
Thermoreversible Gelation with Two-Component Mixed Cross-Link Junctions of Variable Multiplicity in Ternary Polymer Solutions


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
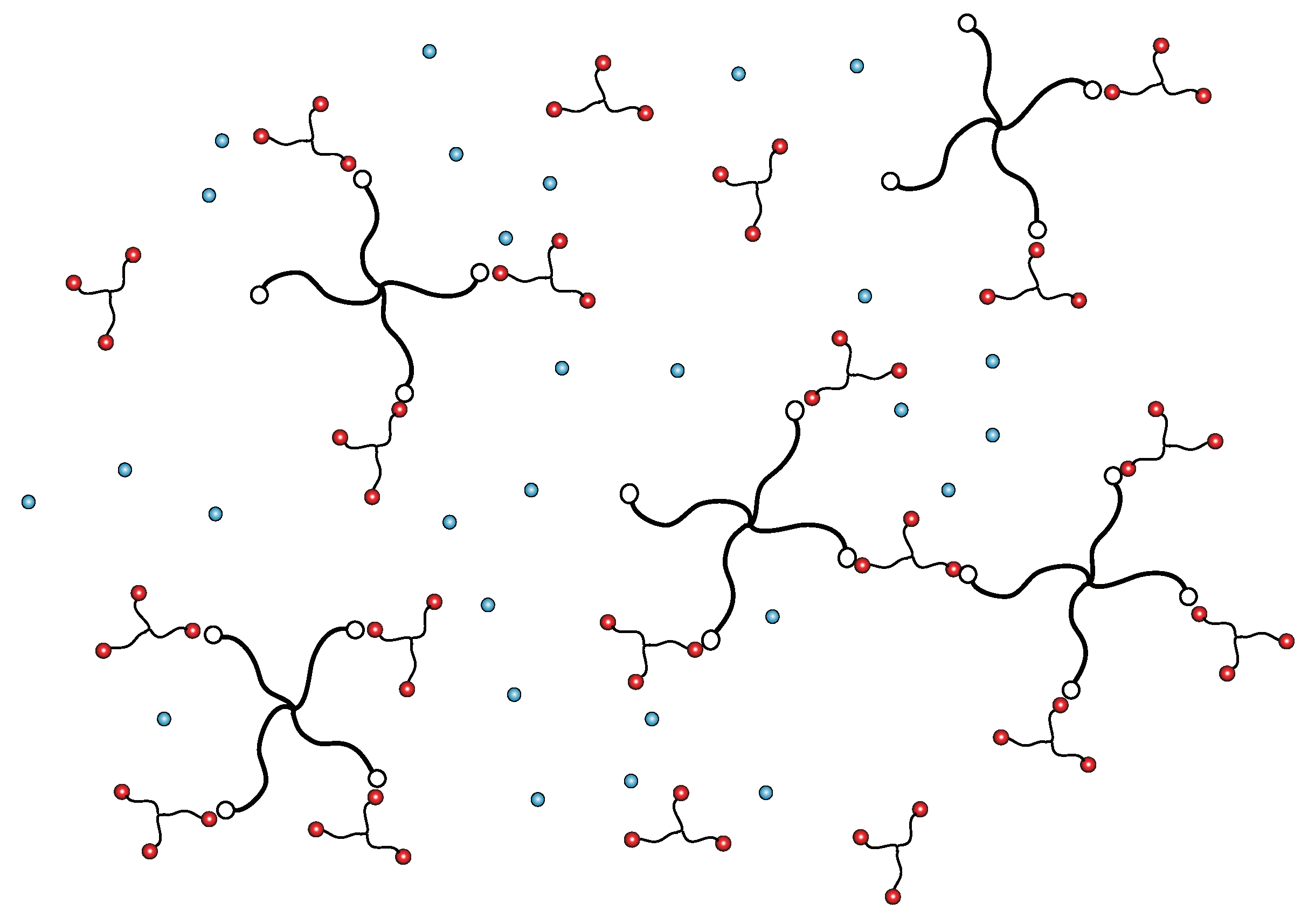
:1. Introduction: Binary Gels with Mixed Cross-Links
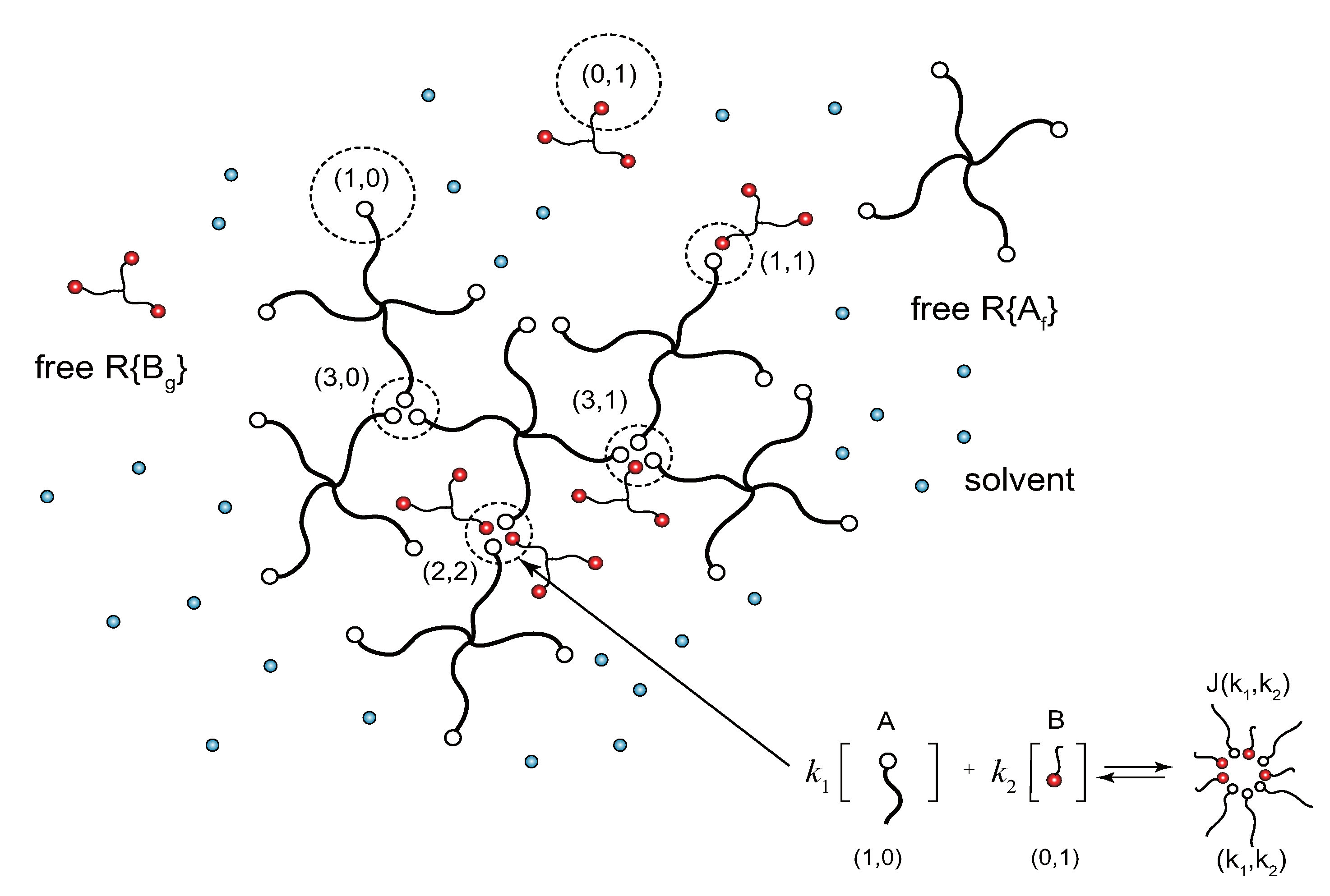
2. Stoichiometric Definition of the Model Solution
3. Equilibrium Condition for the Formation of Cross-Link Junctions
4. The Average Branching Number and Response to the Concentration Fluctuations
5. The Weight Average Molecular Weight of the Cross-Linked Clusters and the Gel Point Condition
6. The Average Number of Molecules in the Cross-Linked Clusters and the Spinodal Condition for the Solution
6.1. Chemical Potentials Described in Terms of the Number-Average Degree of Polymerization
6.2. Average Degree of Polymerization and Number Concentration of the Cross-Linked Clusters
6.3. Spinodal Condition
6.4. Treatment of the Temperature
7. Fixed Multiplicity Model
7.1. Sol-Gel Transition (SGT) and Spinodals for the Fixed Multiplicity Model
7.2. Optimal Gelation Concentration
7.3. Specific Models of the Cross-Link Junctions with Fixed Multiplicity
7.3.1. Pairwise Cross-Links
7.3.2. Mononuclear A Cross-Links
7.3.3. Mononuclear B Cross-Links
7.3.4. Monofunctional B Molecules
8. Variable Multiplicity Model
8.1. Completely Immiscible Cross-Link Junctions
8.2. Completely Miscible Cross-Link Junctions
8.2.1. Pairwise Cross-Links
8.2.2. Fixed Multiplicity Model
8.2.3. Mini-Max Model
8.3. Partially Miscible Cross-Link Junctions
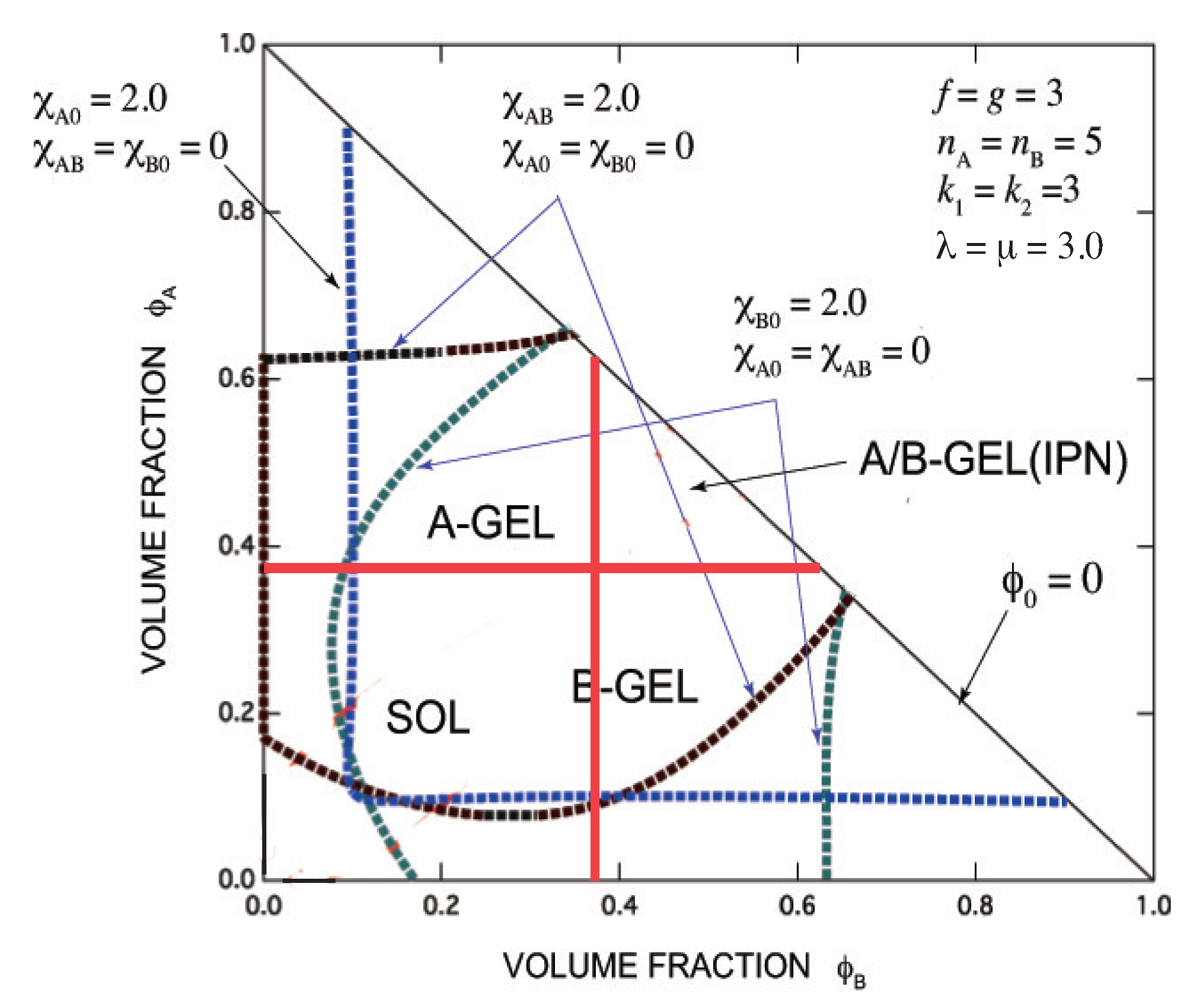
9. Conclusions and Discussion
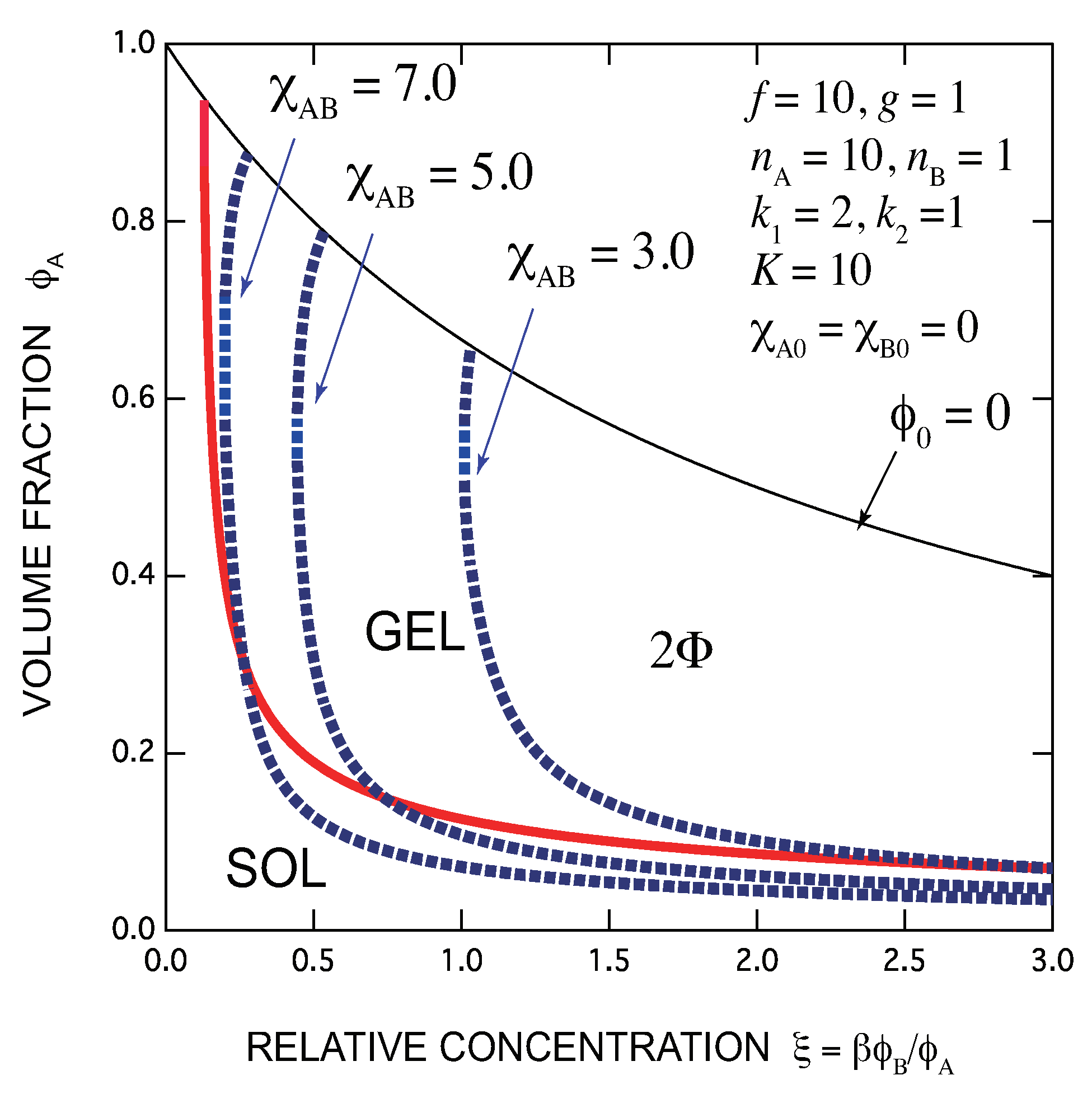
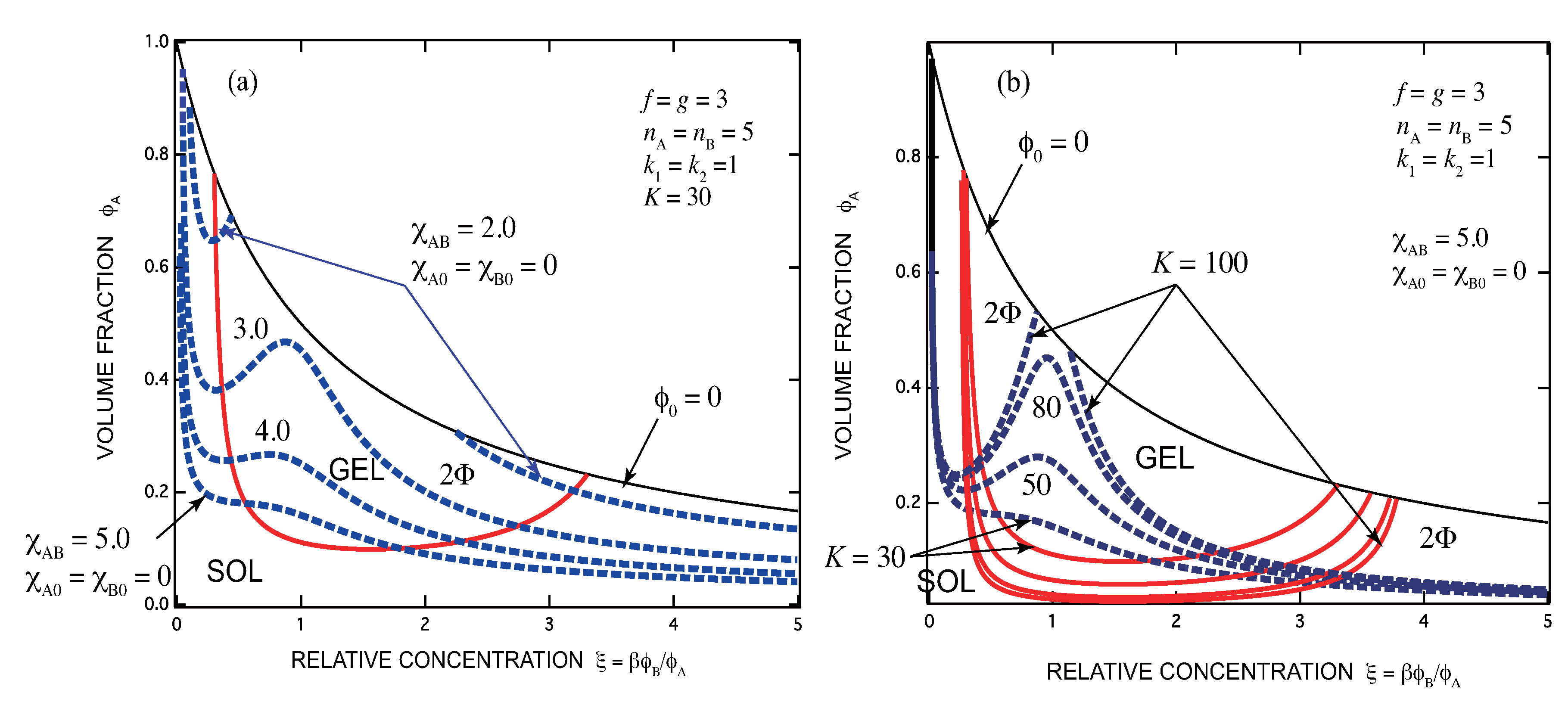
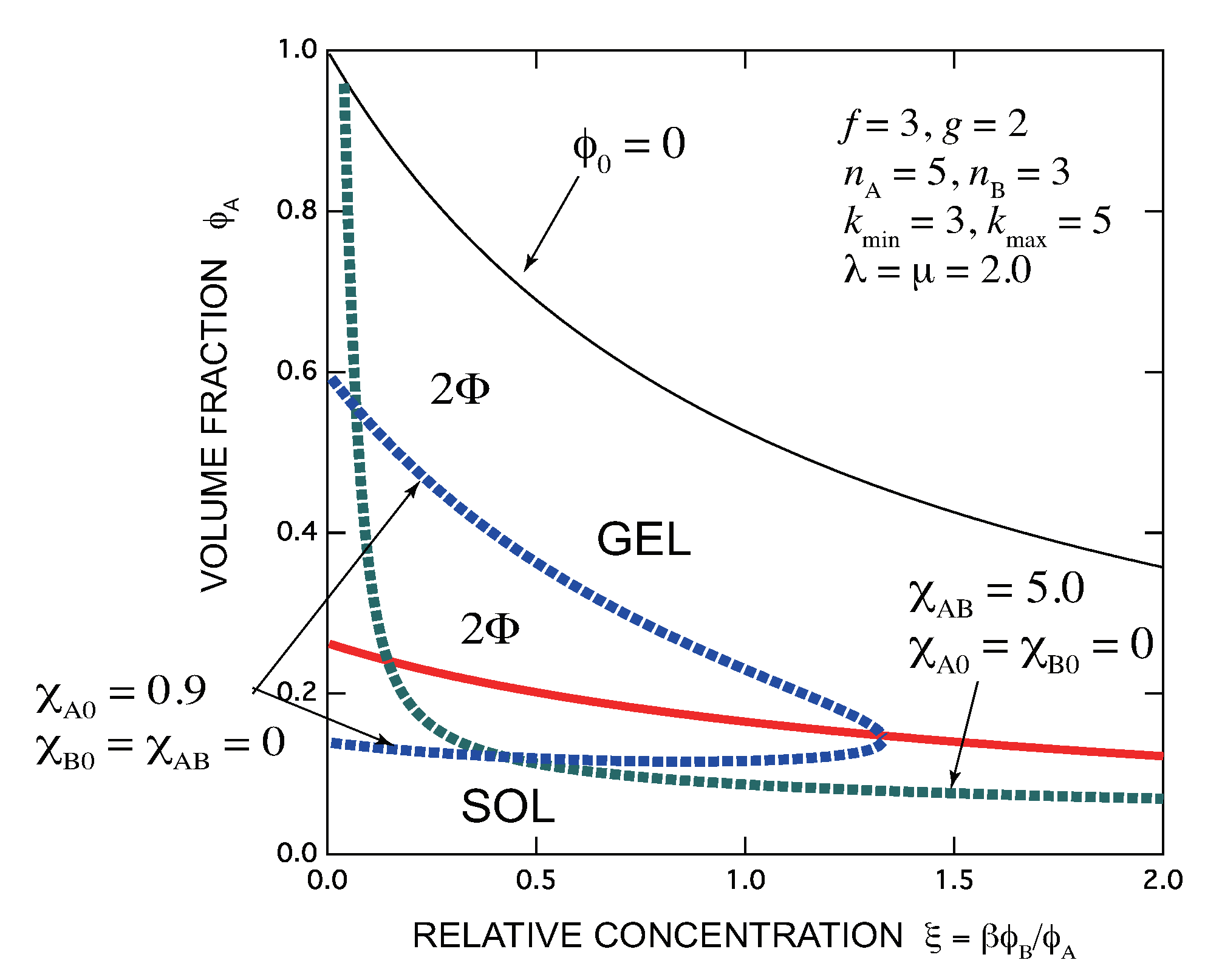
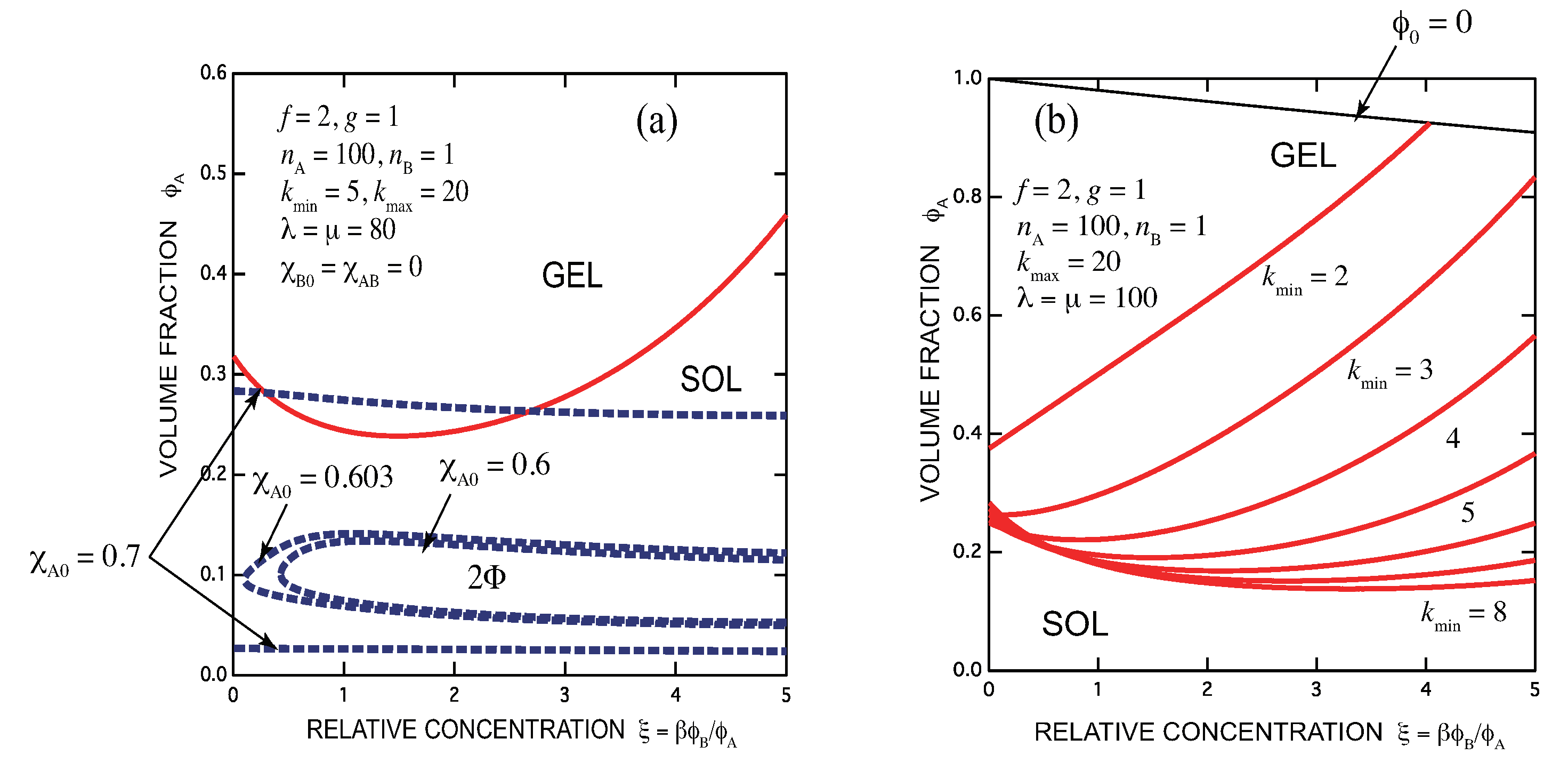
- There exists an optimal concentration of B (secondary) molecules for gelation if monocomplexes (surface coverage by B molecules) are formed at its high concentration region. The condition for optimization depends on the functionalities of both components, the multiplicity of the cross-links, and the association constants of complexes.
