The Twofold Role of 12-Hydroxyoctadecanoic Acid (12-HOA) in a Ternary Water—Surfactant—12-HOA System: Gelator and Co-Surfactant


Abstract
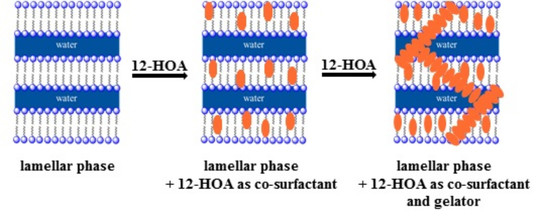
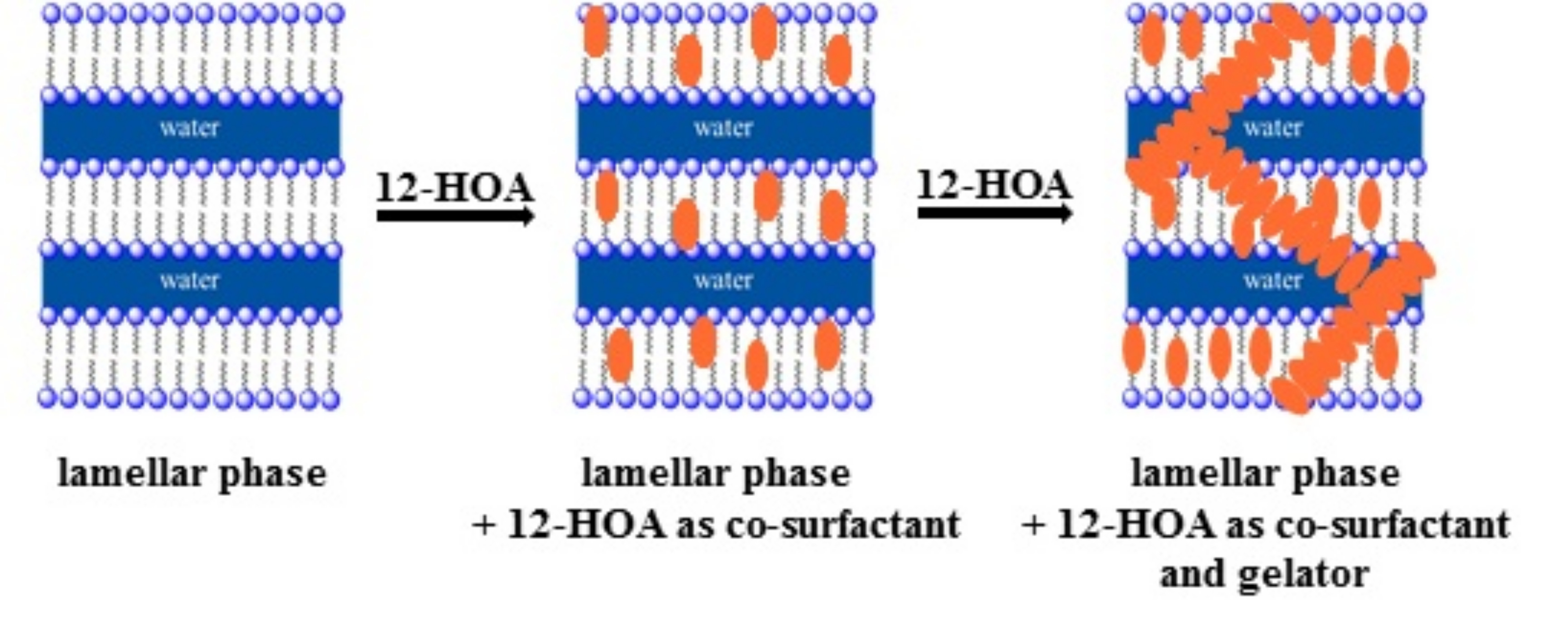{kind=link}
{kind=link}
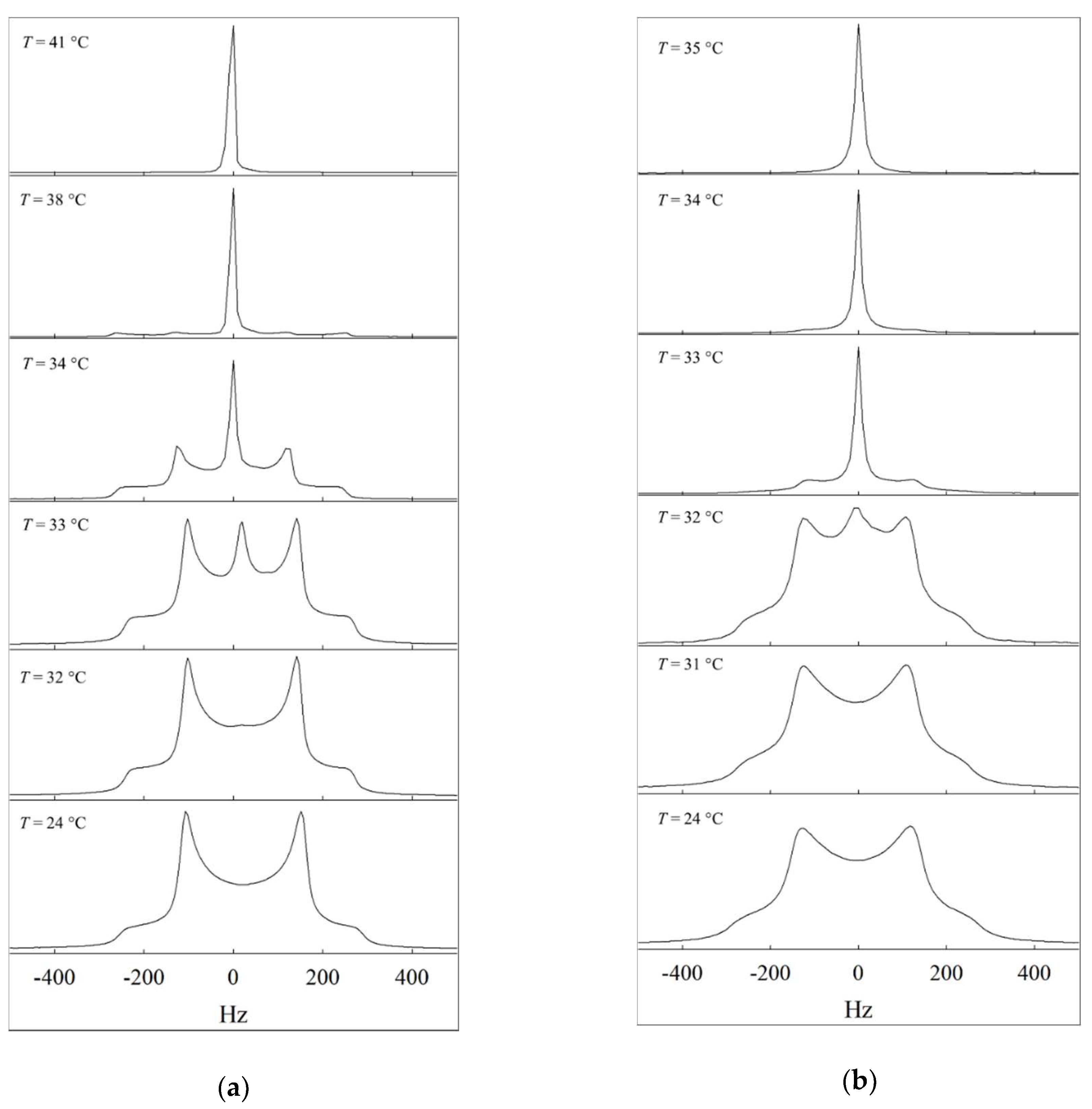{kind=link}
{kind=link}
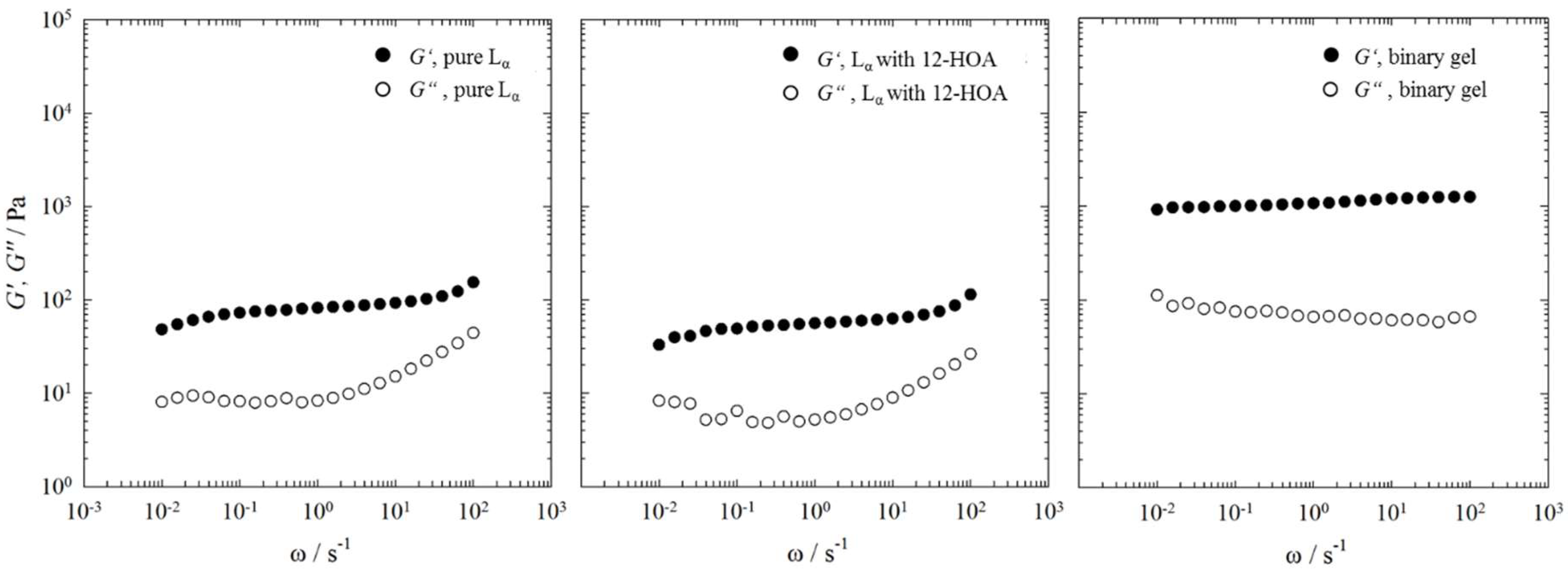{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
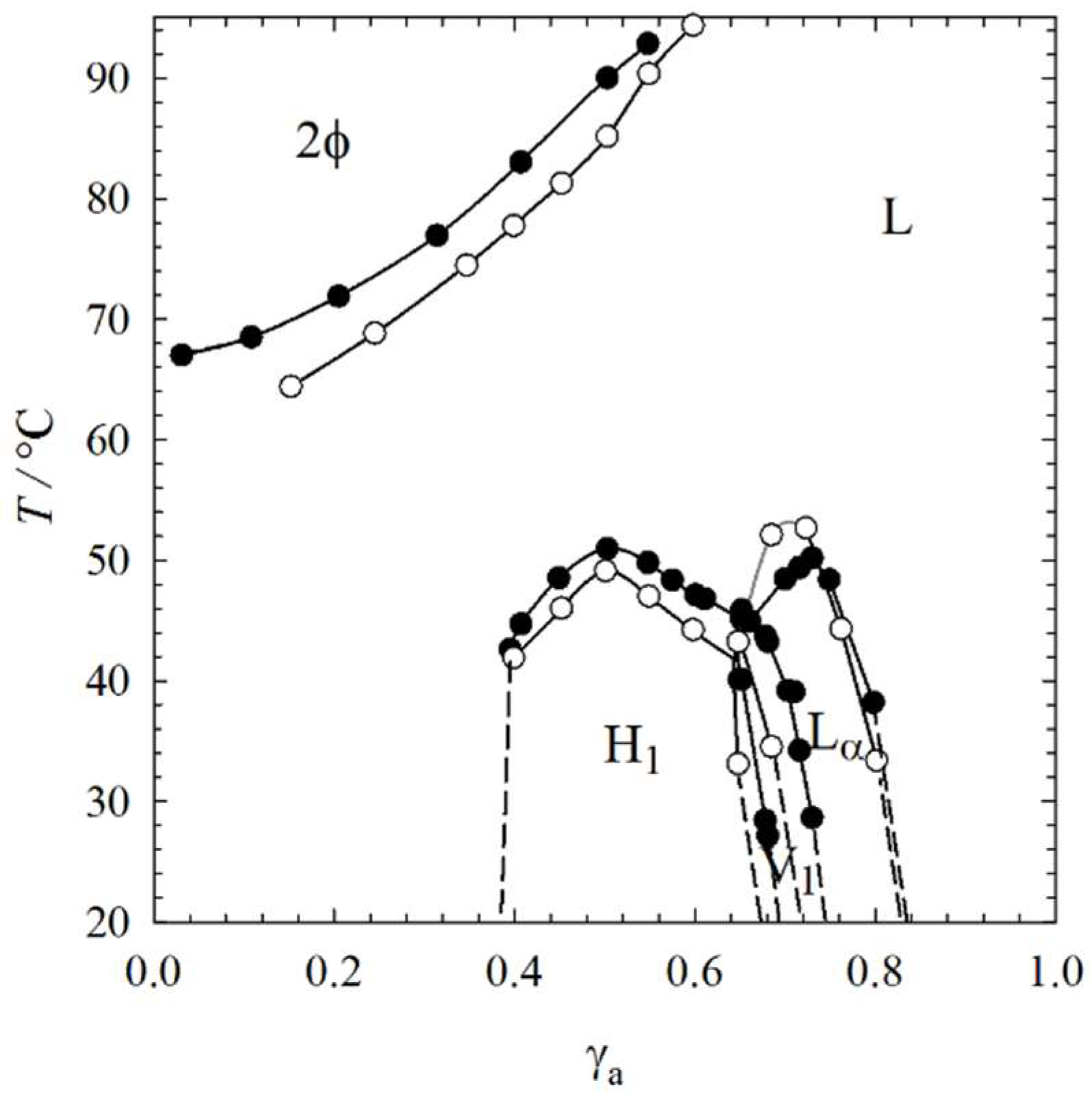
2.1. Visual Phase Studies
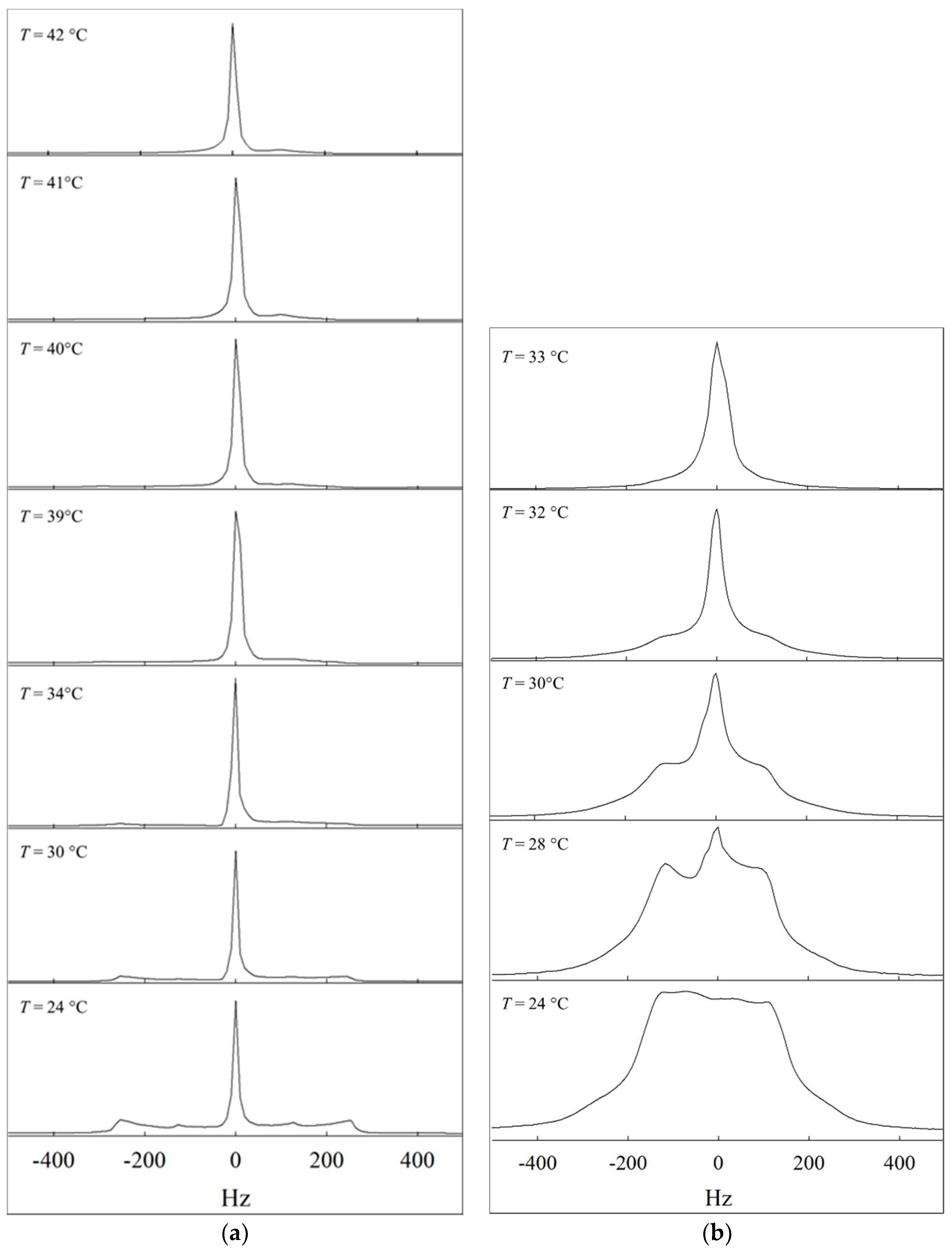
2.2. 2H NMR
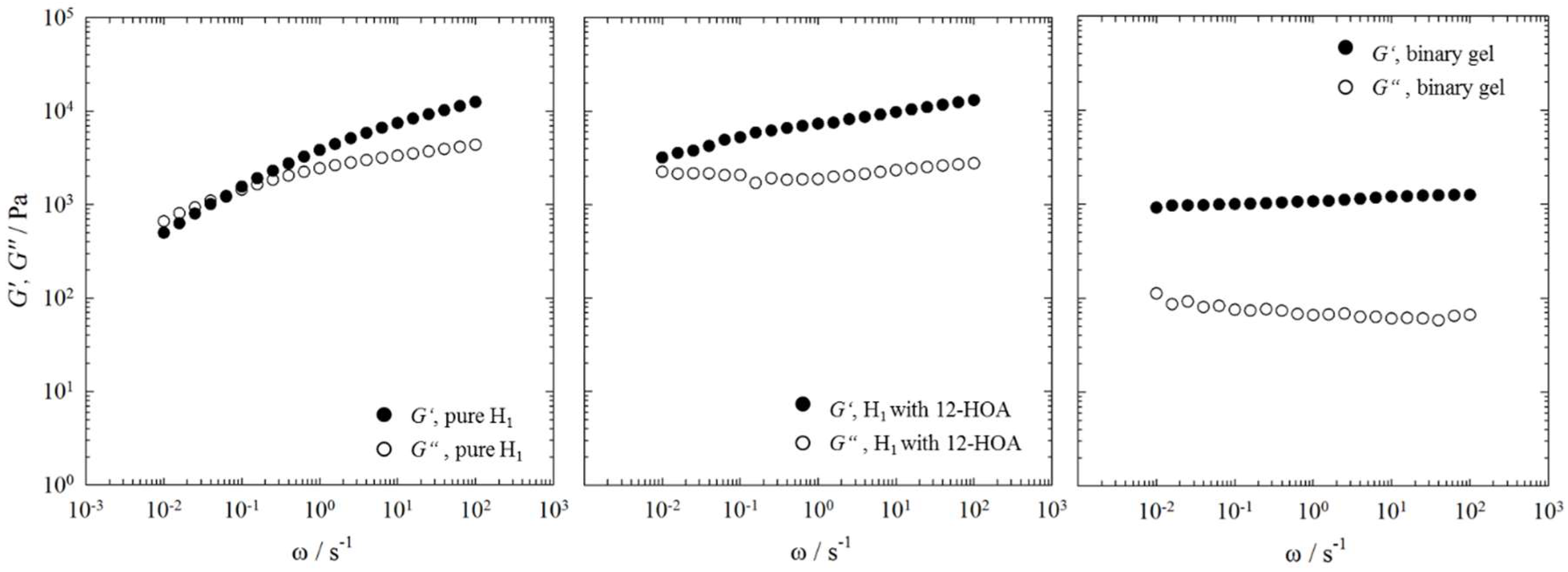
2.3. Rheometry
3. Conclusions
4. Materials and Methods
4.1. Materials and Sample Preparation
4.2. Visual Phase Studies
4.3. 2H NMR
4.4. Rheometry
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A


References
- Stubenrauch, C.; Gießelmann, F. Gelled Complex Fluids: Combining Unique Structures with Mechanical Stability. Angew. Chem. Int. Ed. 2016, 55, 3268–3275. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-H.; Jahn, A.; Cho, S.-J.; Kim, J.S.; Ki, M.-H.; Kim, D.-D. Lyotropic liquid crystal systems in drug delivery: A review. J. Pharm. Investig. 2015, 45, 1–45. [Google Scholar] [CrossRef]
- Kelly, C.V.-D. Perturbations of Cellular Membranes with Synthetic Polymers and Ultrafast Lasers. Ph.D. Thesis, University of Michigan, Ann Arbor, MI, USA, 2009. [Google Scholar]
