2.1. Preparation and Characterisation of Ti3C2Tx MXene
In the Ti
3AlC
2 MAX phases, where the Ti-C bond is stronger than the Ti-Al bond, the Al layer can be removed by suitable etching reagents, producing multilayered stacked Ti
3C
2T
x MXenes, which are then stabilised through H-bonds and van der Waals forces between the 2D nano-thin flakes of Ti
3C
2T
x [
43]. Multilayered MXenes can be delaminated further and intercalated to thinner single/few-layered Ti
3C
2T
x MXenes to increase their surface area and produce electrochemically more attractive materials.
The preparation of Ti
3C
2T
x MXene and their delamination efficacy from the Ti
3AlC
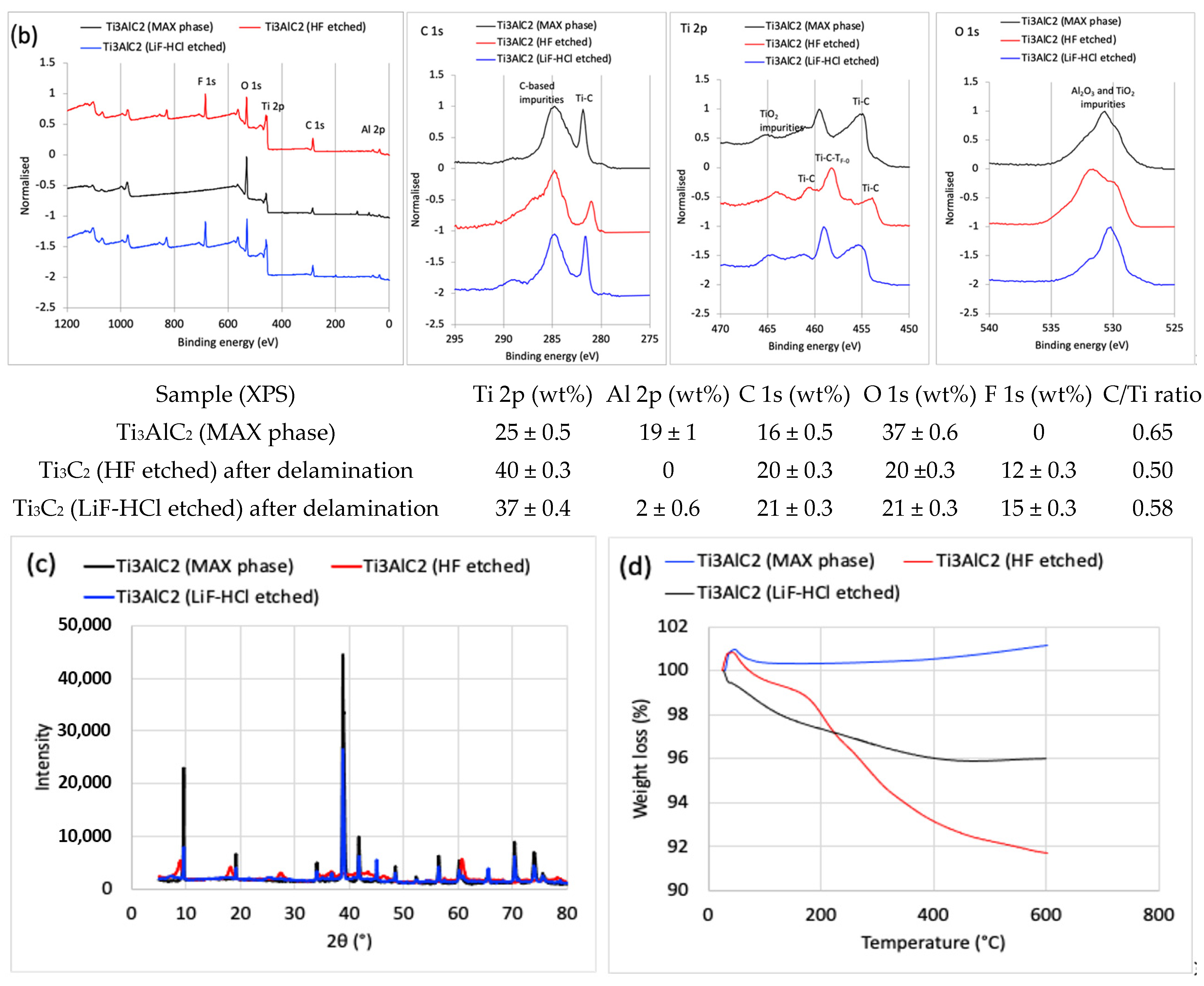
2 MAX phase powder was studied by SEM imaging connected to EDX, as well as XPS and XRD spectroscopies, and TGA analysis. The SEM images (
Figure 1a) and EDX spectra (elemental composition in the inserted Table), performed within the same view field, showed that most of the Al in the Ti
3AlC
2 MAX phase was removed after the HF etching, resulting in a typical accordion-like morphology of the multi-layered (up to ca. 20) and few 10-nm thick Ti
3C
2T
x MXene sheets, with a distance between successive layers of around 100–200 nm after delamination and the MXene of several microns in lateral sizes. The total weight percent of Al was reduced from ~27% in the MAX phase to ~1.9% after etching with HF, and reduced further to ~1.7% after the delamination step, while Ti increased by ~24% and the carbon remained constant, confirming the almost complete elimination of the Ti-Al bonds and optionally present C-Al bonds in the so obtained Ti
3C
2T
x MXene. XPS (
Figure 1b) analysis also confirmed the presence of terminated (T
x) groups (such as =O, –OH, and –F) on the surfaces of the MXene layers by the peaks of O 1s at ~529 eV (LiF-HCl etched) and ~531 eV (HF etched), and the peak of F 1s at ~684 eV, respectively [
34]. The Ti–C components, whose binding energy positions depend on the local bonding of the terminal –F and oxygen (=O, –OH) species, are also confirmed by the Ti 2p spectra. The Ti
3C
2T
x MXene obtained instead had the expected C/Ti atomic concentration ratio of ~0.6 (3:2). The presence of C-based impurities, such as hydrocarbons (CH), alcohol (C–OH), and carboxyl (COO) components, can also be confirmed by the C 1s spectra [
20]. From a relatively high percentage of oxygen components in O 1s and shoulder peak at ~463 eV for the Ti 2p spectra, some of them also have to be in the form of TiO
2 (and possible Al
2O
3), probably caused by exposure of samples to the laboratory atmosphere, strong ultrasonication and centrifugation during the preparation phase; it could be expected that the oxidised Ti and Al were also present on the interface of the layered MXene structure (where they interacted with the conductive Ti–C), as estimated from the EDX analysis. Enhanced oxidation might also be the reason for the higher adhesion between the MXene layers, inhibiting their delamination. No Li, but a small Cl residual in the MXene exfoliated by LiF-HCl was detected, indicating its incomplete removal during washing. The XRD patterns (
Figure 1c) additionally confirmed the selective removal of Al and the formation of a Ti
3C
2T
x crystal structure, characterised by the presence of characteristic diffraction peaks at 2θ at ~8.7°, ~18°, ~29°, and ~36°, ~45° and ~61°, corresponding the to (002), (004), (006), and (008), (010) and (110) planes of the Ti
3C
2T
x MXene, respectively [
20]. A noticeable downshift in the (002) diffraction of Ti
3AlC
2 from ~9.5° to ~8.7° in the Ti
3C
2T
x also indicates the introduction of surface terminal groups in the structural composition of the Ti
3C
2T
x, which corresponds to an increase in the spacing between the 2D layer stacks and a decline in their thickness, as is evident from the SEM images. The large spacing after drying could also be due to the remaining water molecules in between the MXene layers, which was confirmed by the TGA thermograms (
Figure 1d), showing a multi-stage reduction in weight by increasing temperature. The first two stages are due to the loss of physically and chemically (via –OH groups on Ti
3C
