

Point-of-Injury Treatment with Hydrogel Containing Dexamethasone Improves Cognitive Function and Reduces Secondary Injury Response After TBI

 ,
, 
Abstract
1. Introduction
2. Results
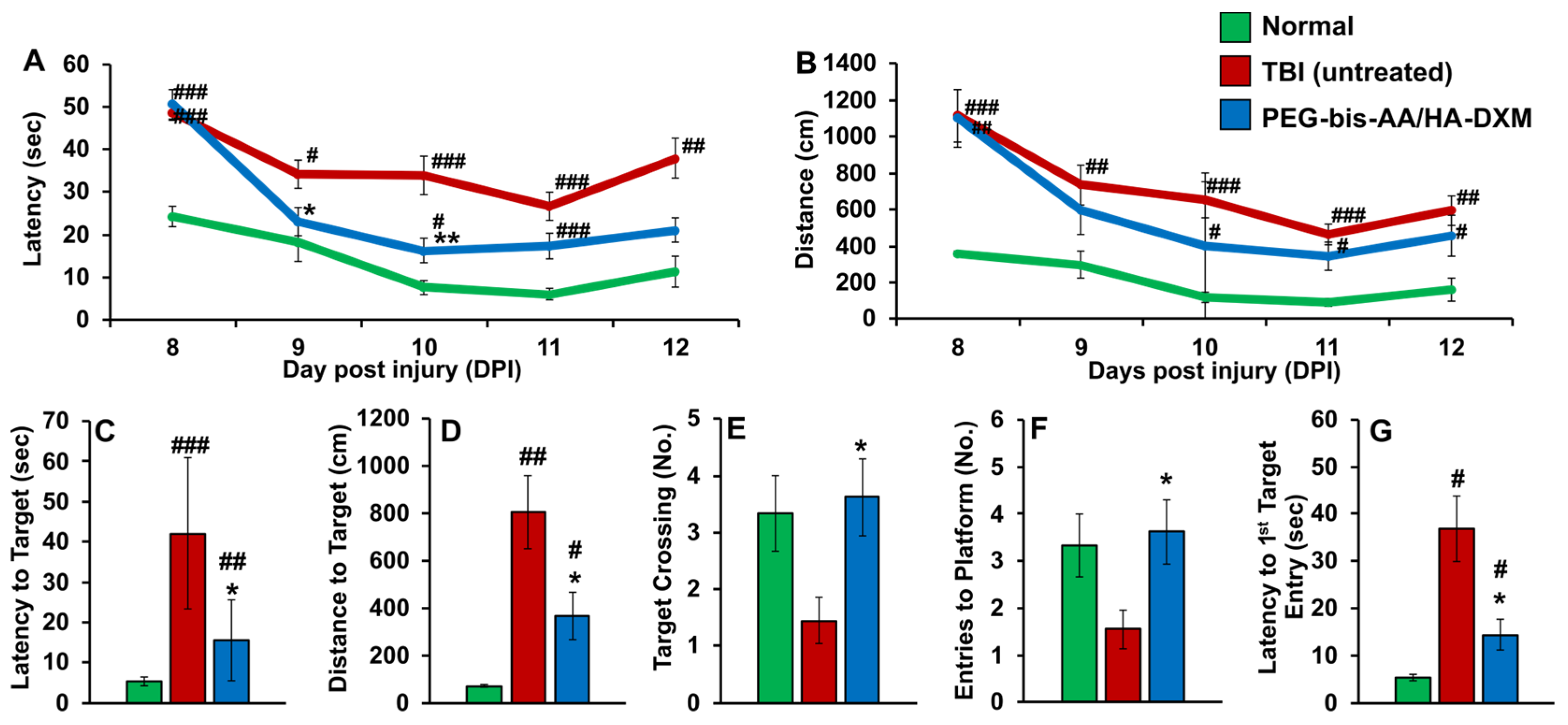
2.1. PEG-Bis-AA/HA-DXM Improves Cognitive Function
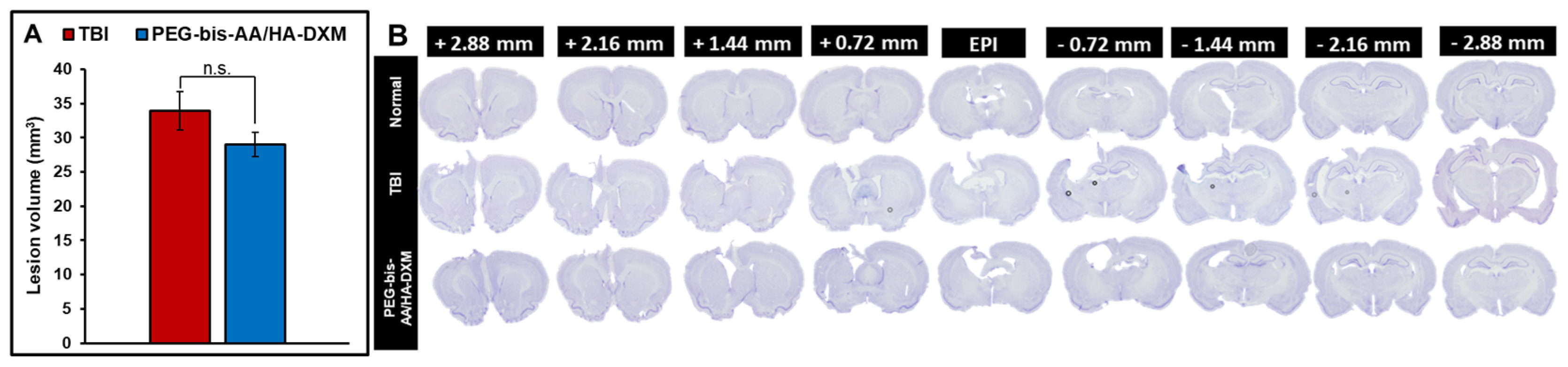
2.2. PEG-Bis-AA/HA-DXM Reduces Lesion Volume
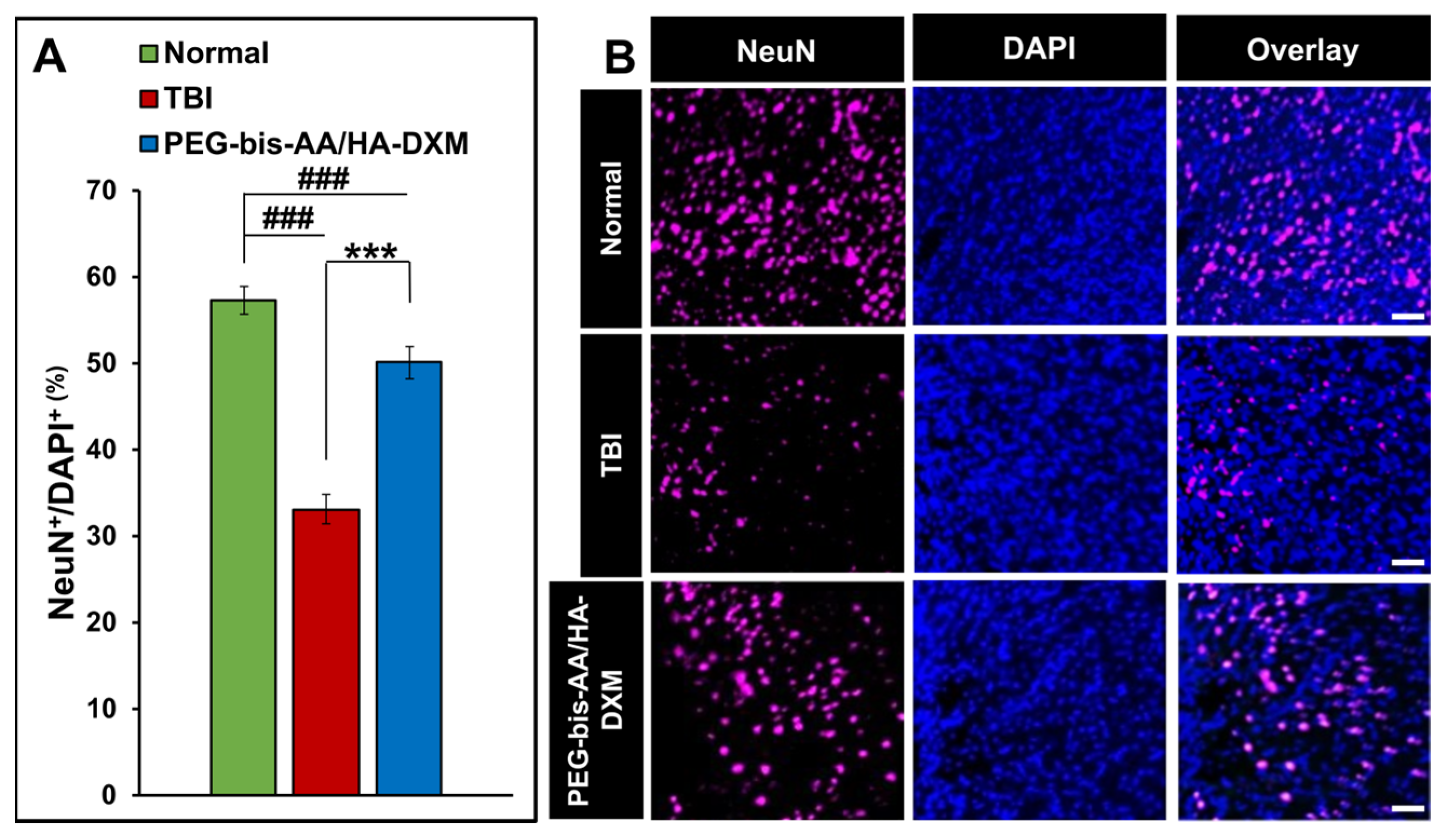
2.3. PEG-Bis-AA/HA-DXM Improves Neuronal Cell Survival
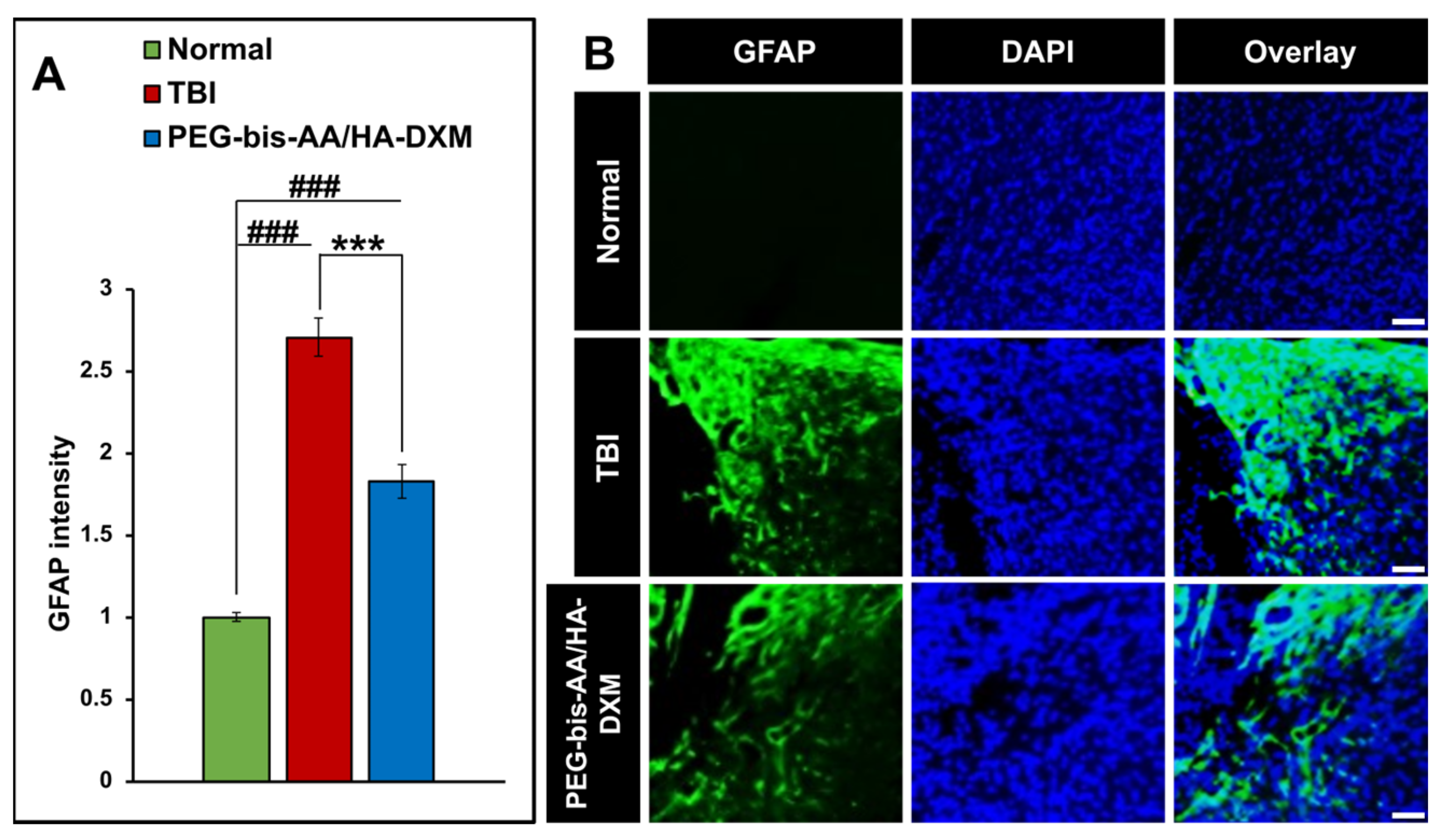
2.4. PEG-Bis-AA/HA-DXM Reduces Astrogliosis
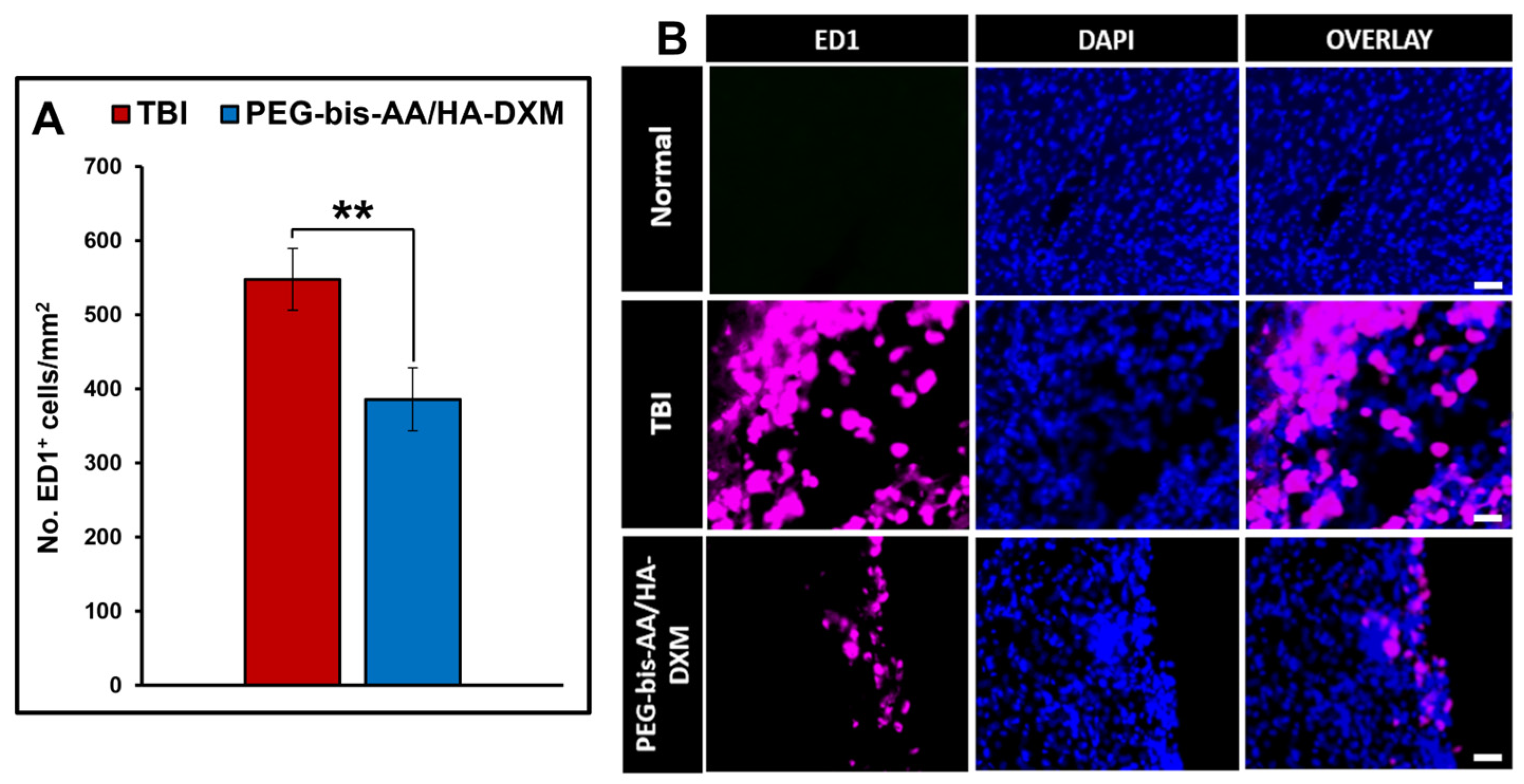
2.5. Effect of PEG-Bis-AA/HA-DXM on the Inflammatory Response
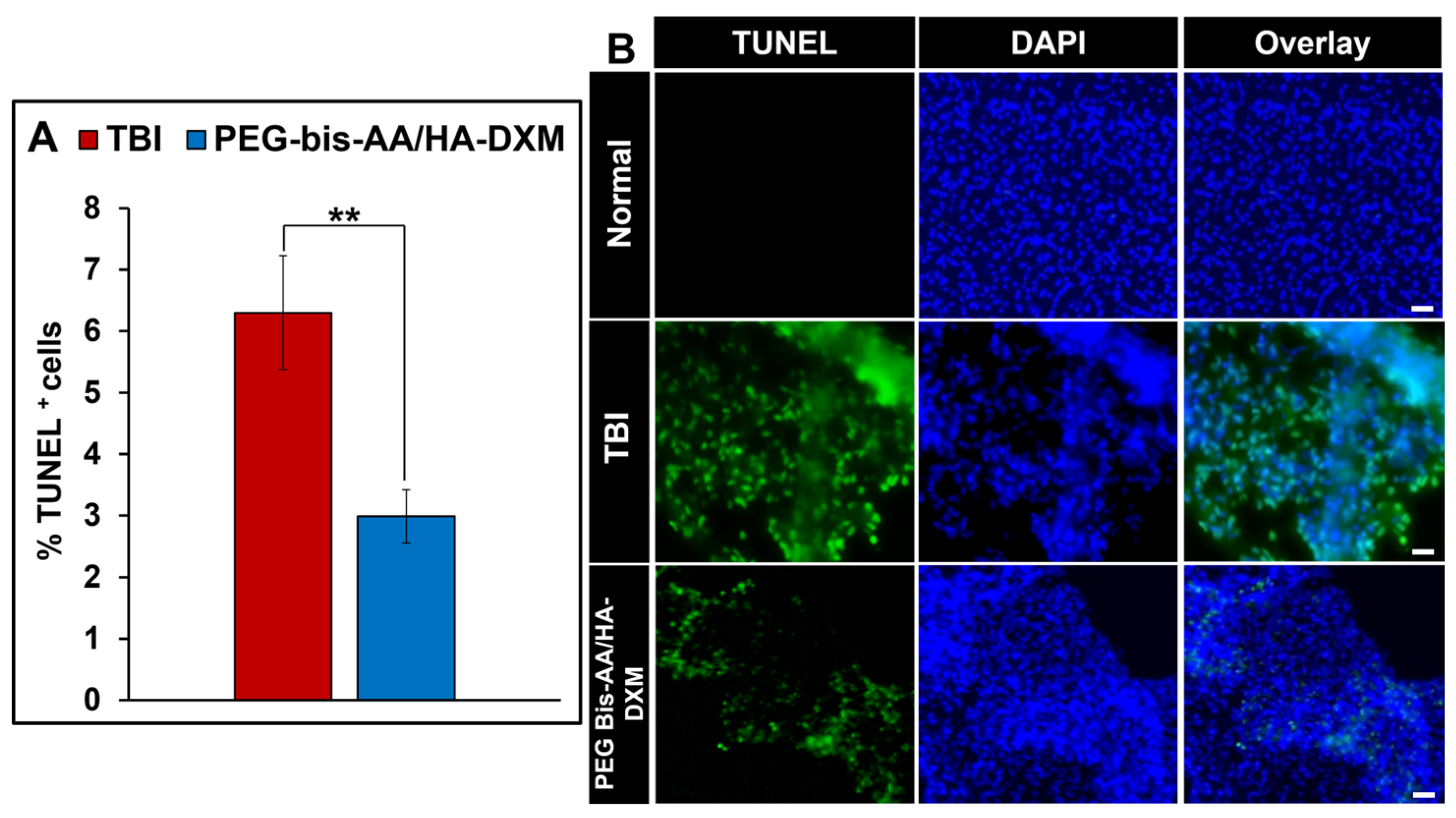
2.6. Effect of PEG-Bis-AA/HA-DXM on Apoptosis
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. PEG-Bis-AA/HA-DXM Hydrogel Preparation
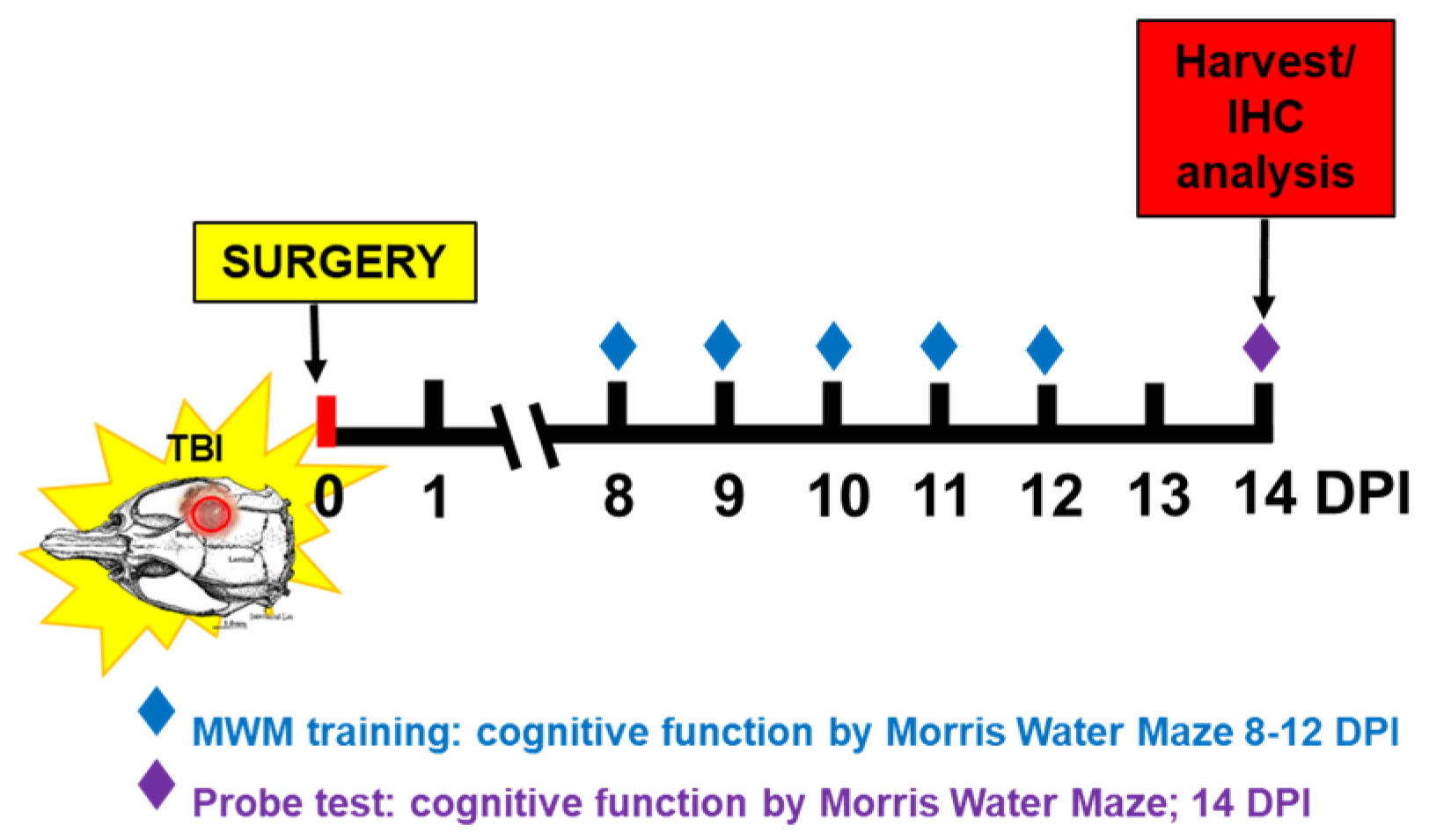
5.2. Animal Care and Surgical Procedure
5.3. Cognitive Function Using Morris Water Maze (MWM) Test
5.4. Tissue Harvest and Histology Preparation
5.5. Histological Analysis
5.5.1. Lesion Volume Analysis
5.5.2. Assessment of Inflammatory Response and Astrogliosis via Immunohistochemistry
5.5.3. Apoptosis by TUNEL Assay
5.6. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| TBI | Traumatic Brain Injury |
| DX | Dexamethasone |
| PEG-bis-AA | Polyethylene glycol-bis-(acryloyloxy acetate) |
| HA-DXM | Dexamethasone-conjugated hyaluronic acid |
| CCI | Controlled cortical impact |
| BBB | Blood–brain barrier |
| GFAP | Glial fibrillar acidic protein |
| NeuN | Neuronal nuclei |
| DPI | Days post-injury |
| MWM | Morris Water Maze |
| IHC | Immunohistochemistry |
| PBS | Phosphate-buffered saline |
| ECM | Extracellular matrix |
| IACUC | Institutional Animal Care and Use Committee |
| IP | Intraperitoneal |
| PFA | Paraformaldehyde |
| DI H2O | Deionized H2O |
| DPX | Dibutylphthalate polystyrene xylene |
| BSA | Bovine serum albumin |
| TUNEL | Terminal deoxynucleotidyl transferase dUTP nick-end |
| DAPI | 4′,6-diamidino-2-phenylindole |
References
- Ng, S.Y.; Lee, A.Y.W. Traumatic Brain Injuries: Pathophysiology and Potential Therapeutic Targets. Front. Cell. Neurosci. 2019, 13, 484040. [Google Scholar] [CrossRef]