- Warriner, H.E.; Idziak, S.H.J.; Slack, N.L.; Davidson, P.; Safinya, C.R. Lamellar Biogels: Fluid-Membrane-Based Hydrogels Containing Polymer Lipids. Science 1996, 271, 969–973. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Laupheimer, M.; Preisig, N.; Sottmann, T.; Schmidt, C.; Stubenrauch, C. Gelled Lyotropic Liquid Crystals. Langmuir 2015, 31, 8589–8598. [Google Scholar] [CrossRef] [PubMed]
- Koitani, S.; Dieterich, S.; Preisig, N.; Aramaki, K.; Stubenrauch, C. Gelling Lamellar Phases of the Binary System Water–Didodecyldimethylammonium Bromide with an Organogelator. Langmuir 2017, 33, 12171–12179. [Google Scholar] [CrossRef] [PubMed]
- Laibinis, P.E.; Hickman, J.J.; Wrighton, M.S.; Whitesides, G.M. Orthogonal Self-Assembled Monolayers: Alkanethiols on Gold and Alkane Carboxylic Acids on Alumina. Science 1989, 245, 845–847. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Xiao, T.; Lin, C.; Huang, F.; Wang, L. Dynamic Supramolecular Complexes Constructed by Orthogonal Self-Assembly. Acc. Chem. Res. 2014, 47, 2041–2051. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.K.; Steed, J.W. Supramolecular gel phase crystallization: Orthogonal self-assembly under non-equilibrium conditions. Chem. Soc. Rev. 2014, 43, 2080–2088. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Xiao, T.; Lin, C.; Wang, L. Advanced supramolecular polymers constructed by orthogonal self-assembly. Chem. Soc. Rev. 2012, 41, 5950–5968. [Google Scholar] [CrossRef] [PubMed]
- Saha, M.L.; De, S.; Pramanik, S.; Schmittel, M. Orthogonality in discrete self-assembly—Survey of current concepts. Chem. Soc. Rev. 2013, 42, 6860–6909. [Google Scholar] [CrossRef] [PubMed]
- Wei, P.; Yan, X.; Huang, F. Supramolecular polymers constructed by orthogonal self-assembly based on host–guest and metal–ligand interactions. Chem. Soc. Rev. 2015, 44, 815–832. [Google Scholar] [CrossRef] [PubMed]
- Heeres, A.; van der Pol, C.; Stuart, M.; Friggeri, A.; Feringa, B.L.; van Esch, J. Orthogonal self-assembly of Low Molecular Weight Hydrogelators and Surfactants. J. Am. Chem. Soc. 2003, 125, 14252–14253. [Google Scholar] [CrossRef] [PubMed]
- Brizard, A.; Stuart, M.; van Bommel, K.; Friggeri, A.; de Jong, M.; van Esch, J. Preparation of Nanostructures by Orthogonal Self-Assembly of Hydrogelators and Surfactants. Angew. Chem. Int. Ed. 2008, 47, 2063–2066. [Google Scholar] [CrossRef] [PubMed]
- Brizard, A.M.; van Esch, J.H. Self-assembly approaches for the construction of cell architecture mimics. Soft Matter 2009, 5, 1320–1327. [Google Scholar] [CrossRef]