2 surface) adsorbed water, and the third is related to the loss of adsorbed –F (also via –OH groups); the latter can be supported by the XPS analysis, according to which the F/O atomic concentration ratio on the MXene surface was ~0.6 for the etched HT and ~0.49 for the LiF-HCl etched MXene.
2.2. Structural and Physico-Chemical Properties of Composite Membranes
The surface and bulk/volumetric densities and total porosity of the selected composite membranes, prepared at both freezing temperatures (−50 °C and −80 °C) and containing different concentrations of HF-etched Ti
3C
2T
x MXene, were evaluated by gravimetric analysis (
Table 1) to characterise their morphological structure. The average volumetric density was decreased from ~289% for the sample prepared with 50 wt% of MXene at −50 °C to ~229% for the same prepared at −80 °C, at similar (99.8–99.3%) porosities. This effect was even more significant in membranes containing 90 wt% MXene. The results are supported by the SEM images of representative samples, presented in
Figure 2, that indicated a fully open web-like net microstructure, with disorderly arranged bundles and single cellulose nanofibrils in the case of membranes prepared with pure CNF. On the other hand, a denser microstructure was observed for the sample with the presence of MXene, also indicating an orientation of CNFs along the Mxene sheets and their high adhesion. Accordingly, an increase in the MXene content resulted in an even denser micro-porous structure, being more pronounced for samples prepared at lower temperatures (−80 °C), having also a slightly lower porosity than those prepared at −50 °C. The process is related to the different freezing kinetics of the water molecules (i.e., water crystallisation growth and structuring), and, thus, the creation of differently sized and distributed ice crystals around the CNFs and MXenes, which became smaller and more ordered at lower temperature, therefore also influencing their assembling and rearrangement dynamics.
The elemental compositions obtained by EDX elementary analysis (and some of the elements’ ratios,
Figure 2, inserted Table) of the representative CNF/MXene hybrid membranes also confirmed the integration of Ti
3C
2T
x MXene in the fibrillated cellulose network by the appearance of Ti, and significant reduction of C and O content and its C/O ratio from ~1 to ~0.86. The XRD spectra of such a hybrid membrane (
Figure 3a) additionally revealed a slight shifting of the main characteristic diffraction peak of MXene to a higher angle (from ~8.7° to ~9.8°), and a significant drop in the intensity of the others after the introduction of CNF. At the same time, the amorphous cellulose peaks at 2θ of ~16° and ~18° (attributed to the (1–10) and (110) planes), was reduced more strongly than the peak for crystalline cellulose at ~22.8° (corresponding to the (200) plane, of cellulose I
β); [
44] indicated the covering/interactions of the Ti
3C
2T
x MXene surface by CNFs [
43], and possible intercalation between the Ti
3C
2T
x interlayers by smaller CNFs fractions, that can also prevent the restacking of delaminated MXene flakes [
45], and, consequently, enhanced accessibility of the electrolyte and accommodation of more ions. The chemical bonding states between the components on the outside of the hybrid membranes surface was confirmed additionally by FTIR and XPS spectroscopy. The IR spectrum of the CNF membrane (
Figure S1 showed characteristic transmittance bands for cellulose, with a broad peak at ~3337 cm
1 (assigned to –OH stretching), ~2900 cm
1 (–CH
2 stretching), ~1410–1420 cm
−1 (–CH
2 bending), ~1360–1310 cm
−1 and ~1150–1110 cm
−1 (–CH stretching and bending), ~1030 cm
−1 (C-O-C stretching and −OH bending), ~890 cm
−1 (cellulosic
β-glycosidic linkages), and ~660 cm
−1 (−OH bending); a small shoulder at ~1600 cm
−1, attributed to the stretching vibrations of C=O, also indicated the presence of −COOH groups. The spectra of the Ti
3C
2T
x MXene powder had small shoulders at ~3400 cm
−1, ~1000 cm
−1, and more intensive ones at ~600–500 cm
−1, corresponding to the stretching vibrations of the –OH, C–F, and Ti–O bonds, respectively [
46]. All the characteristic peaks for the composite membranes were shifting slightly and obviously decreased as compared to the solvent-casted films and CNFs, indicating the formation of H-bonding between the −OH, −O and C=O atoms in the CNF with the surface groups of Ti
3C
2T
