A Diffusion-Reaction Model for Predicting Enzyme-Mediated Dynamic Hydrogel Stiffening



Abstract
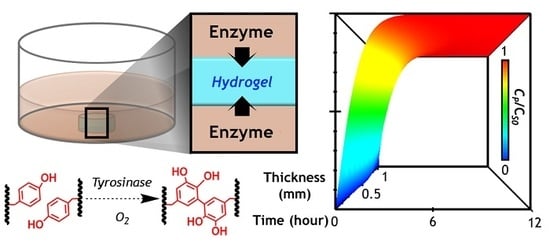
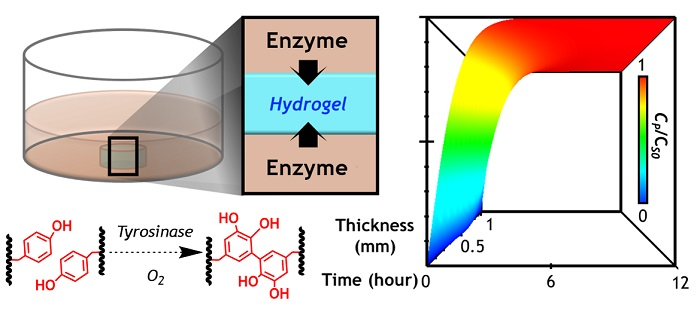
1. Introduction
2. Results and Discussion
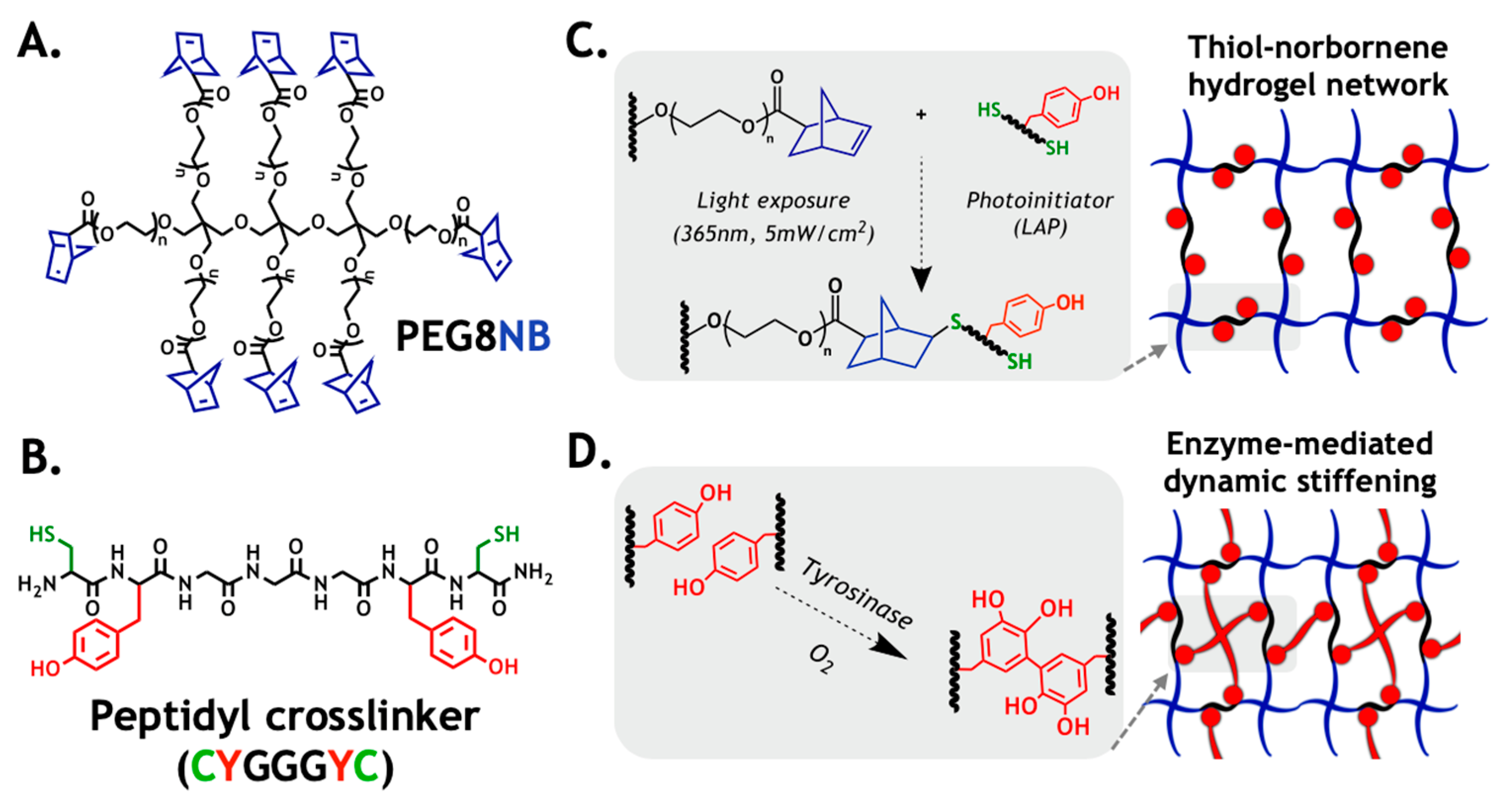
2.1. Design Principle of the Primary Hydrogel Network
2.2. Correlation of Gel Crosslinking Density, Mesh Size, and Enzyme Diffusivity
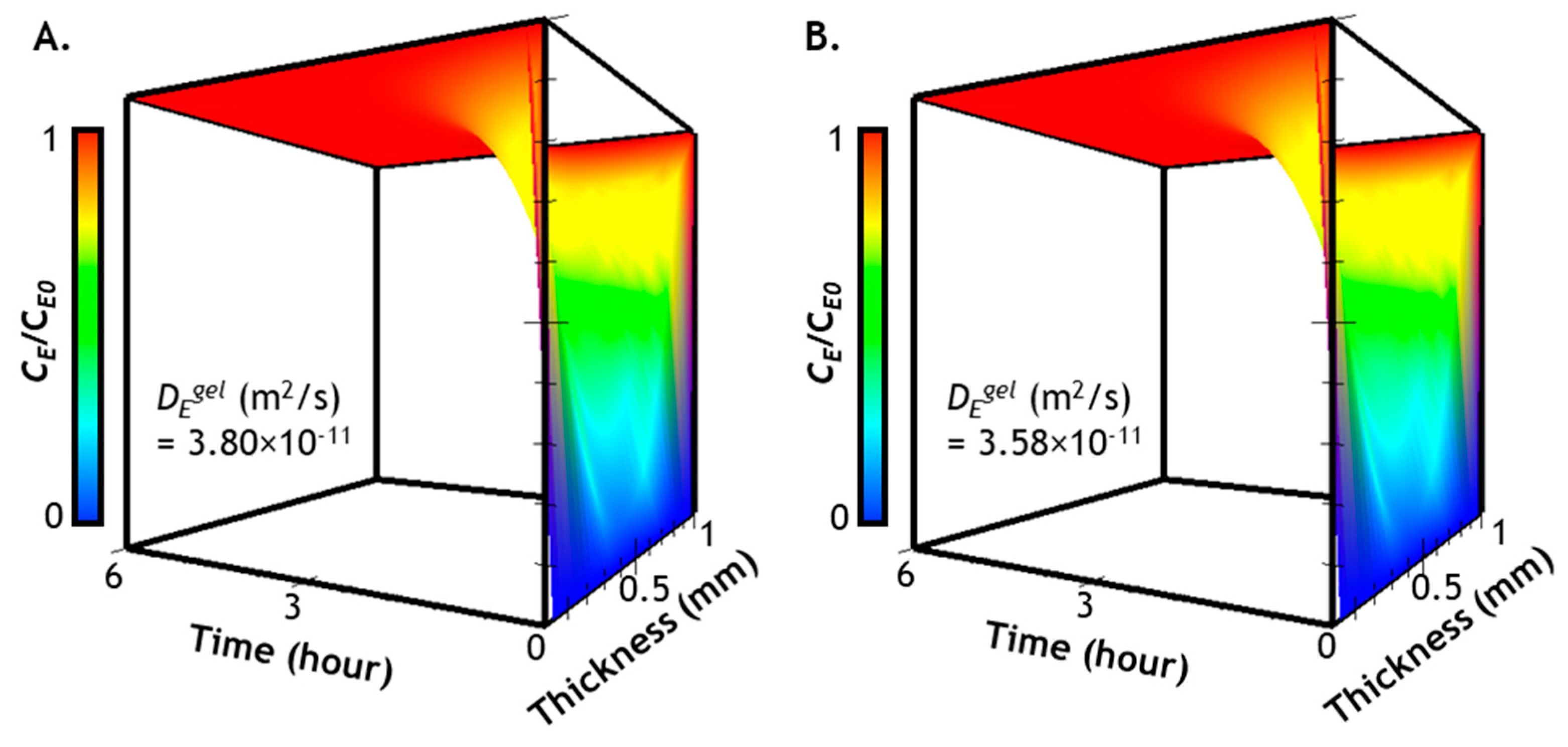
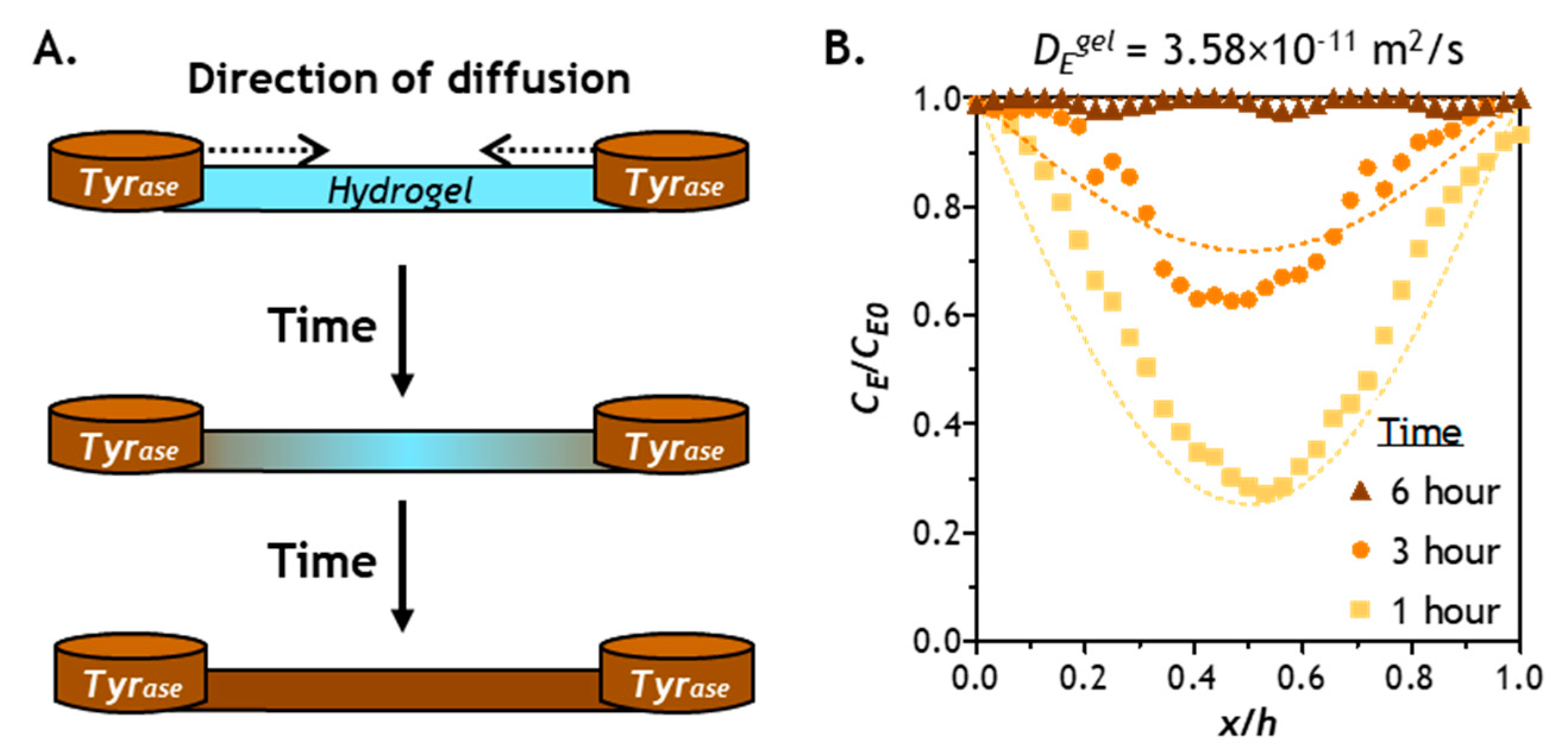
2.3. Prediction of Enzyme Diffusion in Hydrogels with Different Crosslinking Density
2.4. Verification of Enzyme Diffusion in Non-Stiffening Hydrogels
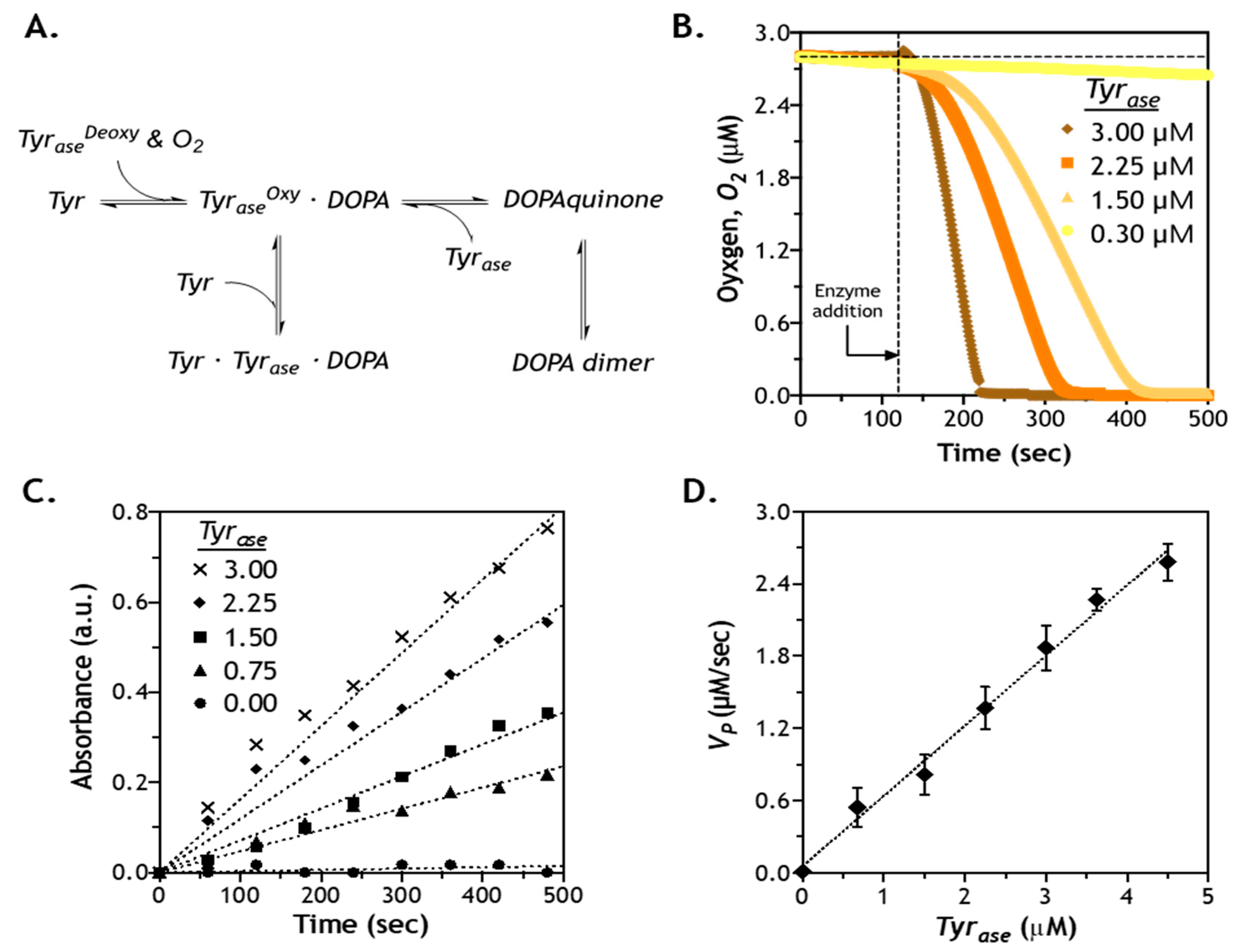
2.5. Effect of Enzyme Concentration on Reaction Velocity
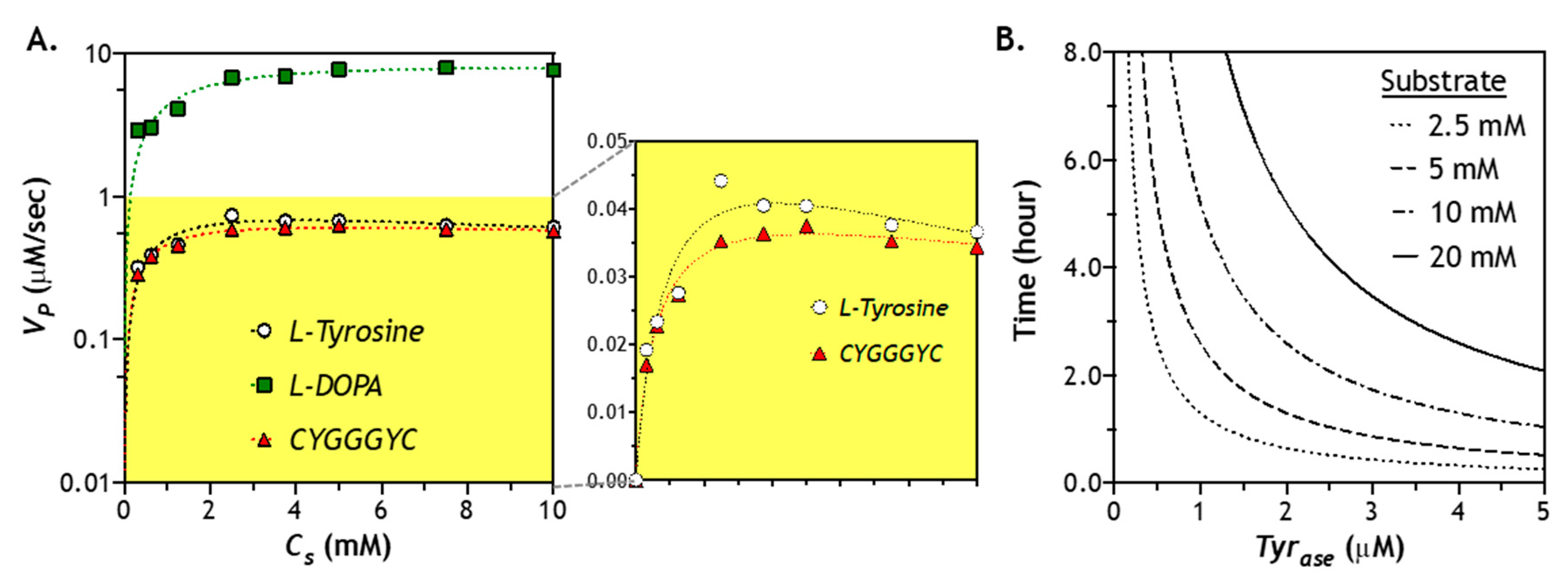
2.6. Effect of Substrate Concentration of Enzymatic Reaction
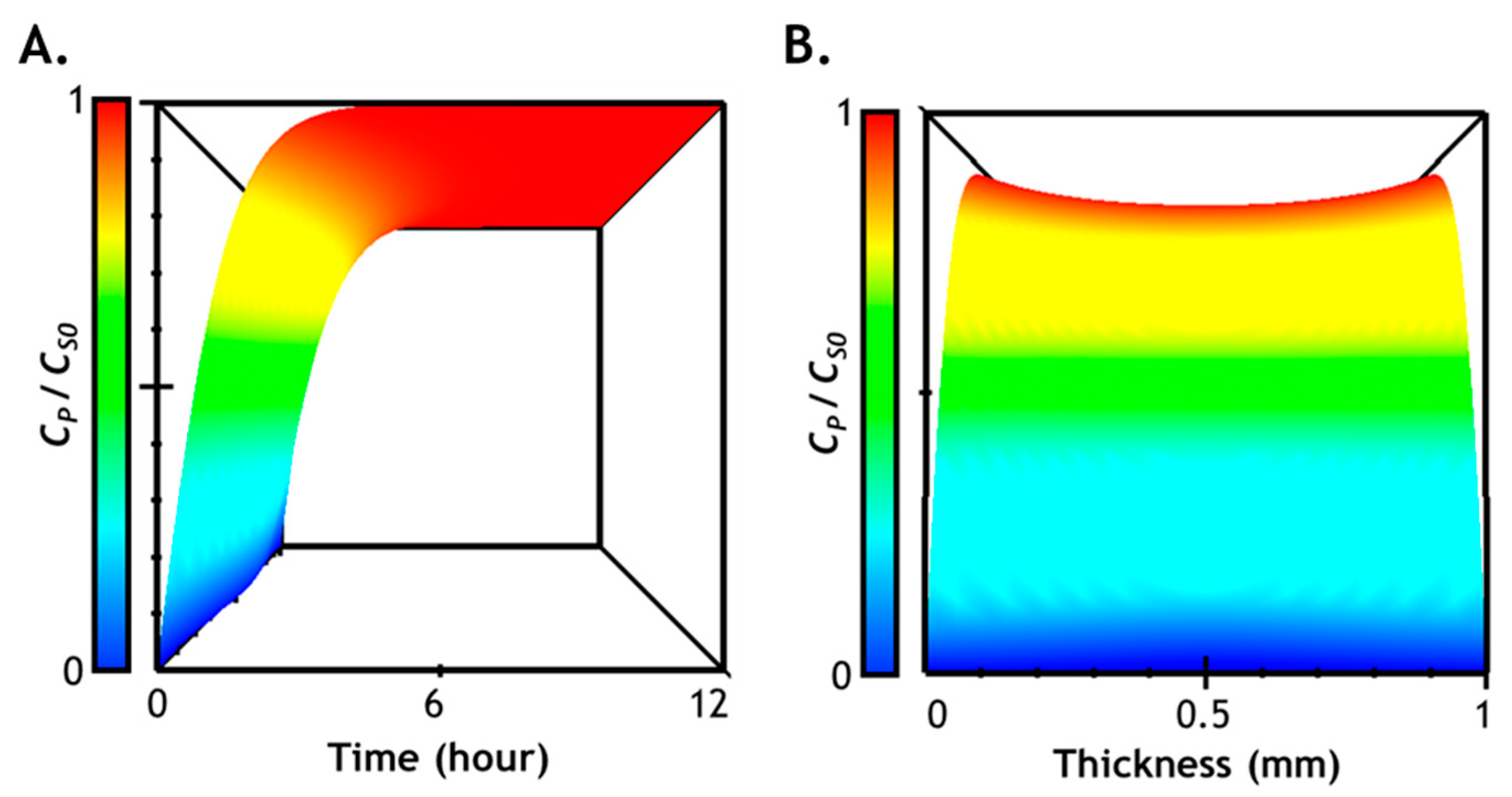
2.7. Numerical Simulation of Diffusion-Reaction in Hydrogel
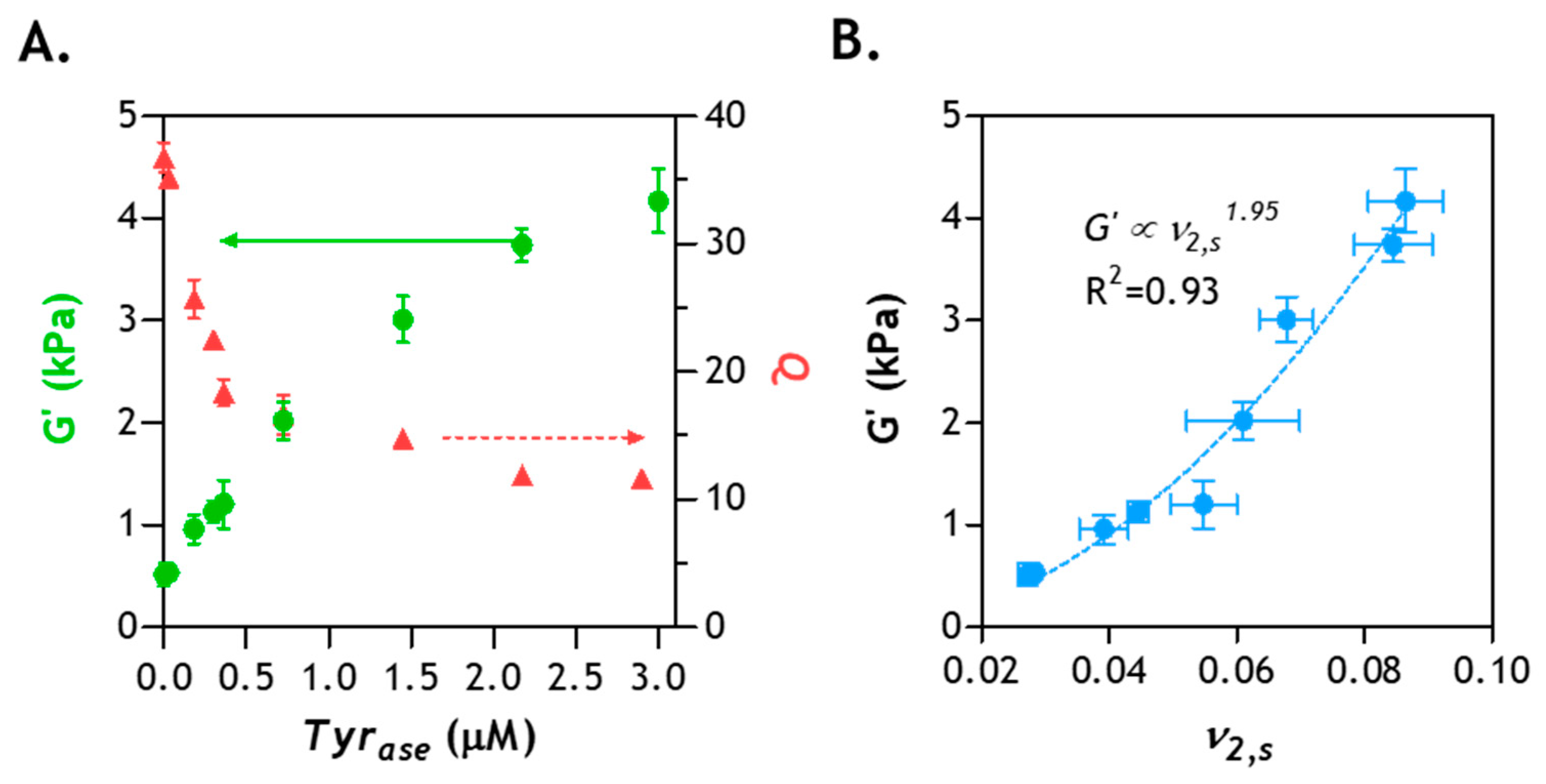
2.8. Correlation of Hydrogel Mechanical Property and Its Microstructure
3. Materials and Methods
3.1. Materials
3.2. Macromer Preparation and Peptide Synthesis
3.3. Modeling of Enzyme Diffusion into Hydrogels
3.4. Characterization of Oxygen Consumption
3.5. Tyrase-Mediated Reaction Kinetics
3.6. Fabrication and Characterization of the Step-Growth PEG-Peptide Hydrogels
3.7. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Lin, C.-C.; Metters, A.T. Hydrogels in controlled release formulations: Network design and mathematical modeling. Adv. Drug Deliv. Rev. 2006, 58, 1379–1408. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K.; Anseth, K.S. Hydrogel scaffolds to study cell biology in four dimensions. MRS Bull. 2013, 38, 260–268. [Google Scholar] [CrossRef]