- Brizard, A.M.; Stuart, M.C.A.; van Esch, J.H. Self-assembled interpenetrating networks by orthogonal self-assembly of surfactants and hydrogelators. Faraday Discuss. 2009, 143, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Boekhoven, J.; Brizard, A.M.; Stuart, M.C.A.; Florusse, L.; Raffy, G.; Del Guerzo, A.; van Esch, J.H. Bio-inspired supramolecular materials by orthogonal self-assembly of hydrogelators and phospholipids. Chem. Sci. 2016, 00, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Laupheimer, M. Gelled Bicontinuous Micoremulsions: A New Type of Orthogonal Self-Assembled Systems. Ph.D. Thesis, University of Stuttgart, Stuttgart, Germany, 2013. [Google Scholar]
- Laupheimer, M.; Jovic, K.; Antunes, F.E.; da Graça Martins Miguel, M.; Stubenrauch, C. Studying orthogonal self-assembled systems: Phase behaviour and rheology of gelled microemulsions. Soft Matter 2013, 9, 3661–3670. [Google Scholar] [CrossRef]
- Laupheimer, M.; Sottman, T.; Schweins, R.; Stubenrauch, C. Studying orthogonal self-assembled systems: Microstructure of gelled bicontinuous microemulsions. Soft Matter 2014, 10, 8744–8757. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Matsuda, M.; Nibu, Y.; Misono, Y.; Suzuki, M. Phase Behavior of Heptaethylene Glycol Dodecyl Ether and Its Aqueous Mixture Revealed by DSC and FT-IR Spectroscopy. Langmuir 2001, 17, 1833–1840. [Google Scholar] [CrossRef]
