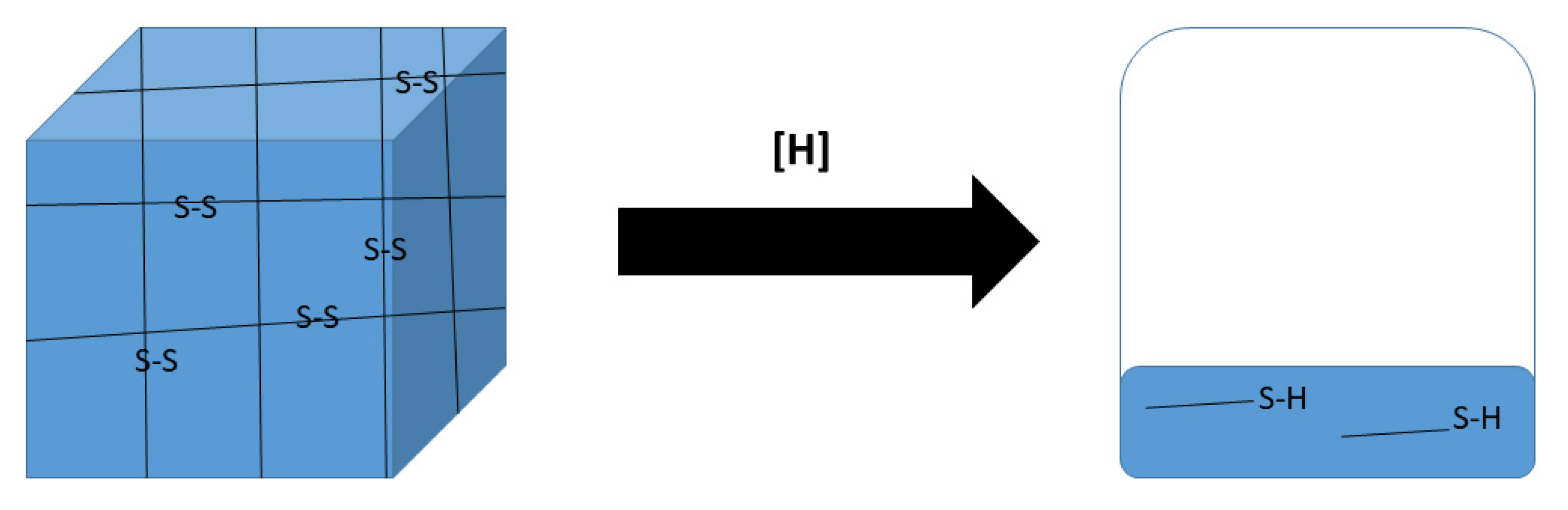
2.1. Rational Design
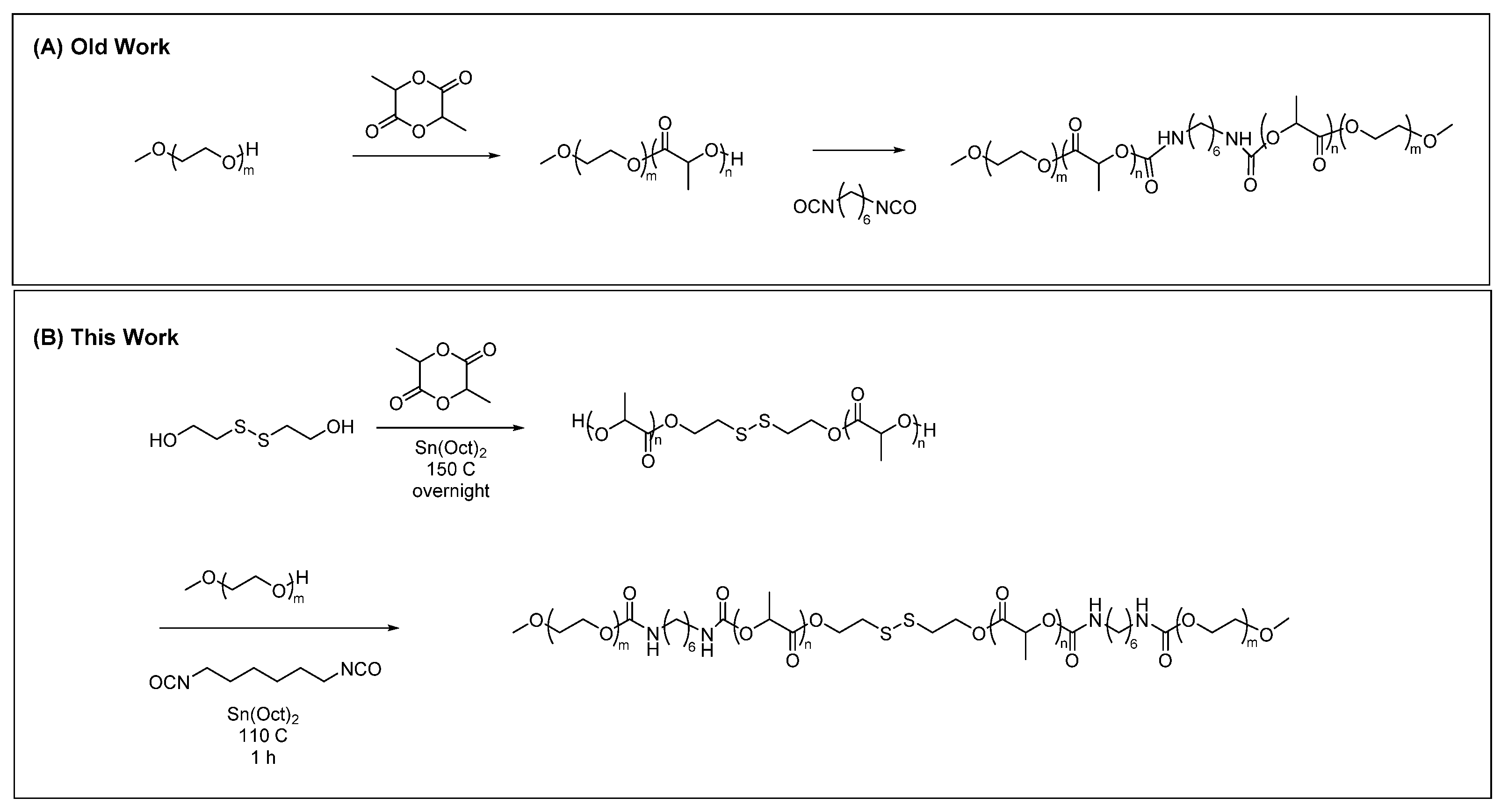
2.2. Synthesis
2.3. Characterization
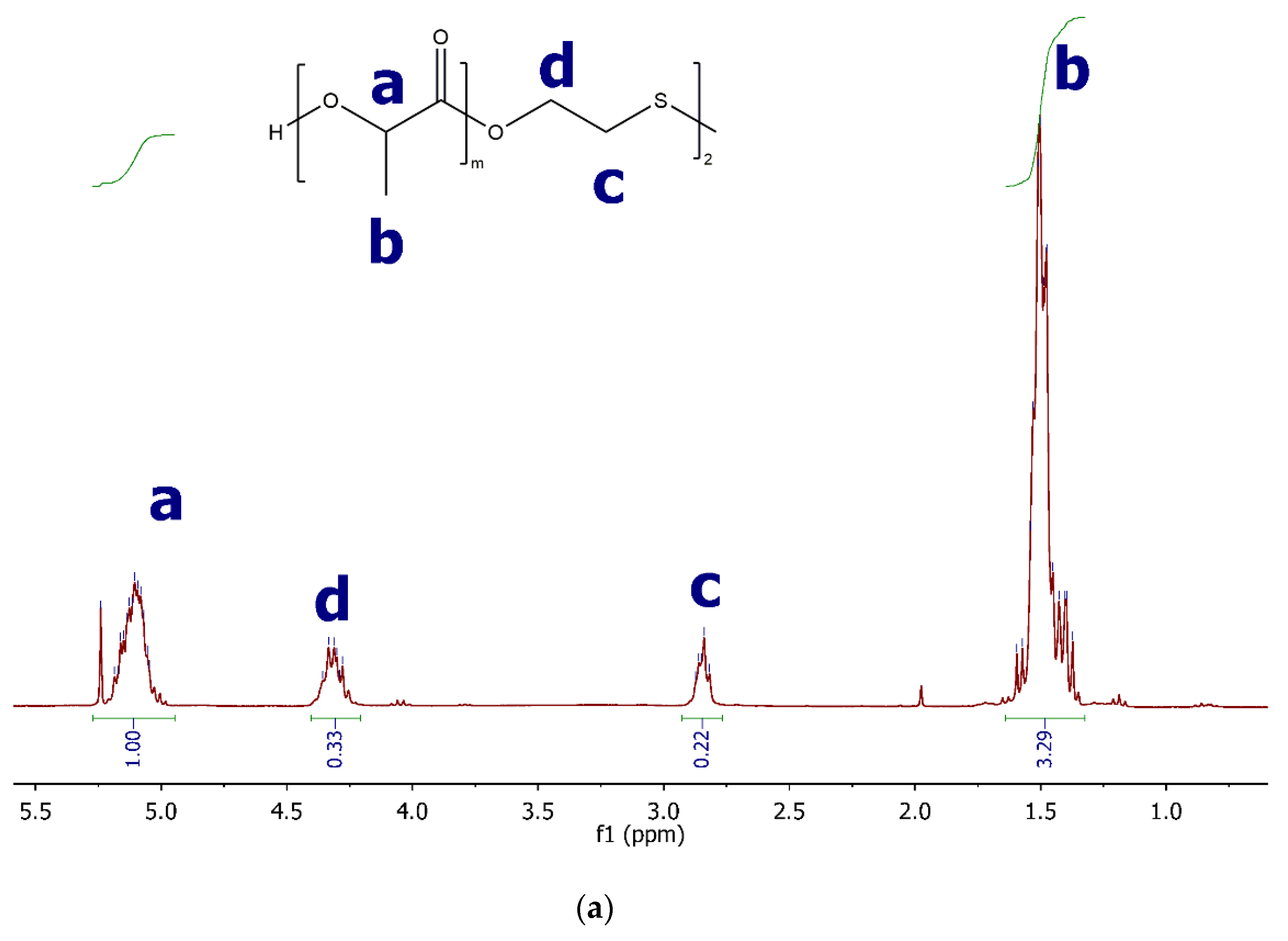
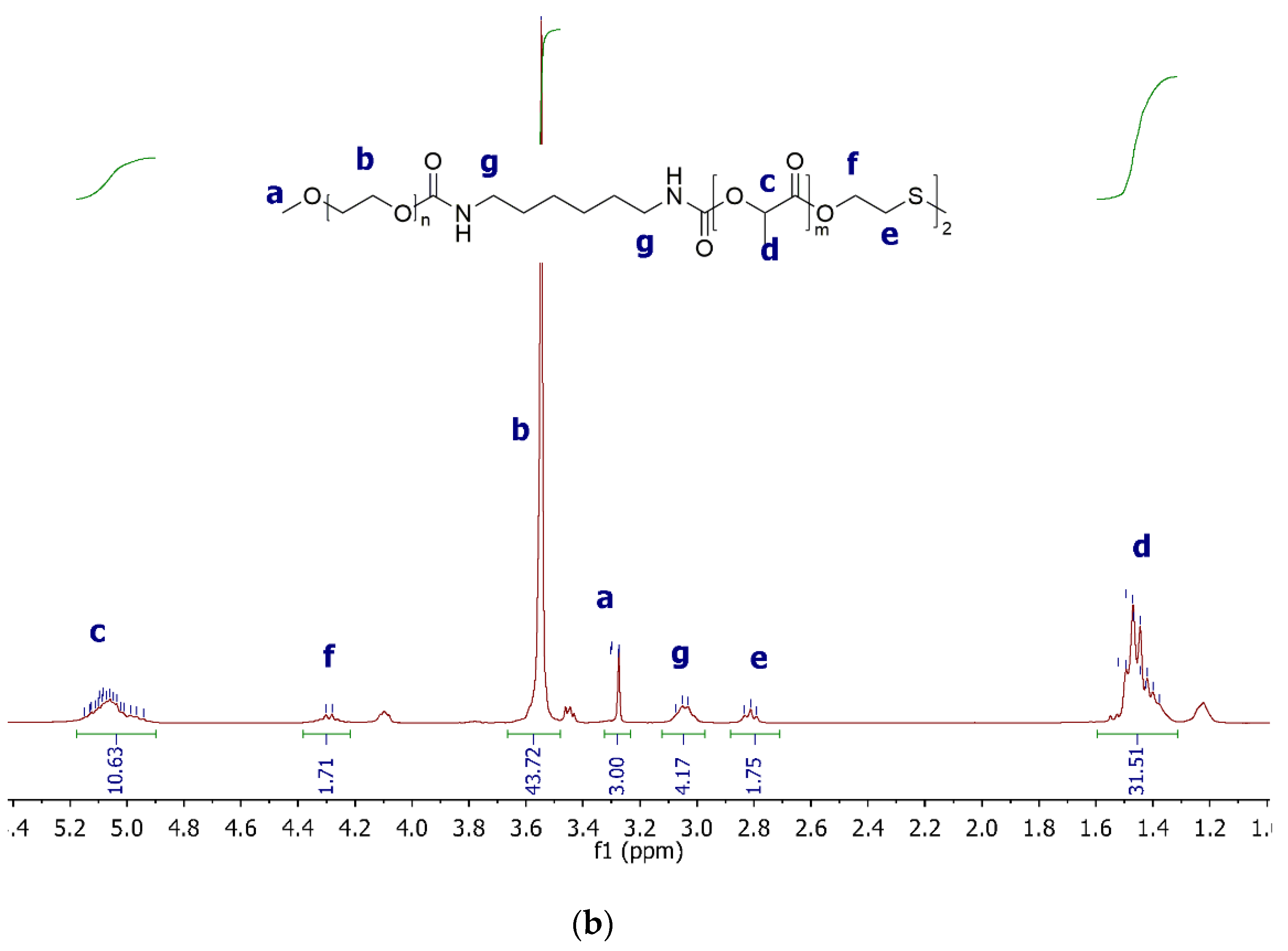
2.3.1. 1H NMR
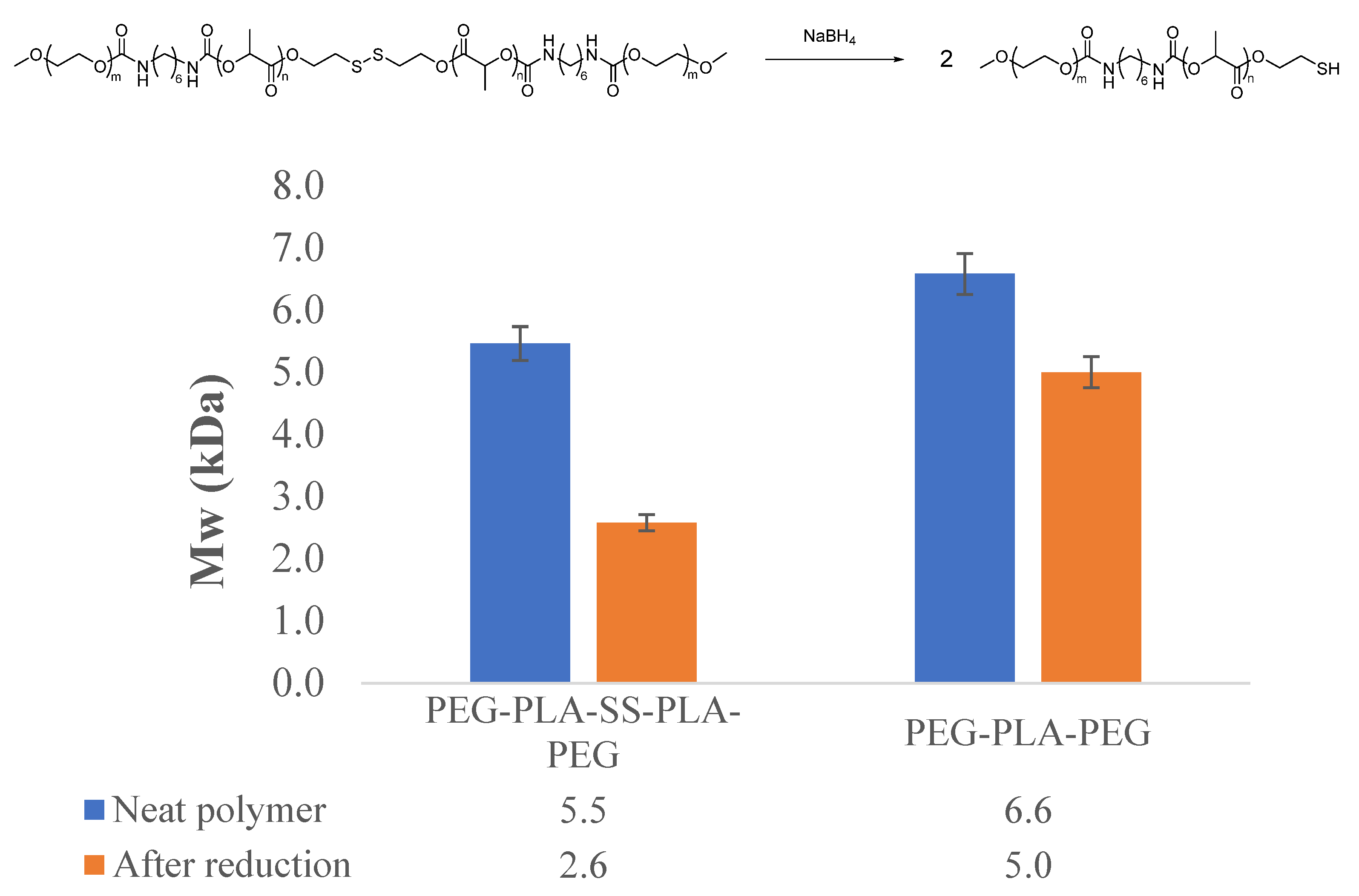
2.3.2. Molecular Weight Determination
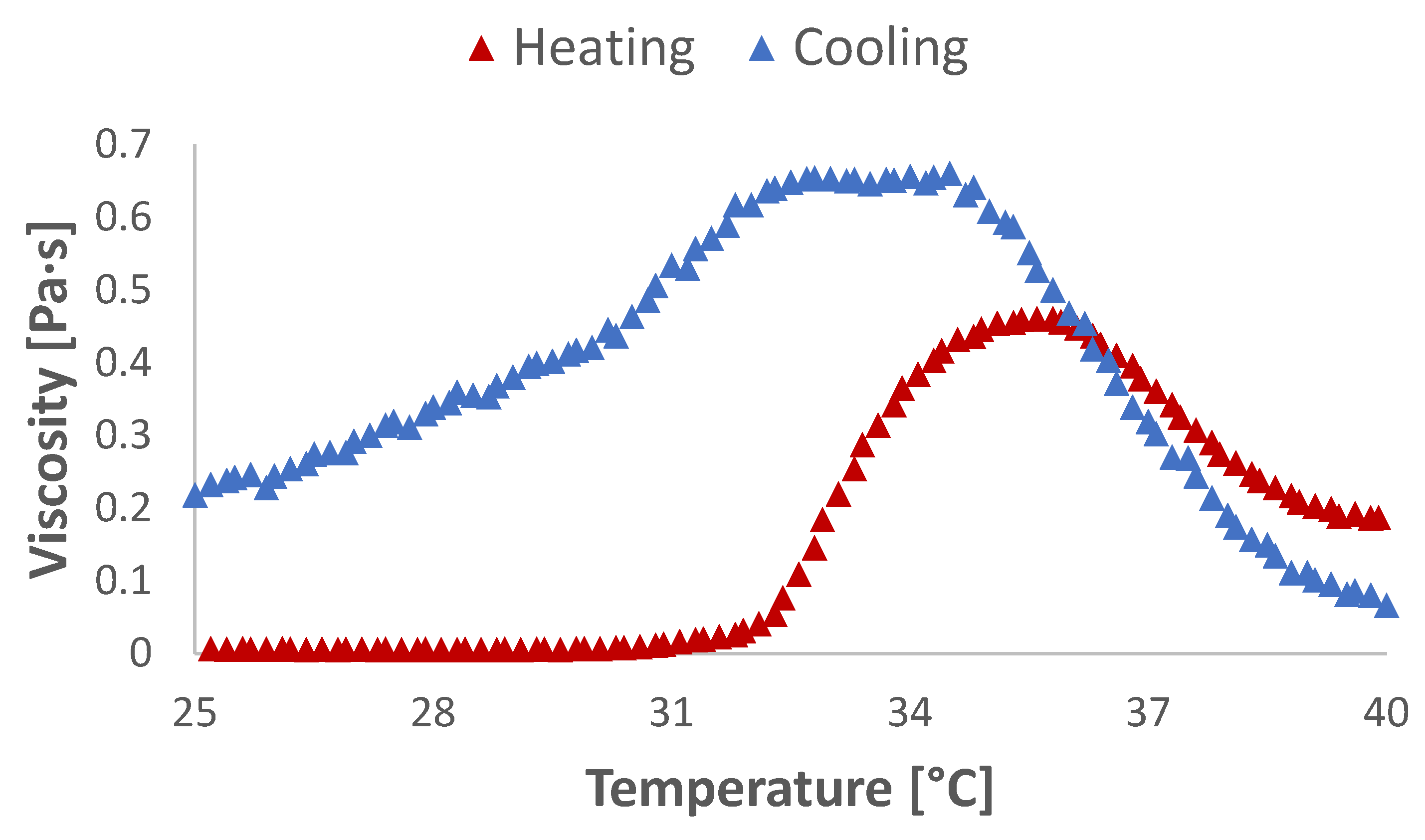
2.4. Rheometry Studies
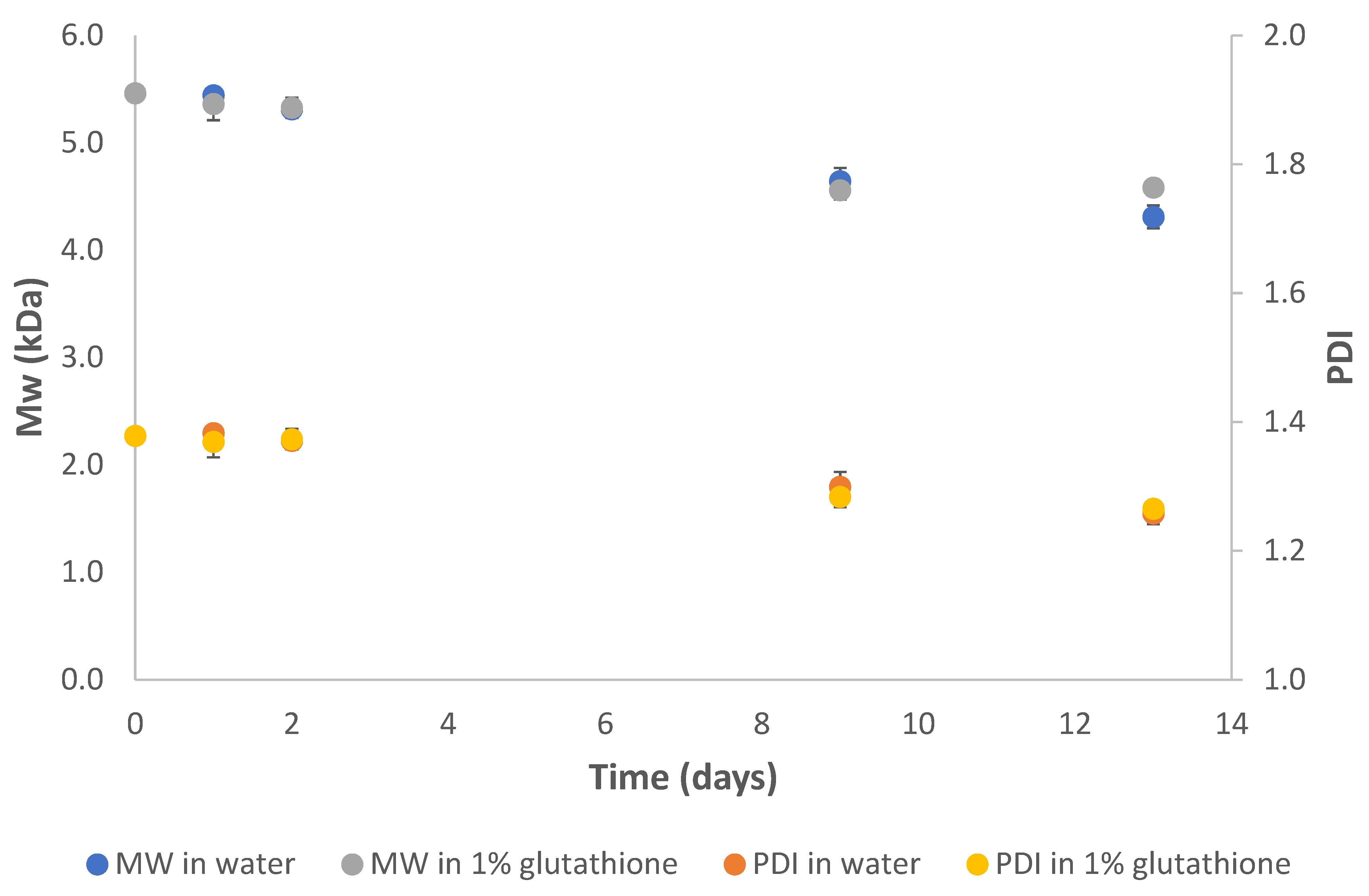
2.5. Degradation Studies
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Materials
5.2. General Methods
5.3. Synthesis
5.4. Rheological Studies
5.5. Degradation Studies
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Priya James, H.; John, R.; Alex, A.; Anoop, K.R. Smart polymers for the controlled delivery of drugs—A concise overview. Acta Pharm. Sin. B 2014, 4, 120–127. [Google Scholar] [CrossRef] [Green Version]
- Jeong, B.; Kim, S.W.; Bae, Y.H. Thermosensitive sol–gel reversible hydrogels. Adv. Drug Deliv. Rev. 2012, 64, 154–162. [Google Scholar] [CrossRef]