- In general, there are lower and upper bound in the concentration of B molecules between which the gel phase appears (reentrant sol-gel-sol transition). Both bounds depend on the functionalities of both components, the multiplicity of the cross-links, and the association constants of complexes.
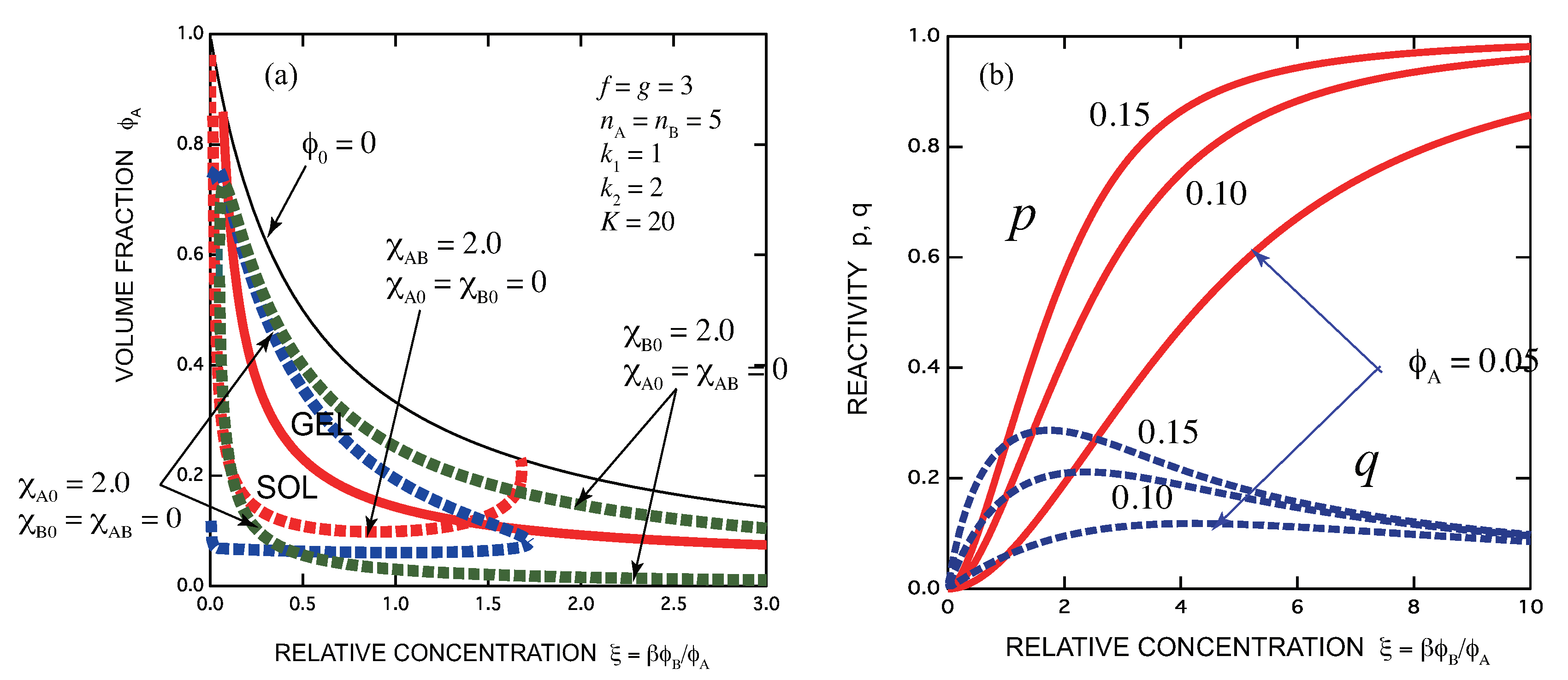
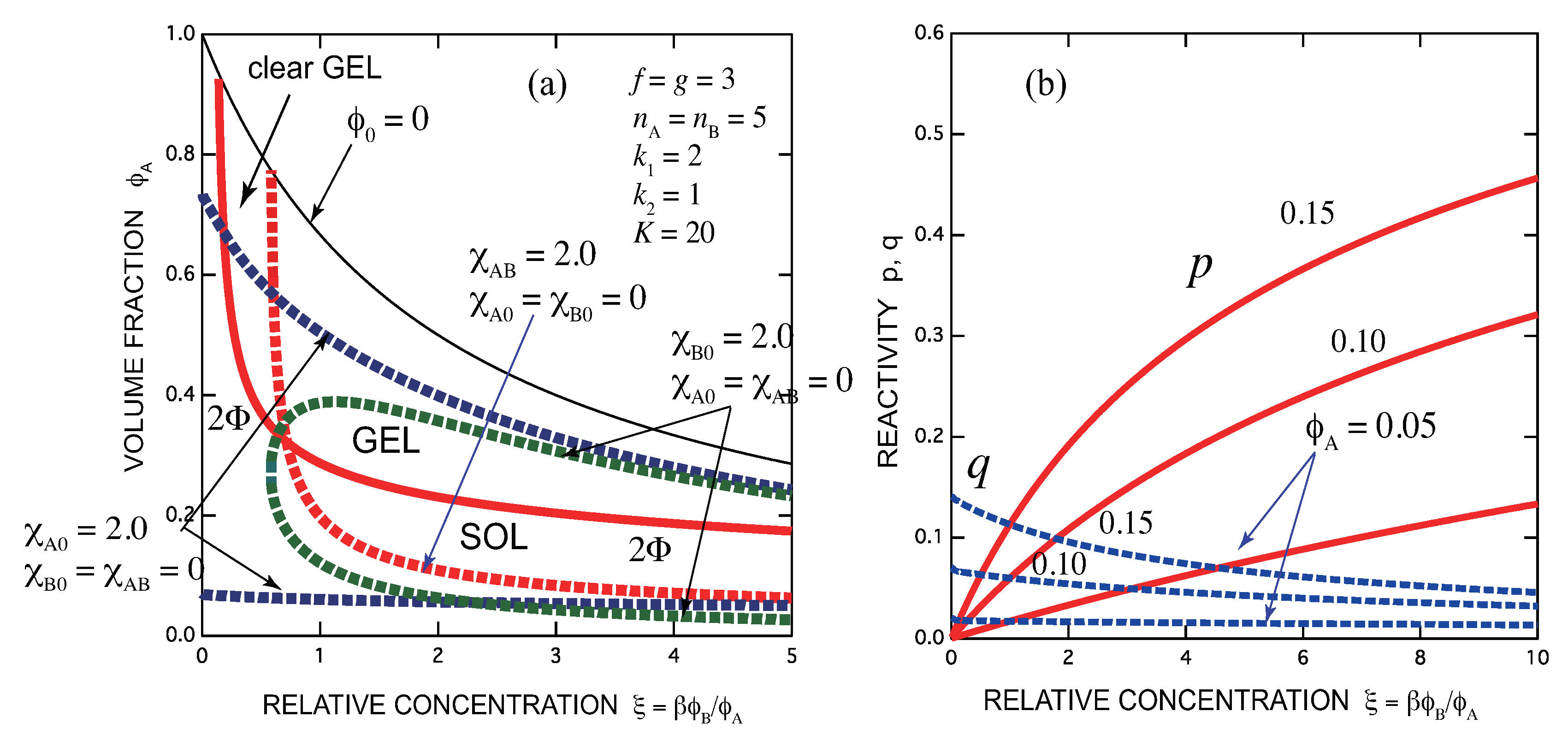
- Gelation interferes with liquid–liquid phase separation. The relative position of the gel region and the two-phase region systematically shifts with the association constants of the cross-links and Flory’s interaction parameters. In one case, gelation takes place before phase separation (from clear gel to turbid gel), and in another vice versa (only turbid gel) with increase in the concentration of B molecules. The crossing point between the sol-gel transition line and the phase separation line is a higher order critical point.
- Addition of B molecules to the phase separated solutions of A molecules in a poor solvent may induce homogenization of the solution above a critical concentration and form a clear gel if the solvent is sufficiently good for B molecules. Oppositely, an already formed clear gel may turn into turbid one if the solvent is poor for B molecules.
Funding
Data Availability Statement
Conflicts of Interest
References
- Guenet, J.M. Thermoreversible Gelation of Polymers and Biopolymers; Academic Press, Harcourt Brace Jovanovich Pub.: London, UK, 1992. [Google Scholar]
- Te Nijenhuis, K. Thermoreversible Networks. Adv. Polym. Sci. 1997, 130, 1–261. [Google Scholar]
- Zhang, J.; Hu, Y.; Li, Y. Gel Chemistry: Interactions, Structures and Properties; Springer: Heidelberg, Germany, 2018. [Google Scholar]
- Thakur, V.K.; Thakur, M.K. Gels Horizons: From Science to Smart Materials. In Polymer Gels: Science and Fundamentals; Springer: Heidelberg, Germany, 2018. [Google Scholar]