- van Reekum, R.; Cohen, T.; Wong, J. Can Traumatic Brain Injury Cause Psychiatric Disorders? J. Neuropsychiatry Clin. Neurosci. 2000, 12, 316–327. [Google Scholar] [CrossRef]
- Kennedy, J.E.; Jaffee, M.S.; Leskin, G.A.; Stokes, J.W.; Felix, O.; Fitzpatrick, P.J. Posttraumatic Stress Disorder and Posttraumatic Stress Disorder-like Symptoms and Mild Traumatic Brain Injury. J. Rehabil. Res. Dev. 2007, 44, 895–919. [Google Scholar] [CrossRef]
- Dikmen, S.; Machamer, J.; Fann, J.R.; Temkin, N.R. Rates of Symptom Reporting Following Traumatic Brain Injury. J. Int. Neuropsychol. Soc. 2010, 16, 401–411. [Google Scholar] [CrossRef] [PubMed]
- de Macedo Filho, L.; Figueredo, L.F.; Villegas-Gomez, G.A.; Arthur, M.; Pedraza-Ciro, M.C.; Martins, H.; Kanawati Neto, J.; Hawryluk, G.J.; Amorim, R.L.O. Pathophysiology-Based Management of Secondary Injuries and Insults in TBI. Biomedicines 2024, 12, 520. [Google Scholar] [CrossRef] [PubMed]
- Masel, B.E.; DeWitt, D.S. Traumatic Brain Injury: A Disease Process, Not an Event. J. Neurotrauma 2010, 27, 1529–1540. [Google Scholar] [CrossRef]
- Finkelstein, E.A.; Corso, P.S.; Miller, T.R. The Incidence and Economic Burden of Injuries in the United States; Oxford University Press: Oxford, UK, 2009. [Google Scholar]
- Grutza, M.; Unterberg, A.; Younsi, A. Neurosurgical Treatment of Traumatic Brain Injury and the Role of Decompressive Hemicraniectomy. In Hot Topics in Acute Care Surgery and Trauma: Traumatic Brain Injury; Springer: Cham, Switzerland, 2024; pp. 363–377. [Google Scholar] [CrossRef]
- Carney, N.; Totten, A.M.; O’Reilly, C.; Ullman, J.S.; Hawryluk, G.W.J.; Bell, M.J.; Bratton, S.L.; Chesnut, R.; Harris, O.A.; Kissoon, N.; et al. Guidelines for the Management of Severe Traumatic Brain Injury, Fourth Edition. Neurosurgery 2017, 80, 6–15. [Google Scholar] [CrossRef]
- Burton, D.; Aisen, M. Traumatic Brain Injury. Handb. Second. Dement. 2006, 26, 83–118. [Google Scholar] [CrossRef]
- Li, S.; Xu, J.; Qian, Y.; Zhang, R. Hydrogel in the Treatment of Traumatic Brain Injury. Biomater. Res. 2024, 28, 0085. [Google Scholar] [CrossRef]