x (=O, –OH, and –F). The presence of a small shoulder for the −OH groups (at ~3334 cm
−1 and ~1030 cm
−1) confirmed their availability for interaction with the electrolyte cations; no differences were observed between the upper and bottom sides of the samples. The XPS spectra (
Figure 3b) reconfirmed the presence of Ti, C, O, and F for the composite membrane, while the deconvoluted Ti 2p spectra exhibited an additional peak for Ti-O at ~461.8 eV as compared to the Ti
3C
2T
x sample after mixing with CNF, additionally confirming their interaction with the surface-decorated −OH and rare −COOH groups of CNF [
2]. The deconvoluted C 1s spectra contained a typical peak at ~286.2 eV (Ti–C) in the composite membrane, where the C/O-enriched structure of pure CNF was well-maintained by showing a broadened peak at ~287.2 eV corresponding to the C=C/C–C in aromatic rings, C–O and C=O, and a small shoulder at ~289 eV of O–C=O. On the other hand, a shifting of a broadened O peak to higher binding energy (~535 eV and ~533 eV) in the O 1s spectra indicates the formation of new—via O and C atoms—linked structures, although the broadened peak at ~532.6 eV can also be assigned to the –OH of the bound water. The results of the XPS were in line with the elemental mapping obtained by EDX spectrometry, and both results confirmed the structural integrity of Ti
3C
2T
x MXene within the CNF matrix and their intercalations. Besides the H-bonding and stronger van der Waals forces, electrostatic repulsions might also be present, influencing their morphological structure.
2.3. Structural and Physico-Chemical Stability of Composite Membranes in Different Electrolyte Solutions
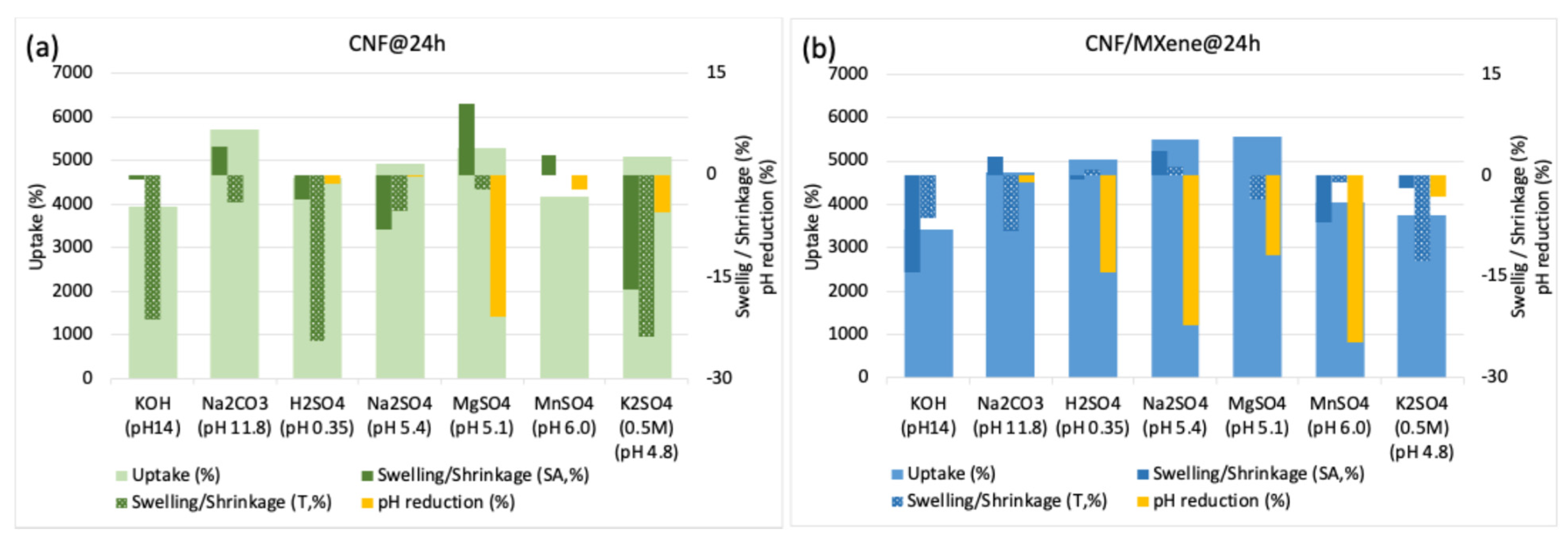
In addition to the conductivity and hydrodynamic size of the electrolyte ions, the wetting behaviour of the membrane in the electrolyte solution also facilitated cation diffusion and thus affected their electrochemical properties. The membranes prepared without and with (HT-etched) MXene at both freezing temperatures were thus immersed into different electrolyte solutions to evaluate their swelling, electrolyte uptake, and interaction with CNFs/MXene, as well as their stability. The membranes prepared with the highest (90 wt%) MXene loading disintegrated after 24 h of incubation in all the aqueous electrolyte solutions (
Figure S2), so they were unsuitable for further analysis. The 1M H
2SO
4 solution was also proved to be a mechanically unsuitable electrolyte for all the CNF-based membranes due to cellulose depolymerisation in such a highly acidic medium (pH~0.35) [
35]; this was confirmed by significant shrinkage (
Figure S3) of the pure CNF sample after 24 h of incubation, and its complete degradation after one week, resulting in glucose and sulphone-acid-decorated MXene particles, as observed from the photo and SEM image presented in
Figure S3 (the spectroscopic elemental analysis of this sample could thus not be performed). On the other hand, the pure CNF membranes behaved differently in the other types of electrolytes (
Figure 4a). While they floated in Na
2SO
4 and MnSO
4 solutions of pH~5.2 and got slightly colourised/stained in MnSO
4 (pH~6.0) due to Mn
2+ adsorption, they did not show any optical difference generally in any of these electrolytes, including MgSO
4 and K
2SO
4, even after 24 h of incubation (
Figure S1). The solution (and, consequently, ion) uptake (
Figure 4a) was also significantly lower for KOH (pH~14) and MnSO
4 (pH~6.0) (~4000%) as compared to the other electrolytes (~4500–5700%). Still, it showed a significant shrinkage (areal and volumetric, ~16 and ~24%) in K
2SO
4 (pH~4.8) and slight shrinkage (~8 and ~5%) in Na
2SO
4 (pH~5.4), and in electrolytes with monovalent cations, a swelling (~10%) in MgSO
4 (pH~5.1), while it did not affecting the dimensional change in MnSO
4 (pH ~6.0). While the swelling in MgSO
4 was related to high adsorption of hygroscopic Mg salts, resulting in a ~21% reduction of pH (from ~5.1 to ~4.0, the uptake of the solution (~5700%) without dimensional and pH change was the highest and most stable in Na
2CO