- Steinman, N.Y.; Haim-Zada, M.; Goldstein, I.A.; Goldberg, A.H.; Haber, T.; Berlin, J.M.; Domb, A.J. Effect of PLGA block molecular weight on gelling temperature of PLGA-PEG-PLGA thermoresponsive copolymers. J. Polym. Sci. Part A Polym. Chem. 2019, 57, 35–39. [Google Scholar] [CrossRef] [Green Version]
- Steinman, N.Y.; Bentolila, N.Y.; Domb, A.J. Effect of Molecular Weight on Gelling and Viscoelastic Properties of Poly (caprolactone)–b-Poly (ethylene glycol)–b-Poly (caprolactone)(PCL–PEG–PCL) Hydrogels. Polymers 2020, 12, 2372. [Google Scholar] [CrossRef]
- Maeda, T. Structures and Applications of Thermoresponsive Hydrogels and Nanocomposite-Hydrogels Based on Copolymers with Poly (Ethylene Glycol) and Poly (Lactide-Co-Glycolide) Blocks. Bioengineering 2019, 6, 107. [Google Scholar] [CrossRef] [Green Version]
- Jeong, B.; Bae, Y.H.; Kim, S.W. In situ gelation of PEG-PLGA-PEG triblock copolymer aqueous solutions and degradation thereof. J. Biomed. Mater. Res. 2000, 50, 171–177. [Google Scholar] [CrossRef]