- Li, Y.; Kumacheva, E. Hydrogel microenvironments for cancer spheroid growth and drug screening. Sci. Adv. 2018, 4. [Google Scholar] [CrossRef] [PubMed]
- Choe, G.; Park, J.; Park, H.; Lee, J. Hydrogel Biomaterials for Stem Cell Microencapsulation. Polymers 2018, 10, 997. [Google Scholar] [CrossRef]
- Cushing, M.C.; Anseth, K.S. Hydrogel Cell Cultures. Science 2007, 316, 1133–1134. [Google Scholar] [CrossRef] [PubMed]
- Trappmann, B.; Baker, B.M.; Polacheck, W.J.; Choi, C.K.; Burdick, J.A.; Chen, C.S. Matrix degradability controls multicellularity of 3D cell migration. Nat. Commun. 2017, 8, 371. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Tong, X.; Jiang, X.; Yang, F. Effect of matrix metalloproteinase-mediated matrix degradation on glioblastoma cell behavior in 3D PEG-based hydrogels. J. Biomed. Mater. Res. Part A 2017, 105, 770–778. [Google Scholar] [CrossRef] [PubMed]
- Brown, T.E.; Silver, J.S.; Worrell, B.T.; Marozas, I.A.; Yavitt, F.M.; Günay, K.A.; Bowman, C.N.; Anseth, K.S. Secondary Photocrosslinking of Click Hydrogels To Probe Myoblast Mechanotransduction in Three Dimensions. J. Am. Chem. Soc. 2018, 140, 11585–11588. [Google Scholar] [CrossRef] [PubMed]
- Cipitria, A.; Salmeron-Sanchez, M. Mechanotransduction and Growth Factor Signalling to Engineer Cellular Microenvironments. Adv. Healthc. Mater. 2017, 6, 1700052. [Google Scholar] [CrossRef] [PubMed]
- Anseth, K.S.; Bowman, C.N.; Brannon-Peppas, L. Mechanical properties of hydrogels and their experimental determination. Biomaterials 1996, 17, 1647–1657. [Google Scholar] [CrossRef]