- Thakur, V.K.; Thakur, M.K. Gels Horizons: From Science to Smart Materials. In Hydrogels: Recent Advances; Springer: Heidelberg, Germany, 2018. [Google Scholar]
- Patrickios, C.S. (Ed.) Amphiphilic Polymer Co-Networks: Synthesis, Properties, Modelling and Applications; Royal Society of Chemistry: Cambridge, UK, 2020. [Google Scholar]
- Fan, H.; Wang, J.; Jin, Z. Tough, Swelling-Resistant, Self-Healing, and Adhesive Dual-Cross-Linked Hydrogels Based on Polymer-Tannic Acid Multiple Hydrogen Bonds. Macromolecules 2018, 51, 1696–1705. [Google Scholar] [CrossRef]
- Zhang, C.; Yang, Z.; Duong, N.T.; Li, X.; Nishiyama, Y.; Wu, Q.; Zhang, R.; Sun, P. Using Dynamic Bonds to Enhance the Mechanical Performance: From Microscopic Molecular Interactions to Macroscopic Properties. Macromolecules 2019, 52, 5014–5025. [Google Scholar] [CrossRef]
- Yu, H.C.; Li, C.Y.; Du, M.; Song, Y.; Wu, Z.L.; Zheng, Q. Improved toughness and stability of κ-carrageenan/polyacrylamide double-network hydrogels by dual cross-linking of the first network. Macromolecules 2019, 52, 629–638. [Google Scholar] [CrossRef]
- Cristiani, T.R.; Filippidi, E.; Behrens, R.L.; Valentine, M.T.; Eisenbach, C.D. Tailoring the toughness of elastomers by incorporating Ionic cross-linking. Macromolecules 2020, 53, 4099–4109. [Google Scholar] [CrossRef]
- Stockmayer, W.H. Molecular distribution in condensation polymers. J. Polym. Sci. 1952, 4, 69–71. [Google Scholar] [CrossRef]
- Piepenbrock, M.-O.M.; Lloyd, G.O.; Clarke, N.; Steed, J.W. Metal- and Anion-Binding Supramolecular Gels. Chem. Rev. 2010, 110, 1960–2004. [Google Scholar] [CrossRef] [PubMed]
- Christensen, T.; Gooden, D.M.; Kung, J.E.; Toone, E.J. Additivity and the Physical Basis of Multivalency Effects: A Thermodynamic Investigation of the Calcium EDTA Interaction. J. Am. Chem. Soc. 2003, 125, 7357–7366. [Google Scholar] [CrossRef] [PubMed]