- Bush, T.G.; Puvanachandra, N.; Horner, C.H.; Polito, A.; Ostenfeld, T.; Svendsen, C.N.; Mucke, L.; Johnson, M.H.; Sofroniew, M.V.; Site, F.; et al. Leukocyte Infiltration, Neuronal Degeneration, and Neurite Outgrowth after Ablation of Scar-Forming, Reactive Astrocytes in Adult Transgenic Mice. Neuron 1999, 23, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Alam, A.; Thelin, E.P.; Tajsic, T.; Khan, D.Z.; Khellaf, A.; Patani, R.; Helmy, A. Cellular Infiltration in Traumatic Brain Injury. J. Neuroinflamm. 2020, 17, 328. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate Mapping Analysis Reveals that Adult Microglia Derive from Primitive Macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Alvarez-Croda, D.M.; Stoica, B.A.; Faden, A.I.; Loane, D.J. Microglial/Macrophage Polarization Dynamics Following Traumatic Brain Injury. J. Neurotrauma 2016, 33, 1732–1750. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic Reactive Astrocytes Are Induced by Activated Microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Adams, K.L.; Gallo, V. The Diversity and Disparity of the Glial Scar. Nat. Neurosci. 2018, 21, 9–15. [Google Scholar] [CrossRef]
- Wilhelmsson, U.; Li, L.; Pekna, M.; Berthold, C.; Blom, S.; Eliasson, C.; Renner, O.; Bushong, E.; Ellisman, M.; Morgan, T.E.; et al. Absence of Glial Fibrillary Acidic Protein and Vimentin Prevents Hypertrophy of Astrocytic Processes and Improves Post-Traumatic Regeneration. J. Neurosci. 2004, 24, 5016–5021. [Google Scholar] [CrossRef]
- Herrmann, J.E.; Imura, T.; Song, B.; Qi, J.; Ao, Y.; Nguyen, T.K.; Korsak, R.A.; Takeda, K.; Akira, S.; Sofroniew, M.V. STAT3 Is a Critical Regulator of Astrogliosis and Scar Formation after Spinal Cord Injury. J. Neurosci. 2008, 28, 7231–7243. [Google Scholar] [CrossRef]
- Pekny, M.; Johansson, C.B.; Eliasson, C.; Stakeberg, J.; Wallén, Å.; Perlmann, T.; Lendahl, U.; Betsholtz, C.; Berthold, C.; Frisén, J. Abnormal Reaction to Central Nervous System Injury in Mice Lacking Glial Fibrillary Acidic Protein and Vimentin. J. Cell Biol. 1999, 145, 503–514. [Google Scholar] [CrossRef]