- Anseth, K.S.; Bowman, C.N. Kinetic Gelation model predictions of crosslinked polymer network microstructure. Chem. Eng. Sci. 1994, 49, 2207–2217. [Google Scholar] [CrossRef]
- Chaudhuri, O. Viscoelastic hydrogels for 3D cell culture. Biomater. Sci. 2017, 5, 1480–1490. [Google Scholar] [CrossRef] [PubMed]
- Caccavo, D.; Cascone, S.; Lamberti, G.; Barba, A.A. Hydrogels: Experimental characterization and mathematical modelling of their mechanical and diffusive behaviour. Chem. Soc. Rev. 2018. [Google Scholar] [CrossRef] [PubMed]
- Kloxin, A.M.; Kloxin, C.J.; Bowman, C.N.; Anseth, K.S. Mechanical Properties of Cellularly Responsive Hydrogels and Their Experimental Determination. Adv. Mater. 2010, 22, 3484–3494. [Google Scholar] [CrossRef]
- Chaudhuri, O.; Gu, L.; Klumpers, D.; Darnell, M.; Bencherif, S.A.; Weaver, J.C.; Huebsch, N.; Lee, H.-P.P.; Lippens, E.; Duda, G.N.; et al. Hydrogels with tunable stress relaxation regulate stem cell fate and activity. Nat. Mater. 2016, 15, 326–334. [Google Scholar] [CrossRef]
- Shih, H.; Lin, C.-C. Cross-Linking and Degradation of Step-Growth Hydrogels Formed by Thiol–Ene Photoclick Chemistry. Biomacromolecules 2012, 13, 2003–2012. [Google Scholar] [CrossRef]
- Falcone, N.; Kraatz, H.-B.B. Stimuli-Responsive Peptide Gels and some Bio-Applications. Chemistry 2018. [Google Scholar] [CrossRef]
- Schweller, R.M.; West, J.L. Encoding Hydrogel Mechanics via Network Cross-Linking Structure. ACS Biomater. Sci. Eng. 2015, 1, 335–344. [Google Scholar] [CrossRef]