- Shibayama, M.; Yoshizawa, H.; Kurokawa, H.; Fujiwara, H.; Nomura, S. Sol-gel transition of poly(vinyl alcohol)-borate complex. Polymer 1988, 29, 2066–2071. [Google Scholar] [CrossRef]
- Kurokawa, H.; Shibayama, M.; Ishimaru, T.; Nomura, S.; Wu, W.-L. Phase behaviour and sol-gel transition of poly (vinyl alcohol)-borate complex in aqueous solution. Polymer 1992, 33, 2182–2188. [Google Scholar] [CrossRef]
- Keita, G.; Ricard, A.; Audebert, R.; Pezron, E.; Leibler, L. The poly(vinyl alcohol)-borate system: Influence of polyelectrolyte effects on phase diagrams. Polymer 1995, 36, 49–54. [Google Scholar] [CrossRef]
- Shibayama, M.; Ikkai, F.; Moriwaki, R.; Nomura, S. Complexation of Poly(vinyl alcohol)-Congo Red Aqueous Solutions. 1. Viscosity Behavior and Gelation Mechanism. Macromolecules 1994, 27, 1738–1743. [Google Scholar] [CrossRef]
- Shibayama, M.; Ikkai, F.; Nomura, S. Complexation of Poly(vinyl alcohol)-Congo Red Aqueous Solutions. 2. SANS and SAXS Studies on Sol-Gel Transition. Macromolecules 1994, 27, 6383–6388. [Google Scholar] [CrossRef]
- Kohri, M.; Yanagimoto, K.; Kohaku, K.; Shiomoto, S.; Kobayashi, M.; Imai, A.; Shiba, F.; Taniguchi, T.; Kishikawa, K. Magnetically Responsive Polymer Network Constructed by Poly(acrylic acid) and Holmium. Macromolecules 2018, 51, 6740–6745. [Google Scholar] [CrossRef]
- Pezron, E.; Ricard, A.; Lafuma, F.; Audebert, R. Reversible Gel Formation Induced by Ion Comlexation. 1. Borax-Galactomannan Interactions. Macromolecules 1988, 21, 1121–1125. [Google Scholar] [CrossRef]
- Pezron, E.; Leibler, L.; Ricard, A.; Audebert, R. Reversible Gel Formation Induced by Ion Comlexation. 2. Phase Diagram. Macromolecules 1988, 21, 1126–1131. [Google Scholar] [CrossRef]
- Pezron, E.; Leibler, L.; Ricard, A.; Lafuma, F.; Audebert, R. Complex Formation in Polymer-Ion Solutions. 1. Polymer Concentration Effects. Macromolecules 1989, 22, 1169–1174. [Google Scholar] [CrossRef]