- Furman, J.L.; Sompol, P.; Kraner, S.D.; Pleiss, M.M.; Putman, E.J.; Dunkerson, J.; Abdul, H.M.; Roberts, K.N.; Scheff, S.W.; Norris, C.M. Blockade of Astrocytic Calcineurin/NFAT Signaling Helps to Normalize Hippocampal Synaptic Function and Plasticity in a Rat Model of Traumatic Brain Injury. J. Neurosci. 2016, 36, 1502–1515. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, J.; Shi, Y.; Pu, H.; Leak, R.K.; Liou, A.K.F.; Badylak, S.F.; Liu, Z.; Zhang, J.; Chen, J.; et al. Implantation of Brain-Derived Extracellular Matrix Enhances Neurological Recovery after Traumatic Brain Injury. Cell Transplant. 2017, 26, 1224–1234. [Google Scholar] [CrossRef]
- Zhang, B.; Bai, M.; Yang, M.; Wang, Y.; Chen, X.; Liu, B.; Shi, G. Injectable Nanocomposite Hydrogel for Localized Precision Delivery of Dexamethasone after Traumatic Brain Injury: Dual Modulation of Neuroinflammation and Blood-Brain Barrier Restoration. J. Transl. Med. 2025, 23, 579. [Google Scholar] [CrossRef]
- Silver, J.; Miller, J.H. Regeneration beyond the Glial Scar. Nat. Rev. Neurosci. 2004, 5, 146–156. [Google Scholar] [CrossRef]
- Ren, Z.; Iliff, J.J.; Yang, L.; Yang, J.; Chen, X.; Chen, M.J.; Giese, R.N.; Wang, B. ‘Hit & Run’ Model of Closed-Skull Traumatic Brain Injury (TBI) Reveals Complex Patterns of Post-Traumatic AQP4 Dysregulation. J. Cereb. Blood Flow Metab. 2013, 33, 834–845. [Google Scholar] [CrossRef]
- Gyoneva, S.; Ransohoff, R.M. Inflammatory Reaction after Traumatic Brain Injury: Therapeutic Potential of Targeting Cell–Cell Communication by Chemokines. Trends Pharmacol. Sci. 2015, 36, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Shlosberg, D.; Benifla, M.; Kaufer, D.; Friedman, A. Blood–Brain Barrier Breakdown as a Therapeutic Target in Traumatic Brain Injury. Nat. Publ. Group 2010, 6, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.W.; Mcgeachy, M.J.; Bayır, H.; Clark, R.S.B.; Loane, D.J.; Kochanek, P.M. The Far-Reaching Scope of Neuroinflammation after Traumatic Brain Injury. Nat. Rev. Neurol. 2017, 13, 171–191. [Google Scholar] [CrossRef] [PubMed]
- Tourdias, T.; Mori, N.; Dragonu, I.; Cassagno, N.; Boiziau, C.; Aussudre, J.; Brochet, B.; Moonen, C.; Petry, K.G.; Dousset, V. Differential Aquaporin 4 Expression during Edema Build-up and Resolution Phases of Brain Inflammation. J. NeuroInflamm. 2011, 8, 143. [Google Scholar] [CrossRef]
- Akerblom, I.E.; Slater, E.P.; Beato, M.; Baxter, J.D.; Mellon, L. Negative Regulation by Glucocorticoids through Interference with a CAMP Responsive Enhancer. Science 1988, 241, 350–353. [Google Scholar] [CrossRef]