- Blackburn, J.C.; Kilpatrick, P.K. Using Deuterium NMR Line Shapes to Analyze Lyotropic Liquid Crystalline Phase Transitions. Langmuir 1992, 8, 1679–1687. [Google Scholar] [CrossRef]
- Davis, J.H. The description of membrane lipid conformation, order and dynamics by 2H-NMR. Biochim. Biophys. Acta 1983, 737, 117–171. [Google Scholar] [CrossRef]
- Dufourc, E.J.; Parish, E.J.; Chitrakorn, S.I.C.P. Smith Structural and Dynamical Details of Cholesterol-Lipid Interaction as Revealed by Deuterium NMR. Biochemistry 1984, 23, 6062–6071. [Google Scholar] [CrossRef]
- Jelinski, L.W. Deuterium NMR of Solid Polymers. In High resolution NMR Spectroscopy of Synthetic Polymers in Bulk; Komorowski, R.A., Ed.; VHC Publishers: New York, NY, USA, 1986; pp. 335–364. [Google Scholar]
- Lukaschek, M.; Grabowski, D.A.; Schmidt, C. Shear-Induced Alignment of a Hexagonal Lyotropic Liquid Crystal as Studied by Rheo-NMR. Langmuir 1995, 11, 3590–3594. [Google Scholar] [CrossRef]
- Seelig, J. Deuterium magnetic resonance: Theory and application to lipid membranes. Q. Rev. Biophys. 1977, 10, 353–418. [Google Scholar] [CrossRef] [PubMed]