- Pezron, E.; Leibler, L.; Lafuma, F. Complex Formation in Polymer-Ion Solutions. 2. Polyelectrolyte Effects. Macromolecules 1989, 22, 2656–2662. [Google Scholar] [CrossRef]
- Lips, A.; Clark, A.H.; Cutler, N.; Durand, D. Measurement of cooperativity of binding of calcium to neutral sodium pectate. Food Hydrocoll. 1991, 5, 87–99. [Google Scholar] [CrossRef]
- Zhang, W.; Piculell, L.; Nilsson, S. Effects of Specific Anion Binding on the Helix-Coil Transition of Lower Charged Carrageenans. NMR Data and Conformational Equilibria Analyzed within the Poisson-Boltzman Cell Model. Macromolecules 1992, 25, 6165–6172. [Google Scholar] [CrossRef]
- Tanaka, F.; Nakagawa, Y.; Ohta, S.; Ito, T. Thermoreversible gelation with ion-binding cross-links of varible multiplicity. J. Chem. Phys. 2019, 150, 174904. [Google Scholar] [CrossRef]
- Norioka, C.; Okita, K.; Mukada, M.; Kawamura, A.; Miyata, T. Biomolecularly stimuli-responsive tetra-poly (ethylene glycol) that undergoes sol-gel transition in response to a target biomolecule. Polym. Chem. 2017, 8, 6378–6385. [Google Scholar] [CrossRef]
- Dubin, D. (Ed.) Microdomains in Polymer Solutions; Plenum Press: New York, NY, USA, 1985. [Google Scholar]
- Goddard, E.D.; Ananthapadmanabhan, K.P. (Eds.) Interactions of Surfactants with Polymers and Proteins; CRC Press: Boca Raton, FL, USA, 1993. [Google Scholar]
- Cabane, B. Structure of Some Polymer-Detergent Aggregates in Water. J. Phys. Chem. 1977, 81, 1639–1645. [Google Scholar] [CrossRef]
- Brown, G.; Chakrabarti, A. Structure formation in self-associationg polymer and surfactant systems. J. Chem. Phys. 1992, 96, 3251–3254. [Google Scholar] [CrossRef]
- Feitosa, E.; Brown, W.; Hansson, P. Interactions between the Non-Ionic Surfactant C12E5 and Poly(ethylene oxide) Studied Using Dynamic Light Scattering and Fluorescsnce Quenching. Macromolecules 1996, 29, 2169–2178. [Google Scholar] [CrossRef]