3 (pH~11.8). Such a behaviour might be due to different interactions of electrolyte cations with anionic (abundant -OH and rare -COOH) surface groups on the CNFs, where, besides hydrogen bonding and electrostatic attractive interactions (the latter is highly dependent on the electrolyte pH that affect both the ionization of functional groups on CNF and the dissociation degree of ions), the internal bonding of the fibril-to-fibril network could thus also be present in the case of electrolytes with divalent cations (above all, hydrodynamically larger Mg
2+) that influenced the membrane’s physical properties. In highly basic electrolytes, the diffusion of cations is thus significantly reduced due their predominant electrostatic interactions with CNF functional groups already on the outer surface of the membrane walls, and the formation of salt aggregates or protective layers (
Figure 2a) with their anions, which actually affects the high value of electrolyte uptake.
The membranes prepared with 50 wt% MXene loading (
Figure 4b; for 10 wt% MXene loading the results are presented in
Figure S4), followed a similar trend, but showed generally lower (by between 5 and 30%) electrolyte uptake, and, consequently, much lower shrinkage (areal and volumetric, ~0–14%) for samples prepared at −80 °C than those prepared at −50 °C (showing more intensive shrinkage in thickness, ~16–43%), which is related to the slightly higher density of those membranes, also influencing the accessibility and diffusivity of the ions (and, hence, shorter diffusion distances within the electroactive MXene). As already discussed above, besides pH (influencing the ionic form of –OH and rare –COOH groups on the CNF and ionization stage of electrolyte), such an effect could also be related to the size and form of the hydrated ions, and their ionic conductivity and mobility in an aqueous environment. Among them, the hydrodynamically smallest H
+ ions (with a radius of 0.28 nm) had the highest conductivity (~350 Scm
2/mol) and ionic mobility (~36.23 10
−8 m
2/sV), followed by OH
− (0.3 nm, ~198 Scm
2/mol, ~20.6 10
−8 m
2/sV), K
+ (0.331 nm, ~73.5 Scm
2/mol, ~7.62 10
−8 m
2/sV), SO
42− (0.379 nm, ~160 Scm
2/mol, ~8.3 10
−8 m
2/sV), Na
+ (0.358 nm, ~50.11 Scm
2/mol, ~5.19 10
−8 m
2/sV), and Mg
2+ (0.427 nm, ~106 Scm
2/mol) [
3,
47]. A larger swelling and, thus, capacitance, can, therefore, be expected for electrolyte ions with different sizes, e.g., large anions and much smaller cations, where the valence of the ions and electrolyte pH also determines their interactions at the membrane–electrolyte interfaces and its stability [
48]. For slightly acidic sulphated electrolytes, the sensitivity of cellulose -OH and rare -COOH groups (covering the MXenes) are relatively higher for hydrodynamically larger and slower divalent cations (Mg
2+) than for the monovalent cations (as K
+ and Na
+) that can interact with hydrogen bonding, but also diffuse easier and faster to the MXenes’ surface where weakly adsorbed on their −F/−O/=O terminal groups (as confirmed by the XPS analysis presented in
Figure 3 where rare Ti atoms and no F could be detected anymore on its surface) and, by this, also influence on ion selectivity. A higher pH reductio in the case of all sulphate electrolytes (Na
2SO
4, MgSO
4, MnSO
4, KSO
4), except H
2SO
4, can thus be related to a much higher diffusion ability of the monovalent cations (Na
+ > Mn
2+ > Mg
2+) and their interaction with available and rare ionically charged (at such a slightly acidic pH 4.8–5.6) functional groups on CNF, and their further diffusion to the MXene interface, or conjugation with SO
42− during swelling and ion transfer. On the contrary, the diffusion of Na
+ cations in highly basic Na
2CO
3 (pH~11.8) is more limited on the membrane outer surface due their predominant interactions with fully ionized functional surface groups on CNFs, and therefore also more prone to the formation of an electroactive layer on the membrane interface with larger CO
3−2 anions, as is clearly evident from
Figure 2b. The pronounced shrinkage across the thickness of samples with a more open structure prepared at −50 °C in all sulphate electrolytes, independent of the amount of MXen added (
Figure S4b), thus causes stacking of diffused ions and limits the mass transfer during incubation, which actually affects the high value of electrolyte uptake, membrane density and pH reduction. In contrast, other slightly acidic electrolytes with divalent cations enable a higher rate of ion diffusion into the membrane structure.