- Stubenrauch, C.; Frank, C.; Strey, R.; Burgmeister, D.; Schmidt, C. Lyotropic Mesophases Next to Highly Efficient Microemulsions: A 2H NMR Study. Langmuir 2002, 18, 5027–5030. [Google Scholar] [CrossRef]
- Stubenrauch, C.; Burauer, S.; Strey, R. A new approach to lamellar phases (Lα) in water–non-ionic surfactant systems. Liq. Cryst. 2004, 31, 39–53. [Google Scholar] [CrossRef]
- Stockton, G.W.; Polnaszek, C.F.; Tulloch, A.P.; Hasan, F.; Smith, I.C.P. Molecular Motion and Order in Single-Bilayer Vesicles and Multilamellar Dispersions of Egg Lecithin and Lecithin-Cholesterol Mixtures. A Deuterium Nuclear Magnetic Resonance Study of Specifically Labeled Lipids. Biochemistry 1976, 15, 954–966. [Google Scholar] [CrossRef] [PubMed]
- Israelachvili, J.N.; Mitchell, D.J.; Ninham, B.W. Theory of Self-Assembly of Hydrocarbon Amphiphiles into Micelles and Bilayers. J. Chem. Soc. Faraday Trans. 2 1976, 72, 1525–1568. [Google Scholar] [CrossRef]
- Mitchell, D.J.; Tiddy, G.J.T.; Waring, L.; Bostock, T.; McDonald, M.P. Phase behaviour of Polyoxyethylene Surfactants with Water. J. Chem. Soc. Faraday Trans. 1 1983, 79, 975–1000. [Google Scholar] [CrossRef]
- Hoffmann, H.; Ulbricht, W. Faszinierende Phänomene in Tensidlösungen. Chemie Unserer Zeit 1995, 29, 76–86. [Google Scholar] [CrossRef]