- Feitosa, E.; Brown, W.; Vasilescu, M.; Swason-Vethamuthu, M. Effect of Temperature on the Interaction between the Nonionic Surfactant C12E5 and Poly(ethylene oxide) Investigated by Dynamic Light Scattering and Fluorescence Methods. Macromolecules 1996, 29, 6837–6846. [Google Scholar] [CrossRef]
- Annable, T.; Buscall, R.; Ettelaie, R.; Shepherd, P.; Whittlestone, D. Influence of Surfactants on the Rheology of Associating Polymers in Solution. Langmuir 1994, 10, 1060–1070. [Google Scholar] [CrossRef]
- Nilsson, S. Interactions between Water-Soluble Cellulose Derivatives and Surfactants. 1. The HPMC/SDS/Water System. Macromolecules 1995, 28, 7837–7844. [Google Scholar] [CrossRef]
- Piculell, L.; Thuresson, K.; Ericsson, O. Surfactant Binding and Micellization in Polymer Solutions and Gels: Binding Isotherms and their Consequences. Faraday Disc. 1995, 101, 307–318. [Google Scholar] [CrossRef]
- Flory, P.J. Molecular Size Distribution in Three Dimensional Polymers I. Gelation. J. Am. Chem. Soc. 1941, 63, 3083–3090. [Google Scholar] [CrossRef]
- Flory, P.J. Molecular Size Distribution in Three Dimensional Polymers II. Trifunctional Branching Units. J. Am. Chem. Soc. 1941, 63, 3091–3096. [Google Scholar] [CrossRef]
- Flory, P.J. Molecular Size Distribution in Three Dimensional Polymers III. Tetrafunctional Branching Units. J. Am. Chem. Soc. 1941, 63, 3096–3100. [Google Scholar] [CrossRef]
- Flory, P.J. Principles of Polymer Chemistry; Cornell University Press: Ithaca, NY, USA, 1953; Chapter XI. [Google Scholar]
- Stockmayer, W.H. Theory of Molecular Size Distribution and Gel Formation in Branched-Chain Polymers. J. Chem. Phys. 1943, 11, 45–55. [Google Scholar] [CrossRef]
- Stockmayer, W.H. Theory of Molecular Size Distribution and Gel Formation in Branched Polymers II. General Cross Linking. J. Chem. Phys. 1944, 12, 125–131. [Google Scholar] [CrossRef]
- Fukui, K.; Yamabe, T. A General Theory of Gel Formation with Multifunctional Interunit Junctions. Bull. Chem. Soc. Jpn. 1967, 40, 2052–2063. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, F.; Stockmayer, W.H. Thermoreversible Gelation with Junctions of Variable Multiplicity. Macromolecules 1994, 27, 3943–3954. [Google Scholar] [CrossRef]