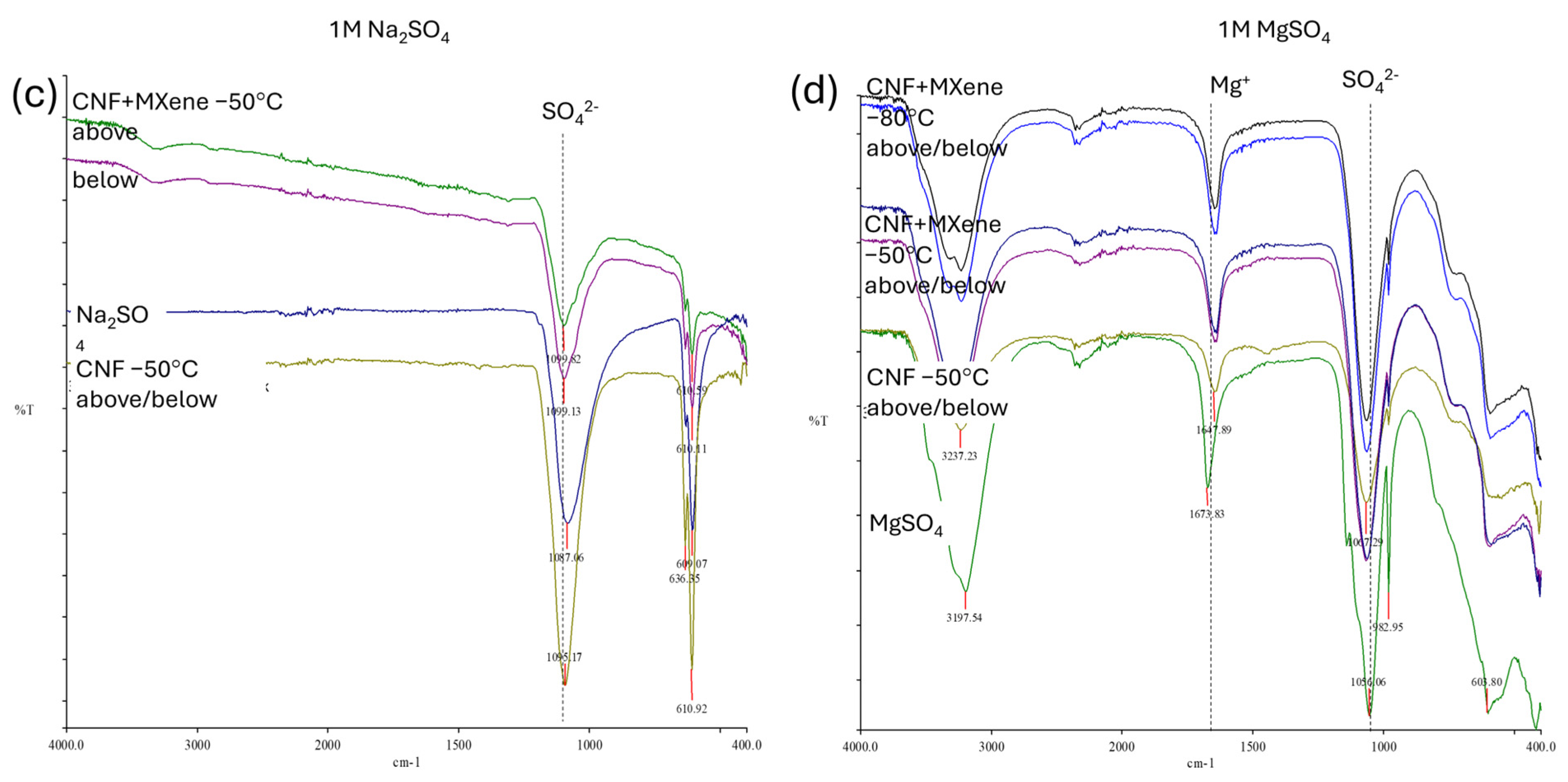
Such an interaction of ions with the CNF surface can also be confirmed by FTIR spectroscopy. As seen from the spectra in
Figure S4c, all the characteristic peaks for CNFs were found in the IR spectra of the pure CNF membranes (prepared at −50 °C) after 7 days of incubation in different electrolyte salts, but with different intensities, while the new peaks—associated with the crystallite salts—appeared, indicating different associations of electrolyte ions with the CNFs’ surface. The bands assigned to the −OH surface groups (above all, those at ~3337 cm
−1 and ~1030 cm
−1) in the membranes after incubation were broadened, weakened, and/or shifted slightly to a lower (~3230–3282 cm
−1) or higher (~1070–1095 cm
−1) wavelength, depending on the electrolyte solution used, and, consequently, H-bonding between the anionic −OH and −O, as well as rare –COOH groups in the CNF with deposited electrolyte cation and the formation of crystal structures after its conjugation with anion on the anchoring sites on the CNF surfaces. In general, two new bands appeared in the IR spectrum of the CNF samples after incubation in sulphate salts, at ~1059–1095 cm
−1 and ~636–613 cm, assigned to the O=S=O stretching and bending vibrations and S–O stretching vibration of the sulphonic acid (–SO
42−) groups of sulphate-based crystallites in accordance with the IR spectra of pure sulphate salts, being accompanied by a decrease in the –OH surface groups, confirming coverage of the CNF surfaces by them. It can also be observed that the presence of a small shoulder for the −OH groups (at ~3337 cm
−1) in the case of samples incubated in MgSO
4, MnSO
4 and K
2SO
4, were still present, indicating their availability, while it was not detected anymore for Na
2SO
4 due its full coverage followed by Na
2CO
3 and KOH, followed by the trends of the ions’ diffusion and swelling limitation of the CNF membranes in such electrolytes (
Figure 3). The IR spectra for the representative CNF/MXene hybrid membranes, presented in
Figure 5, followed the same trend, although they also showed significant differences in chemical structure between the upper and bottom sides after long-term incubation (i.e., several days of electrochemical performance under CV) in alkaline and monovalent electrolytes (KOH and Na
2CO
3). This might be due to even poorer/limited accessibility (rather than smaller penetration) of solvated ions to the anchoring sites on the CNF surface into the morphologically more closed structure on the bottom (freezing-exposed during preparation) side of the membranes prepared at a lower (kinetically faster) freezing temperature (−80 °C). The lowering of the intensities and shifting of the −OH related peaks (centred at ~3334 cm
−1 and ~1030 cm
−1) on the upper sides and peak intensification for both cations (K
+ and Na
+) and anions (CO
32−) for membranes in those electrolytes (
Figure 5a,b), additionally confirming the involvement of these groups in the H-bonding with cations, covered the membrane surface fully by forming an uniform electroactive layer at the membrane–electrolyte interfaces after their further conjugation with anions, as already established by the results presented in
Figure 2b and
Figure S5. The −COOH groups present on the CNFs’ surfaces allow additional electrostatic interactions with the electrolyte cations (
Figure 2). On the other hand, the membranes in the acidic medium did not show any particular chemical differences (
Figure 5c,d), but became fully covered with the electrolyte layer on both membrane sides. The strongly intense peak at ~3200 cm
−1 for the membrane tested in MgSO
4 is due to the high hydroscopicity of Mg ions, forming different crystallites after anchoring with the SO
42− anions.