- Auphan, N.; DiDonato, J.A.; Rosette, C.; Helmberg, A.; Karin, M. Immunosuppression by Glucocorticoids: Inhibition of NF-ΚB Activity through Induction of IκB Synthesis. Science 1995, 270, 286–290. [Google Scholar] [CrossRef]
- Hall, E.D. High-Dose Glucocorticoid Treatment Improves Neurological Recovery in Head-Injured Mice. J. Neurosurg. 1985, 62, 882–887. [Google Scholar] [CrossRef]
- Spataro, L.; Dilgen, J.; Retterer, S.; Spence, A.J.; Isaacson, M.; Turner, J.N.; Shain, W. Dexamethasone Treatment Reduces Astroglia Responses to Inserted Neuroprosthetic Devices in Rat Neocortex. Exp. Neurol. 2005, 194, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Poetker, D.M.; Reh, D.D. A Comprehensive Review of the Adverse Effects of Systemic Corticosteroids. Otolaryngol. Clin. N. Am. 2010, 43, 753–768. [Google Scholar] [CrossRef]
- Edwards, P.; Arango, M.; Balica, L.; Cottingham, R.; El-Sayed, H.; Farrell, B.; Fernandes, J.; Gogichaisvili, T.; Golden, N.; Hartzenberg, B.; et al. Final Results of MRC CRASH, a Randomised Placebo-Controlled Trial of Intravenous Corticosteroid in Adults with Head Injury—Outcomes at 6 Months. Lancet 2005, 365, 1957–1959. [Google Scholar] [CrossRef] [PubMed]
- Alderson, P.; Roberts, I. Corticosteroids in Acute Traumatic Brain Injury: Systematic Review of Randomised Controlled Trials. BMJ 1997, 314, 1855–1859. [Google Scholar] [CrossRef]
- Alderson, P.; Roberts, I. Corticosteroids for Acute Traumatic Brain Injury. Cochrane Database Syst. Rev. 2005, CD000196. [Google Scholar] [CrossRef]
- Vella, M.A.; Crandall, M.L.; Patel, M.B. Acute Management of Traumatic Brain Injury. Surg. Clin. N. Am. 2017, 97, 1015–1030. [Google Scholar] [CrossRef]
- Carola, F.; Silwedel, C.; Golenhofen, N.; Burek, M.; Kietz, S.; Mankertz, J.; Drenckhahn, D. Occludin as Direct Target for Glucocorticoid-Induced Improvement of Blood—Brain Barrier Properties in a Murine in vitro System. J. Physiol. 2005, 565, 475–486. [Google Scholar] [CrossRef]
- Jeong, D.U.; Bae, S.; Macks, C.; Whitaker, J.; Lynn, M.; Webb, K.; Lee, J.S. Hydrogel-Mediated Local Delivery of Dexamethasone Reduces Neuroinflammation after Traumatic Brain Injury. Biomed. Mater. 2021, 16, 035002. [Google Scholar] [CrossRef] [PubMed]