- Tanaka, F. Polymer Physics—Applications to Molecular Association and Thermoreversible Gelation; Cambridge University Press: Cambridge, UK, 2011. [Google Scholar]
- Macosko, C.W.; Miller, D.R. A New Derivation of Average Molecular Weights of Nonlinear Polymers. Macromolecules 1976, 9, 199–205. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, F. Thermodynamic Theory of Network-Forming Polymer Solutions. 1. Macromolecules 1990, 23, 3784–3789. [Google Scholar] [CrossRef]
- Tanaka, F. Thermodynamic Theory of Network-Forming Polymer Solutions. 2. Equilibrium Gelation by Conterminous Cross-Linking. Macromolecules 1990, 23, 3790–3795. [Google Scholar] [CrossRef]
- Tanaka, F.; Ishida, M. Phase Formation of Two-Component Physical Gels. Phys. A 1994, 204, 660–672. [Google Scholar] [CrossRef]
- Tanaka, F.; Ishida, M. Thermoreversible Gelation with Two-Component Networks. Macromolecules 1999, 32, 1271–1283. [Google Scholar] [CrossRef]
- Tanaka, F. Gel Formation with Multiple Interunit Junctions. I—Molecules Carrying Different Functional Groups. J. Polym. Sci. Part B Polym. Phys. 2003, 41, 2405–2412. [Google Scholar] [CrossRef]
- Tanaka, F. Gel Formation with Multiple Interunit Junctions. II—Mixture of Different Functional Molecules. J. Polym. Sci. Part B Polym. Phys. 2003, 41, 2413–2421. [Google Scholar] [CrossRef]
- Gordon, M. Good’s Theory of Cascade Processes applied to the Statistics of Polymer Distribution. Proc. R. Soc. 1962, A268, 240–257. [Google Scholar]
- Matsuyama, A.; Tanaka, F. Theory of Solvation-Induced Reentrant Phase Separation in Polymer Solutions. Phys. Rev. Lett. 1990, 65, 341–344. [Google Scholar] [CrossRef] [Green Version]
- Ishida, M.; Tanaka, F. Theoretical Study of Hydrogen-Bonded Supramolecular Liquid Crystals. Macromolecules 2002, 35, 7460–7472. [Google Scholar]