Such results are also in line with the structural analysis supported with SEM-EDX (
Figure 2), as well as chemical analysis performed with XRD and XPS (
Figure 3), proving the structurally different integrity of electrolytes within the CNF matrix and their intercalations. As is evident from
Figure 2, after the incubation of the pure CNF membrane sample in alkaline electrolyte solutions (1M KOH and 1M Na
2CO
3), the C content was reducing much more significantly than O, resulting in the reduction of the C/O ratio from ~1 to ~0.48 (KOH) and to ~0.28 (Na
2CO
3), while showing the presence of a similar quantity of cations (~38 wt% of K
+, ~35 wt% of Na
+), thus confirming the coverage of CNF with electrolytes formed into different crystallite structures. For the CNF-based membranes containing MXene, the content of both Ti and C was reduced significantly for the sample tested in KOH (from ~23 wt% to ~5 wt% for Ti, and from ~25 wt% to ~13 wt% for C) and slightly in Na
2CO
3 (to ~16 wt% and ~10 wt%, respectively), while F was not detected anymore and the content of O increased from ~29 wt% to ~46 wt% and 52 wt%, also confirming the structural changes of Ti
3C
2T
x MXene after incubation in such a highly alkaline medium. Indeed, different from the pure CNF membrane (
Figure 2a), the CNF/MXene membrane (
Figure 2b) had an almost completely covered surface with a salt-specific crystal structure (represented by XRD peaks of very high intensities,
Figure 3a), formed on the interphase of both components at their anchoring sites. The broader peak between ~10 and 30° also indicates that the original crystal structure of cellulose was still retained. The XPS spectra (
Figure 3b) reconfirmed the presence of C, O, Ti, and F atoms for the CNF/Ti
3C
2T
x membrane. The deconvoluted Ti 2p spectra for the same composite membrane exhibited a new peak of Ti-O at ~461.8 eV as compared to the Ti
3C
2T
x sample after mixing with CNF, interacting with the surface-decorated −OH and rare −COOH groups of the CNF [
2], which, however, disappeared or shifted to ~460.5 eV after long-term incubation within KOH or Na
2CO
3, respectively. An intensely increased shoulder peak at ~463 eV in the Ti 2p spectra bound to TiO
2 compounds also confirmed a kinetically faster formation of oxygenated species in KOH than Na
2CO
3. However, the broadened C-O and C=O related peak at ~287.2 eV in the C 1s spectra of the CNF/Ti
3C
2T
x membrane were reduced completely after long-term incubation in both alkaline electrolytes, and new peaks appeared at the binding energy value at ~292 eV and ~295 eV corresponding to the KOH, and at ~289 eV, which, with a peak at ~532 eV in the O 1s spectra, corresponded to the -CO
3 surface decorated groups, thus confirming the formation of an electroactive layer on the surfaces. The H-bonding was again confirmed to improve the stability by enhancing interfacial adhesion between components of such hybrid membranes. From both the SEM and optical analysis, it is also evident that the black colour of MXene was lightening and the membranes yellowing, confirming distinctive changes of their physical and chemical characteristics, above all, the formation of TiO
2 compounds.
2.4. Electrochemical Properties of Composite Membranes in Three-Electrode System
The ions’ diffusion and the capacitive behaviour of Ti
3C
2T
x MXene is influenced by the interlayer spacing (its expansion) and cation migration barrier, above all hydrophilic and negatively charged oxygen termination groups such as =O and −OH [
43] on the MXene surface, which creates an electrostatic attraction with opposite charges in an acidic medium, and, thus, depending on the hydrodynamic size of the electrolyte cations, enables their adsorption and passage through the layers [
28] to generate pseudocapacitance [
49,
50]. The high mass loading of active materials is thus crucial in achieving high capacitance (C) and energy density (E), as the energy density is evaluated at an applied voltage (V) by
E = 1/2 CV
2. However, besides the MXene surface chemistry (the presence of −F, −O, −OH) and stability after delamination, trace impurities (above all the partial removal of Al and presence of TiO
2 and AlO
3) in the nanofibrilated cellulose acting as a binder can thereby reduce the accessible area for ionic transport significantly, and, thus, the area next to their interactions, limiting the utilisation of the active sites of Ti
3C
2T
x MXene and reducing its electrochemical properties [
5]. The size of the hydrated cations and their interaction with dissociated functional groups on CNFs that have to match the electroactive site (Ti
3C
2T
x MXene) in such a hybrid electrode material are thus also crucial, resulting in limited capacity values. The larger ions that cannot penetrate can, therefore, only contribute to the cyclic stability of the intercalation layers by forming EDL capacitance by electrochemical adsorption [
48].