- Laklai, H.; Miroshnikova, Y.A.; Pickup, M.W.; Collisson, E.A.; Kim, G.E.; Barrett, A.S.; Hill, R.C.; Lakins, J.N.; Schlaepfer, D.D.; Mouw, J.K.; et al. Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression. Nat. Med. 2016, 22, 497–505. [Google Scholar] [CrossRef]
- Calvo, F.; Ege, N.; Grande-Garcia, A.; Hooper, S.; Jenkins, R.P.; Chaudhry, S.I.; Harrington, K.; Williamson, P.; Moeendarbary, E.; Charras, G.; et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 2013, 15, 637. [Google Scholar] [CrossRef] [PubMed]
- Burdick, J.A.; Murphy, W.L. Moving from static to dynamic complexity in hydrogel design. Nat. Commun. 2012, 3, 1269. [Google Scholar] [CrossRef] [PubMed]
- Stowers, R.S.; Allen, S.C.; Suggs, L.J. Dynamic phototuning of 3D hydrogel stiffness. Proc. Natl. Acad. Sci. USA 2015, 112, 1953–1958. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Hu, J.; Wang, H.; Huang, J.; Yu, Y.; Zhang, Q.; Cheng, Y. Dynamic softening or stiffening a supramolecular hydrogel by an ultraviolet or near-infrared light. ACS Appl. Mater. Interfaces 2017. [Google Scholar] [CrossRef]
- Guvendiren, M.; Burdick, J.A. Stiffening hydrogels to probe short- and long-term cellular responses to dynamic mechanics. Nat. Commun. 2012, 3, 792. [Google Scholar] [CrossRef] [PubMed]
- Tibbitt, M.W.; Anseth, K.S. Dynamic microenvironments: The fourth dimension. Sci. Transl. Med. 2012, 4. [Google Scholar] [CrossRef]
- Young, J.L.; Engler, A.J. Hydrogels with time-dependent material properties enhance cardiomyocyte differentiation in vitro. Biomaterials 2011, 32, 1002–1009. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.-Y.; Korc, M.; Lin, C.-C. Biomimetic and enzyme-responsive dynamic hydrogels for studying cell-matrix interactions in pancreatic ductal adenocarcinoma. Biomaterials 2018, 160, 24–36. [Google Scholar] [CrossRef]