- Pake, G.E. Nuclear Resonance Absorption in Hydrated Crystals: Fine Structure of the Proton Line. J. Chem. Phys. 1948, 16, 327–336. [Google Scholar] [CrossRef]
- Nishinari, K. Some Thoughts on the Defnition of a Gel. Prog. Colloid Polym. Sci. 2009, 136, 87–94. [Google Scholar] [CrossRef]
- Terech, P.; Rodriguez, V.; Barnes, J.D.; McKenna, G.B. Organogels and Aerogels of Racemic and Chiral 12-Hydroxyoctadecanoic Acid. Langmuir 1994, 10, 3406–3418. [Google Scholar] [CrossRef]
- Terech, P.; Weiss, R.G. Low Molecular Mass Gelators of Organic Liquids and the Properties of Their Gels. Chem. Rev. 1997, 97, 3113–3160. [Google Scholar] [CrossRef]
- Terech, P.; Pasquier, D.; Bordas, V.; Rossat, C. Rheological Properties and Structural Correlations in Molecular Organogels. Langmuir 2000, 16, 4485–4494. [Google Scholar] [CrossRef]
- Weiss, R.G.; Terech, P. Molecular Gels: Materials with Self-Assembled Fibrillar Networks; Springer: Dordrecht, The Netherlands, 2005; ISBN 978-1-4020-3689-7. [Google Scholar]
- Collings, P.J.; Hird, M. Introduction to Liquid Crystals: Chemistry and Physics; Taylor & Francis Ltd.: London, UK, 1997; ISBN 9780748404834. [Google Scholar]
- Kato, T.; Hirai, Y.; Nakaso, S.; Moriyama, M. Liquid Crystalline physical gels. Chem. Soc. Rev. 2007, 36, 1857–1867. [Google Scholar] [CrossRef] [PubMed]