- Macks, C.; Jeong, D.; Bae, S.; Webb, K.; Lee, J.S. Dexamethasone-Loaded Hydrogels Improve Motor and Cognitive Functions in a Rat Mild Traumatic Brain Injury Model. Int. J. Mol. Sci. 2022, 23, 11153. [Google Scholar] [CrossRef]
- Jones, C.; Elliott, B.; Liao, Z.; Johnson, Z.; Ma, F.; Bailey, Z.S.; Gilsdorf, J.; Scultetus, A.; Shear, D.; Webb, K.; et al. PEG Hydrogel Containing Dexamethasone-Conjugated Hyaluronic Acid Reduces Secondary Injury and Improves Motor Function in a Rat Moderate TBI Model. Exp. Neurol. 2023, 369, 114533. [Google Scholar] [CrossRef]
- Blecharz, K.G.; Drenckhahn, D.; Fo, C.Y. Glucocorticoids Increase VE-Cadherin Expression and Cause Cytoskeletal Rearrangements in Murine Brain Endothelial CEND Cells. J. Cereb. Blood Flow Metab. 2008, 28, 1139–1149. [Google Scholar] [CrossRef]
- Rosenberg, G.A. Matrix Metalloproteinases and Their Multiple Roles in Neurodegenerative Diseases. Lancet Neurol. 2009, 8, 205–216. [Google Scholar] [CrossRef]
- Carola, F.; Kahles, T.; Kietz, S.; Drenckhahn, D. Dexamethasone Induces the Expression of Metalloproteinase Inhibitor TIMP-1 in the Murine Cerebral Vascular Endothelial Cell Line CEND. J. Physiol. 2007, 3, 937–949. [Google Scholar] [CrossRef]
- Zeni, P.; Doepker, E.; Topphoff, U.S.; Huewel, S.; Tenenbaum, T.; Galla, H.J. MMPs Contribute to TNF-α-Induced Alteration of the Blood-Cerebrospinal Fluid Barrier in Vitro. Am. J. Physiol.-Cell Physiol. 2007, 293, C855–C864. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.; Kutty, J.K.; Datar, K.; Lee, J.S.; Vyavahare, N.R.; Webb, K. A Novel Synthetic Route for the Preparation of Hydrolytically Degradable Synthetic Hydrogels. J. Biomed. Mater. Res. A 2009, 90, 1073–1082. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.; Lee, H.J.; Lee, J.S.; Webb, K. Cell-Mediated Dexamethasone Release from Semi-IPNs Stimulates Osteogenic Differentiation of Encapsulated Mesenchymal Stem Cells. Biomacromolecules 2015, 16, 2757–2765. [Google Scholar] [CrossRef]