- Shim, M.S.; Lee, H.T.; Shim, W.S.; Park, I.; Lee, H.; Chang, T.; Kim, S.W.; Lee, D.S. Poly(D,L-lactic acid-co-glycolic acid)-b-poly(ethylene glycol)-b-poly (D,L-lactic acid-co-glycolic acid) triblock copolymer and thermoreversible phase transition in water. J. Biomed. Mater. Res. 2002, 61, 188–196. [Google Scholar] [CrossRef]
- Lee, D.S.; Shim, M.S.; Kim, S.W.; Lee, H.; Park, I.; Chang, T. Novel thermoreversible gelation of biodegradable PLGA-block-PEO-block-PLGA triblock copolymers in aqueous solution. Macromol. Rapid Commun. 2001, 22, 587–592. [Google Scholar] [CrossRef]
- Qiao, M.; Chen, D.; Ma, X.; Liu, Y. Injectable biodegradable temperature-responsive PLGA–PEG–PLGA copolymers: Synthesis and effect of copolymer composition on the drug release from the copolymer-based hydrogels. Int. J. Pharm. 2005, 294, 103–112. [Google Scholar] [CrossRef]
- Jeong, B.; Bae, Y.H.; Kim, S.W. Biodegradable thermosensitive micelles of PEG-PLGA-PEG triblock copolymers. Colloids Surf. B Biointerfaces 1999, 16, 185–193. [Google Scholar] [CrossRef]