- Liu, H.-Y.; Greene, T.; Lin, T.-Y.; Dawes, C.S.; Korc, M.; Lin, C.-C. Enzyme-mediated stiffening hydrogels for probing activation of pancreatic stellate cells. Acta Biomater. 2017, 48, 258–269. [Google Scholar] [CrossRef]
- Lee, B.P.; Dalsin, J.L.; Messersmith, P.B. Synthesis and Gelation of DOPA-Modified Poly(ethylene glycol) Hydrogels. Biomacromolecules 2002, 3, 1038–1047. [Google Scholar] [CrossRef]
- Kord Forooshani, P.; Lee, B.P. Recent approaches in designing bioadhesive materials inspired by mussel adhesive protein. J. Polym. Sci. A Polym. Chem. 2017, 55, 9–33. [Google Scholar] [CrossRef]
- Skaalure, S.C.; Akalp, U.; Vernerey, F.J.; Bryant, S.J. Tuning Reaction and Diffusion Mediated Degradation of Enzyme-Sensitive Hydrogels. Adv. Healthc. Mater. 2016, 5, 432–438. [Google Scholar] [CrossRef]
- Rehmann, M.S.; Skeens, K.M.; Kharkar, P.M.; Ford, E.M.; Maverakis, E.; Lee, K.H.; Kloxin, A.M. Tuning and Predicting Mesh Size and Protein Release from Step Growth Hydrogels. Biomacromolecules 2017, 18, 3131–3142. [Google Scholar] [CrossRef]
- Rubiano, A.; Delitto, D.; Han, S.; Gerber, M.; Galitz, C.; Trevino, J.; Thomas, R.M.; Hughes, S.J.; Simmons, C.S. Viscoelastic properties of human pancreatic tumors and in vitro constructs to mimic mechanical properties. Acta Biomater. 2017, 67, 331–340. [Google Scholar] [CrossRef]
- Hearing, V.J.; Ekel, T.M.; Montague, P.M.; Nicholson, J.M. Mammalin tyrosinase. Stoichiometry and measurement of reaction products. Biochim. Biophys. Acta 1980, 611, 251–268. [Google Scholar] [CrossRef]