Since the CNF/MXene membrane’s physico-chemical properties (swelling/shrinkage) and stability are highly dependent on the environmental pH (
Figure 4b), electrochemical measurements of the CNF/MXene membranes prepared with 50 wt% Ti
3C
2T
x MXene at both freeze-casting temperatures (−50 °C and −80 °C) acting as a cathode in a three-electrode setup were investigated using pH and ions-type different aqueous electrolyte solutions. Membranes prepared with higher (90 wt%) MXene content gave surprisingly comparable capacitance values when tested in the generally used H
2SO
4 and KOH (
Figure S6) but demonstrated poorer mechanical stability (
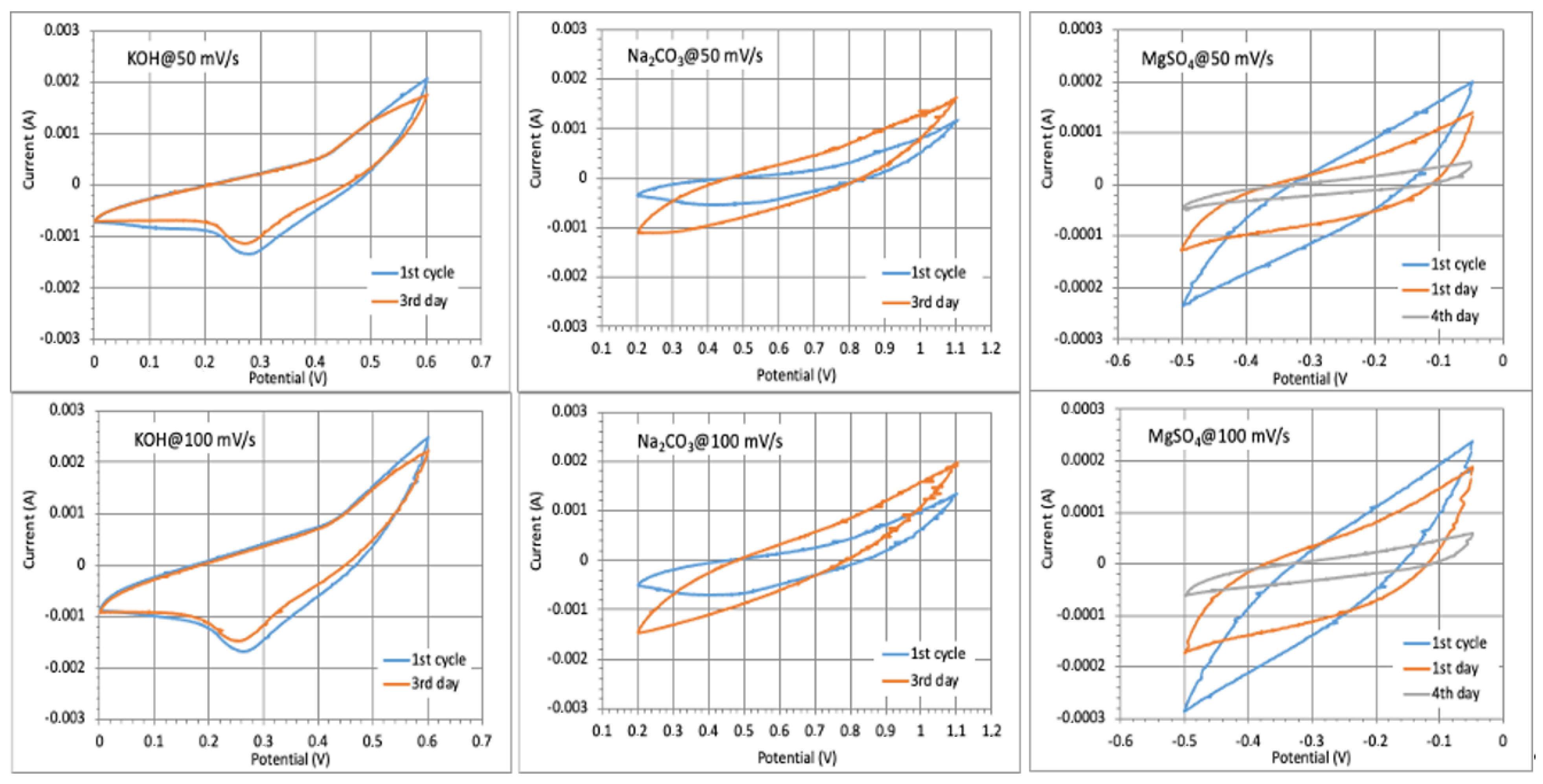
Figure S1). Therefore, they were not selected for further testing. First, cyclic voltammetry (CV) experiments were conducted for different electrolytes to determine their voltage window (vs. Ag/AgCl) and to understand the capacitive behaviours (gravimetrical, areal and volumetric) of CNF/MXene-based electrodes at different scan rates and time intervals. The CV curves (presented in
Figures S7 and S8) showed very similar profiles (redox peaks) by increasing the scan rates, suggesting that the electrodes have a relatively fast ionic response. One broad peak at the cathode and one broad at the anode were observed for all pH slightly acidic electrolytes, which corresponded to the insertion and extraction reactions of specific cations (H
+, K
+, Na
+, Mg
2+) during cycling (charging and discharging). The charge and discharge curves had no visible plateau, indicating that the capacitance originates mainly from the pseudocapacitance, i.e., reversible intercalation/deintercalation of protons due to the change of the Ti oxidation state, which, as expected, was most exposed for the hydrodynamically smallest and fastest H
+ cations using H
2SO
4 [
49,
50]. According to the surface area of CV curves, the pseudocapacitance generated by other electrolyte (larger) cations in a such a slightly acidic sulphated media was much smaller due both their hydrodynamically limited mobility to and through the MXene layers, as well as interactions with the CNF surfaces additionally stabilized by sulphate anions. On the other hand, the weak cathodic peak, ranging from 0.5 to 0.6 V and from 0.9 to 1.1 V for basic electrolytes (KOH and Na
2CO
3, respectively), can be attributed to the formation of an electroactive layer on the interface (as confirmed by the other studies and observed well from the SEM images presented in
Figure 2), which was increasing for the subsequent cathodic scans, due to the intercalation of K
+ and Na
+ ions to/into the Ti
3C
2T
x that formed Ti
3C
2T
xK
x/Ti
3C
2T
xNa
x. In the anodic scans, the broader peak between 0/0.2–0.4 V and 0.2–0.7 V was assigned to the extraction of cations from the Ti
3C
2T
x MXene, being wider and larger for the membranes prepared at −80 °C.