- Jeong, B.; Bae, Y.H.; Kim, S.W. Thermoreversible gelation of PEG− PLGA− PEG triblock copolymer aqueous solutions. Macromolecules 1999, 32, 7064–7069. [Google Scholar] [CrossRef]
- Jian, X.; Hu, Y.; Zhou, W.; Xiao, L. Self-healing polyurethane based on disulfide bond and hydrogen bond. Polym. Adv. Technol. 2018, 29, 463–469. [Google Scholar] [CrossRef]
- Naskar, K.; Dey, A.; Maity, S.; Ray, P.P.; Ghosh, P.; Sinha, C. Biporous Cd (II) Coordination Polymer via in Situ Disulfide Bond Formation: Self-Healing and Application to Photosensitive Optoelectronic Device. Inorg. Chem. 2020, 59, 5518–5528. [Google Scholar] [CrossRef] [PubMed]
- Naoi, K.; Kawase, K.i.; Mori, M.; Komiyama, M. Electrochemistry of Poly (2, 2′-dithiodianiline): A New Class of High Energy Conducting Polymer Interconnected with S‒S Bonds. J. Electrochem. Soc. 1997, 144, L173. [Google Scholar] [CrossRef]
- Liu, L.; Liu, P. Synthesis strategies for disulfide bond-containing polymer-based drug delivery system for reduction-responsive controlled release. Front. Mater. Sci. 2015, 9, 211–226. [Google Scholar] [CrossRef]