- Lustig, S.R.; Peppas, N.A. Solute diffusion in swollen membranes. IX. Scaling laws for solute diffusion in gels. J. Appl. Polym. Sci. 1988, 36, 735–747. [Google Scholar] [CrossRef]
- Aimetti, A.A.; Tibbitt, M.W.; Anseth, K.S. Human neutrophil elastase responsive delivery from poly(ethylene glycol) hydrogels. Biomacromolecules 2009, 10, 1484–1489. [Google Scholar] [CrossRef]
- Espín, J.C.; Varón, R.; Fenoll, L.G.; Gilabert, M.A.; García-Ruíz, P.A.; Tudela, J.; García-Cánovas, F. Kinetic characterization of the substrate specificity and mechanism of mushroom tyrosinase. Eur. J. Biochem. 2000, 267, 1270–1279. [Google Scholar] [CrossRef]
- Kim, Y.; Yun, J.; Lee, C.-K.; Lee, H.; Min, K.; Kim, Y. Oxyresveratrol and Hydroxystilbene Compounds inhibitory effect on tyrosinase and mechanism of Action. J. Biol. Chem. 2002, 277, 16340–16344. [Google Scholar] [CrossRef]
- Fenoll, L.G.; Rodríguez-López, J.; García-Molina, F.; García-Cánovas, F.; Tudela, J. Michaelis constants of mushroom tyrosinase with respect to oxygen in the presence of monophenols and diphenols. Int. J. Biochem. Cell Biol. 2002, 34, 332–336. [Google Scholar] [CrossRef]
- Winder, A.J. A stopped spectrophotometric assay for the dopa oxidase activity of tyrosinase. J. Biochem. Biophys. Methods 1994, 28, 173–183. [Google Scholar] [CrossRef]
- Ferrari, R.; Laurenti, E.; Ghibaudi, E.; Casella, L. Tyrosinase-catecholic substrates in Vitro model: Kinetic studies on the o-quinone/o-semiquinone radical formation. J. Inorganic Biochem. 1997, 68, 61–69. [Google Scholar] [CrossRef]