- Zhang, B.; Zhu, X.; Wang, L.; Hao, S.; Xu, X.; Niu, F.; He, W.; Liu, B. Dexamethasone Impairs Neurofunctional Recovery in Rats Following Traumatic Brain Injury by Reducing Circulating Endothelial Progenitor Cells and Angiogenesis. Brain Res. 2019, 1725, 146469. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, K.L.; Yang, S.Y.; Dong, J.F.; Zhang, J.N. Glucocorticoids Aggravate Retrograde Memory Deficiency Associated with Traumatic Brain Injury in Rats. J. Neurotrauma 2009, 26, 253–260. [Google Scholar] [CrossRef]
- Cho, K.; Yang, L.; Lu, B.; Ma, H.F.; Huang, X.; Pekny, M.; Chen, D.F. Re-Establishing the Regenerative Potential of Central Nervous System Axons in Postnatal Mice. J. Cell Sci. 2005, 118, 863–872. [Google Scholar] [CrossRef] [PubMed]
- Amlerova, Z.; Chmelova, M.; Anderova, M.; Vargova, L. Reactive Gliosis in Traumatic Brain Injury: A Comprehensive Review. Front. Cell. Neurosci. 2024, 18, 1335849. [Google Scholar] [CrossRef]
- Newcomb, J.K.; Zhao, X.; Pike, B.R.; Hayes, R.L. Temporal Profile of Apoptotic-like Changes in Neurons and Astrocytes Following Controlled Cortical Impact Injury in the Rat. Exp. Neurol. 1999, 158, 76–88. [Google Scholar] [CrossRef]
- Holmin, S.; Mathiesen, T. Characterization of Bax and Bcl-2 in Apoptosis after Experimental Traumatic Brain Injury in the Rat. Acta Neuropathol. 2003, 105, 281–288. [Google Scholar] [CrossRef]
- Holmin, S.; Mathiesen, T. Intracerebral Administration of Interleukin-1 β and Induction of Inflammation, Apoptosis, and Vasogenic Edema. J. Neurosurg. 2000, 92, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Vorhees, C.V.; Williams, M.T. Morris Water Maze: Procedures for Assessing Spatial and Related Forms of Learning and Memory. Nat. Protoc. 2006, 1, 848–858. [Google Scholar] [CrossRef] [PubMed]
- Scheff, S.W.; Baldwin, S.A.; Brown, R.W.; Kraemer, P.J. Morris Water Maze Deficits in Rats Following Traumatic Brain Injury: Lateral Controlled Cortical Impact. J. Neurotrauma 1997, 14, 615–627. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MILD TBI | Moderate TBI | |||
|---|---|---|---|---|
| Assessment | 7DPI | 14 DPI | 7DPI | 14 DPI |
| Lesion volume |  | 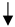 | 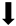 |  |
| Macrophage/Microglia | 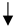 |  | 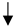 | 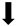 |
| Reactive gliosis (GFAP) |  | N/A |  |  |