- Zhang, P.; Wu, J.; Xiao, F.; Zhao, D.; Luan, Y. Disulfide bond based polymeric drug carriers for cancer chemotherapy and relevant redox environments in mammals. Med. Res. Rev. 2018, 38, 1485–1510. [Google Scholar] [CrossRef]
- Wang, Y.-C.; Wang, F.; Sun, T.-M.; Wang, J. Redox-responsive nanoparticles from the single disulfide bond-bridged block copolymer as drug carriers for overcoming multidrug resistance in cancer cells. Bioconj. Chem. 2011, 22, 1939–1945. [Google Scholar] [CrossRef]
- Wong, D.Y.-K.; Hsiao, Y.-L.; Poon, C.-K.; Kwan, P.-C.; Chao, S.-Y.; Chou, S.-T.; Yang, C.-S. Glutathione concentration in oral cancer tissues. Cancer Lett. 1994, 81, 111–116. [Google Scholar] [CrossRef]
- Hoon Jeong, J.; Christensen, L.V.; Yockman, J.W.; Zhong, Z.; Engbersen, J.F.J.; Jong Kim, W.; Feijen, J.; Wan Kim, S. Reducible poly(amido ethylenimine) directed to enhance RNA interference. Biomaterials 2007, 28, 1912–1917. [Google Scholar] [CrossRef]