- Reed, M.C.; Lieb, A.; Nijhout, F.H. The biological significance of substrate inhibition: A mechanism with diverse functions. BioEssays 2010, 32, 422–429. [Google Scholar] [CrossRef] [PubMed]
- Marumo, K.; Waite, J.H. Optimization of hydroxylation of tyrosine and tyrosine-containing peptides by mushroom tyrosinase. Biochim. Biophys. Acta 1986, 872, 98–103. [Google Scholar] [CrossRef]
- Barry, D.A.; Parlange, J.Y.; Li, L.; Prommer, H.; Cunningham, C.J.; Stagnitti, F. Analytical approximations for real values of the Lambert W-function. Math. Comput. Simul. 2000, 53, 95–103. [Google Scholar] [CrossRef]
- Schleeger, M.; Heberle, J.; Kakorin, S. Simplifying the Analysis of Enzyme Kinetics of Cytochrome Oxidase by the Lambert-W Function. Open J. Biophys. 2012, 2012, 117–129. [Google Scholar] [CrossRef]
- Anseth, K.S.; Metters, A.T.; Bryant, S.J.; Martens, P.J.; Elisseeff, J.H.; Bowman, C.N. In situ forming degradable networks and their application in tissue engineering and drug delivery. J. Control Release 2002, 78, 199–209. [Google Scholar] [CrossRef]
- Flory, P.J. Principles of Polymer Chemistry; Cornell University Press: Ithaca, NY, USA, 1953. [Google Scholar]
- Yazici, I.; Okay, O. Spatial inhomogeneity in poly(acrylic acid) hydrogels. Polymer 2005, 46, 2595–2602. [Google Scholar] [CrossRef]
- Lin, C.-C.; Raza, A.; Shih, H. PEG hydrogels formed by thiol-ene photo-click chemistry and their effect on the formation and recovery of insulin-secreting cell spheroids. Biomaterials 2011, 32, 9685–9695. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-C.; Anseth, K.S. Cell-cell communication mimicry with poly(ethylene glycol) hydrogels for enhancing beta-cell function. Proc. Natl. Acad. Sci. USA 2011, 108, 6380–6385. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.C.; Ali, R. Hydrodynamic properties of mushroom tyrosinase. Phytochemistry 1981, 20, 399–401. [Google Scholar] [CrossRef]
- Duggleby, R.G.; Morrison, J.F. The analysis of progress curves for enzyme-catalysed reactions by non-linear regression. Biochim. Biophys. Acta 1977, 481, 297–312. [Google Scholar] [CrossRef]
- Cleland, W.W. The kinetics of enzyme-catalyzed reactions with two or more substrates or products: I. Nomenclature and rate equations. Biochim. Biophys. Acta 1963, 67, 104–137. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| l-Tyrosine | l-DOPA | CYGGGYC | |
|---|---|---|---|
| kcat (s−1) | 0.93 | 8.63 | 0.60 |
| KM (mM) | 0.85 | 1.02 | 0.58 |
| Ki (mM) | 19.85 | - | - |
| R2 | 0.956 | 0.971 | 0.98 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, H.-Y.; Lin, C.-C. A Diffusion-Reaction Model for Predicting Enzyme-Mediated Dynamic Hydrogel Stiffening. Gels 2019, 5, 17. https://doi.org/10.3390/gels5010017
Liu H-Y, Lin C-C. A Diffusion-Reaction Model for Predicting Enzyme-Mediated Dynamic Hydrogel Stiffening. Gels. 2019; 5(1):17. https://doi.org/10.3390/gels5010017
Chicago/Turabian StyleLiu, Hung-Yi, and Chien-Chi Lin. 2019. "A Diffusion-Reaction Model for Predicting Enzyme-Mediated Dynamic Hydrogel Stiffening" Gels 5, no. 1: 17. https://doi.org/10.3390/gels5010017
APA StyleLiu, H.-Y., & Lin, C.-C. (2019). A Diffusion-Reaction Model for Predicting Enzyme-Mediated Dynamic Hydrogel Stiffening. Gels, 5(1), 17. https://doi.org/10.3390/gels5010017