- Tanaka, F. Polymer-Surfactant Interaction in Thermoreversible Gels. Macromolecules 1998, 31, 384–393. [Google Scholar] [CrossRef]
- Renou, F.; Benyahia, L.; Nicoli, T. Structure and Rheology of Mixed Polymeric Micelles Formed by Hydrophobically End-Capped Poly(ethylene oxide). Macromolecules 2008, 41, 6523–6530. [Google Scholar] [CrossRef]
- Eldridge, J.E.; Ferry, J.D. Studies on the Cross-Linking Process in Gelatin Gels. III. Dependence of Melting Point on Concentration and Molecular Weight. J. Phys. Chem. 1954, 58, 992–995. [Google Scholar] [CrossRef]
- Tanaka, F.; Nishinari, K. Junction Multiplicity in Thermoreversible Gelation. Macromolecules 1996, 29, 3625–3628. [Google Scholar] [CrossRef]
- Ohkura, M.; Kanaya, T.; Kaji, K. Gels of poly(vinyl alcohol) from dimethyl sulphoxide/water solutions. Polymer 1992, 33, 3686–3690. [Google Scholar] [CrossRef]
- Ohkura, M.; Kanaya, T.; Kaji, K. Gelation rates of poly(vinyl alcohol) solution. Polymer 1992, 33, 5044–5048. [Google Scholar] [CrossRef]
- Ziff, R.M.; Stell, G. Kinetics of Polymer Gelation. J. Chem. Phys. 1980, 73, 3492–3499. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tanaka, F. Thermoreversible Gelation with Two-Component Mixed Cross-Link Junctions of Variable Multiplicity in Ternary Polymer Solutions. Gels 2021, 7, 89. https://doi.org/10.3390/gels7030089
Tanaka F. Thermoreversible Gelation with Two-Component Mixed Cross-Link Junctions of Variable Multiplicity in Ternary Polymer Solutions. Gels. 2021; 7(3):89. https://doi.org/10.3390/gels7030089
Chicago/Turabian StyleTanaka, Fumihiko. 2021. "Thermoreversible Gelation with Two-Component Mixed Cross-Link Junctions of Variable Multiplicity in Ternary Polymer Solutions" Gels 7, no. 3: 89. https://doi.org/10.3390/gels7030089
APA StyleTanaka, F. (2021). Thermoreversible Gelation with Two-Component Mixed Cross-Link Junctions of Variable Multiplicity in Ternary Polymer Solutions. Gels, 7(3), 89. https://doi.org/10.3390/gels7030089