- Taranejoo, S.; Chandrasekaran, R.; Cheng, W.; Hourigan, K. Bioreducible PEI-functionalized glycol chitosan: A novel gene vector with reduced cytotoxicity and improved transfection efficiency. Carbohydr. Polym. 2016, 153, 160–168. [Google Scholar] [CrossRef]
- Davoodi, P.; Srinivasan, M.P.; Wang, C.-H. Synthesis of intracellular reduction-sensitive amphiphilic polyethyleneimine and poly(ε-caprolactone) graft copolymer for on-demand release of doxorubicin and p53 plasmid DNA. Acta Biomater. 2016, 39, 79–93. [Google Scholar] [CrossRef]
- Wang, L.-H.; Wu, D.-C.; Xu, H.-X.; You, Y.-Z. High DNA-Binding Affinity and Gene-Transfection Efficacy of Bioreducible Cationic Nanomicelles with a Fluorinated Core. Angew. Chem. Int. Ed. 2016, 55, 755–759. [Google Scholar] [CrossRef]
- Yang, H.; Bai, Y.; Yu, B.; Wang, Z.; Zhang, X. Supramolecular polymers bearing disulfide bonds. Polym. Chem. 2014, 5, 6439–6443. [Google Scholar] [CrossRef]
- Jeong, B.; Bae, Y.H.; Lee, D.S.; Kim, S.W. Biodegradable block copolymers as injectable drug-delivery systems. Nature 1997, 388, 860–862. [Google Scholar] [CrossRef]