- Laupheimer, M.; Preisig, N.; Stubenrauch, C. The Molecular Organogel n-Decane/12-Hydroxyoctadecanoic Acid: Sol-Gel Tranisition, Rheology and Microstructure. COLSUA 2015, 469, 315–325. [Google Scholar] [CrossRef]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Steck, K.; Schmidt, C.; Stubenrauch, C. The Twofold Role of 12-Hydroxyoctadecanoic Acid (12-HOA) in a Ternary Water—Surfactant—12-HOA System: Gelator and Co-Surfactant. Gels 2018, 4, 78. https://doi.org/10.3390/gels4030078
Steck K, Schmidt C, Stubenrauch C. The Twofold Role of 12-Hydroxyoctadecanoic Acid (12-HOA) in a Ternary Water—Surfactant—12-HOA System: Gelator and Co-Surfactant. Gels. 2018; 4(3):78. https://doi.org/10.3390/gels4030078
Chicago/Turabian StyleSteck, Katja, Claudia Schmidt, and Cosima Stubenrauch. 2018. "The Twofold Role of 12-Hydroxyoctadecanoic Acid (12-HOA) in a Ternary Water—Surfactant—12-HOA System: Gelator and Co-Surfactant" Gels 4, no. 3: 78. https://doi.org/10.3390/gels4030078
APA StyleSteck, K., Schmidt, C., & Stubenrauch, C. (2018). The Twofold Role of 12-Hydroxyoctadecanoic Acid (12-HOA) in a Ternary Water—Surfactant—12-HOA System: Gelator and Co-Surfactant. Gels, 4(3), 78. https://doi.org/10.3390/gels4030078