However, the mass-specific (gravimetric) capacitance of the CNF/MXene membranes was generally very low, and it decreased additionally with the increasing scan rate, as illustrated in
Figure 6. As already established in other studies [
5] and reconfirmed in this one, as discussed above, such low capacitance is due to the presence of traces of Al and formed oxidised species (TiO
2 and AlO
3) on the surface of the Ti
3C
2T
x MXene layers, which affects the inhomogeneous and less open interlayer structures, and thereby reduces the interactions of the electrolyte cations with the terminal groups on the MXene (=O, –OH, and –F), limiting their penetration to transport through the MXene nanosheets and thus its electrochemical ability. The accessibility of ionic transport is additionally reduced due to interactions of cations with the negatively charged functional groups (–OH, and –COOH) on the CNF’s outer surface, highly depending on the electrolyte pH, size and valence of hydrated cations, as discussed in previous section. Relatively low measured specific capacitance values were thus obtained for the highly alkaline KOH electrolyte, and even lower for all the sulphate electrolyte solutions, except in highly acidic H
2SO
4 as well as medium alkaline Na
2CO
3 (
Figure S7). At higher current rates, the capacitances additionally decreased significantly, showing still higher behaviour in H
2SO
4 and Na
2CO
3 electrolytes, and using a denser membrane prepared at −80 °C. At a low scan rate (~5 mV/s), the membrane capacitance for the H
2SO
4 electrolyte solution thus showed more than ~66% higher values (~10 F/g) for the membrane prepared at −80 °C as compared to that at −50 °C (~6 F/g), or using Na
2CO
3 as the electrolyte (6–4 F/g), due to both the higher (not interacting with CNFs) ionic mass transfer ability of the hydrodynamically smaller (H
+) cations to the active sites on the Ti
3C
2T
x interfaces and their reduced diffusion path in the case of the denser membrane, which increased both the kinetics and capacitance. This advantage cannot be utilised at higher scan rates due to still-limited access to the interlayer spacing in MXene for larger ion-accessible surface areas, favouring faster ion transport. The excessive amounts of electrolyte in the electrode pores which blocks them (in the case of acidic sulphate electrolytes) or layered structure formed on the membrane interface (in the case of basic electrolytes) impedes the ions’ mobility additionally, and, thus, accessibility to active sites on the Ti
3C
2T
x. The normalised gravimetric capacitances (presented by the inserted graphs), calculated solely based on the weight of Ti
3C
2T
x using the composition ratios of the initial hybrid dispersions, increased almost once (~100%, to ~21 F/g and ~12 F/g, respectively) for these two electrolytes (including KOH for the membrane prepared at −50 °C) at all scan rates, additionally confirming the effect of microstructural composition of the CNF/Ti
3C
2T
x MXene membranes and their interactions with electrolytes, that limits mass transport. It can also be concluded that, compared to highly acidic H
2SO
4, the selected slightly acidic electrolytes with monovalent cations (e.g., K
2SO
4 and Na
2SO
4) display much lower capacitance (~25–17% at 5 mV/s), but still higher than that with divalent Mg
+ (MgSO
4, ~7%), while exhibiting a similarly high current response in a less alkaline electrolyte with monovalent Na
+ (Na
2CO
3, ~85/50%) for the composite membrane prepared with 50 wt% of MXene at −50/80 °C. Although the gravimetric capacitance was not comparable to some of the currently reported NC/MXene-based SCs, summarized in
Table S1, the areal and volumetric performance values were quite similar, which can be due to the shrinkage of membranes across their surface area and/or thickness (
Figure 4) in all electrolytes, which causes stacking of diffused ions and limits mass transfer.
The repeated cyclic performance of the electrode prepared at −80 °C in different electrolyte solutions (
Figure 7) showed almost a 100% CV curve profile after a few days cycling in KOH (comparatively lower electrolyte uptake with intensive membrane shrinkage,
Figure 4b), a slight enlarging of voltammograms in Na
2CO
3 (increased adsorption ability of the monovalent cations with slight membrane swelling), and reduction in MgSO
4 (the lowest diffusion ability of larger Mg
2+), while the long-time testing could not be performed in H
2SO
4 due to membrane instability (CNF degradation) in such a highly acidic medium, as already discussed above (
Figure S3). However, the capacitance values were reduced to ~75% after 1000 numbers of cycling in KOH due to the slow oxidation of MXene to TiO
2 in such a highly basic electrolyte solution (pH 14), as was also evident from the sample whitening (the photograph presented in
Figure 8), while the sample remained quite stable in Na
2CO
3 and retained 90% capacitance. During the first few cycles, the electrolyte reached the active sites easily, and the capacity increased accordingly. Considering that CNF has no electrochemical capability, the changing of voltammogram areas after charging–discharging is thus related to the accessibility or limitation of active sites at the Ti
3C
2T
x for transport of differently large ions due to the interaction with CNFs, their swelling/shrinkage ability in an electrolyte solution and pH-dependent electrostatic attraction interactions with the cations [
37]. These results further show that the type of CNFs and their interactions with electrolyte ions thus have a significant impact on the rate and capacitance of CNF/Ti
3C
2T
x electrodes, i.e., the accessibility to surface active sites on Ti
3C
2T
x to store the ions after redox reactions, exhibiting a pseudocapacitive property, which, however, is also significantly dependent on the type and size of the cations.














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}