- Jeong, B.; Bae, Y.H.; Kim, S.W. Drug release from biodegradable injectable thermosensitive hydrogel of PEG–PLGA–PEG triblock copolymers. J. Control Release 2000, 63, 155–163. [Google Scholar] [CrossRef]
- Li, X.; Wang, Y.; Chen, J.; Wang, Y.; Ma, J.; Wu, G. Controlled release of protein from biodegradable multi-sensitive injectable poly (ether-urethane) hydrogel. ACS Appl. Mater. Interfaces 2014, 6, 3640–3647. [Google Scholar] [CrossRef] [PubMed]
- Doppalapudi, S.; Jain, A.; Khan, W.; Domb, A.J. Biodegradable polymers—An overview. Polym. Adv. Technol. 2014, 25, 427–435. [Google Scholar] [CrossRef]
- Ramot, Y.; Haim-Zada, M.; Domb, A.J.; Nyska, A. Biocompatibility and safety of PLA and its copolymers. Adv. Drug Deliv. Rev. 2016, 107, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Hogan, K.J.; Mikos, A.G. Biodegradable thermoresponsive polymers: Applications in drug delivery and tissue engineering. Polymer 2020, 211, 123063. [Google Scholar] [CrossRef]
- Ghahremankhani, A.A.; Dorkoosh, F.; Dinarvand, R. PLGA-PEG-PLGA Tri-Block Copolymers as In Situ Gel-Forming Peptide Delivery System: Effect of Formulation Properties on Peptide Release. Pharm. Dev. Technol. 2008, 13, 49–55. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | PLA(SS) | PEG-PLA-SS-PLA-PEG |
|---|---|---|
| Expected MW 1,2 | 1.53 | 1.96 |
| Mn (1H NMR) 1,3 | 1.46 | 3.11 |
| Mn (SEC) 1,4 | 2.65 | 4.24 |
| Mw (SEC) 1,4 | 2.85 | 5.97 |
| PDI 5 | 1.07 | 1.41 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Steinman, N.Y.; Domb, A.J. Instantaneous Degelling Thermoresponsive Hydrogel. Gels 2021, 7, 169. https://doi.org/10.3390/gels7040169
Steinman NY, Domb AJ. Instantaneous Degelling Thermoresponsive Hydrogel. Gels. 2021; 7(4):169. https://doi.org/10.3390/gels7040169
Chicago/Turabian StyleSteinman, Noam Y., and Abraham J. Domb. 2021. "Instantaneous Degelling Thermoresponsive Hydrogel" Gels 7, no. 4: 169. https://doi.org/10.3390/gels7040169
APA StyleSteinman, N. Y., & Domb, A. J. (2021). Instantaneous Degelling Thermoresponsive Hydrogel. Gels, 7(4), 169. https://doi.org/10.3390/gels7040169