| Apoptosis |  |  | 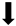 | 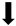 |
| Inflammatory gene expression | 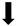 | 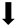 | N/A | N/A |
| Neuronal cell survival | 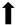 | 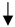 | 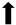 |  |
| Motor function | 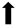 | N/A | 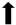 | N/A |
| Cognitive Function | N/A |  | N/A | 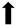 |
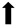 ) and significantly decrease (
) and significantly decrease (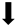 ), qualitative or not statistically significantly decrease (
), qualitative or not statistically significantly decrease ( ), and not assessed (N/A).
), and not assessed (N/A).Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jones, C.E.; Elliott, B.; Ma, F.; Bailey, Z.; Gilsdorf, J.; Scultetus, A.H.; Shear, D.; Webb, K.; Lee, J.S. Point-of-Injury Treatment with Hydrogel Containing Dexamethasone Improves Cognitive Function and Reduces Secondary Injury Response After TBI. Gels 2025, 11, 600. https://doi.org/10.3390/gels11080600
Jones CE, Elliott B, Ma F, Bailey Z, Gilsdorf J, Scultetus AH, Shear D, Webb K, Lee JS. Point-of-Injury Treatment with Hydrogel Containing Dexamethasone Improves Cognitive Function and Reduces Secondary Injury Response After TBI. Gels. 2025; 11(8):600. https://doi.org/10.3390/gels11080600
Chicago/Turabian StyleJones, Claire E., Bradley Elliott, Fuying Ma, Zachary Bailey, Janice Gilsdorf, Anke H. Scultetus, Deborah Shear, Ken Webb, and Jeoung Soo Lee. 2025. "Point-of-Injury Treatment with Hydrogel Containing Dexamethasone Improves Cognitive Function and Reduces Secondary Injury Response After TBI" Gels 11, no. 8: 600. https://doi.org/10.3390/gels11080600
APA StyleJones, C. E., Elliott, B., Ma, F., Bailey, Z., Gilsdorf, J., Scultetus, A. H., Shear, D., Webb, K., & Lee, J. S. (2025). Point-of-Injury Treatment with Hydrogel Containing Dexamethasone Improves Cognitive Function and Reduces Secondary Injury Response After TBI. Gels, 11(8), 600. https://doi.org/10.3390/gels11080600