
Unveiling Fungal Community Structure along Different Levels of Anthropic Disturbance in a South American Subtropical Lagoon

 , , , ,
, , , ,  , , , , ,
, , , , ,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
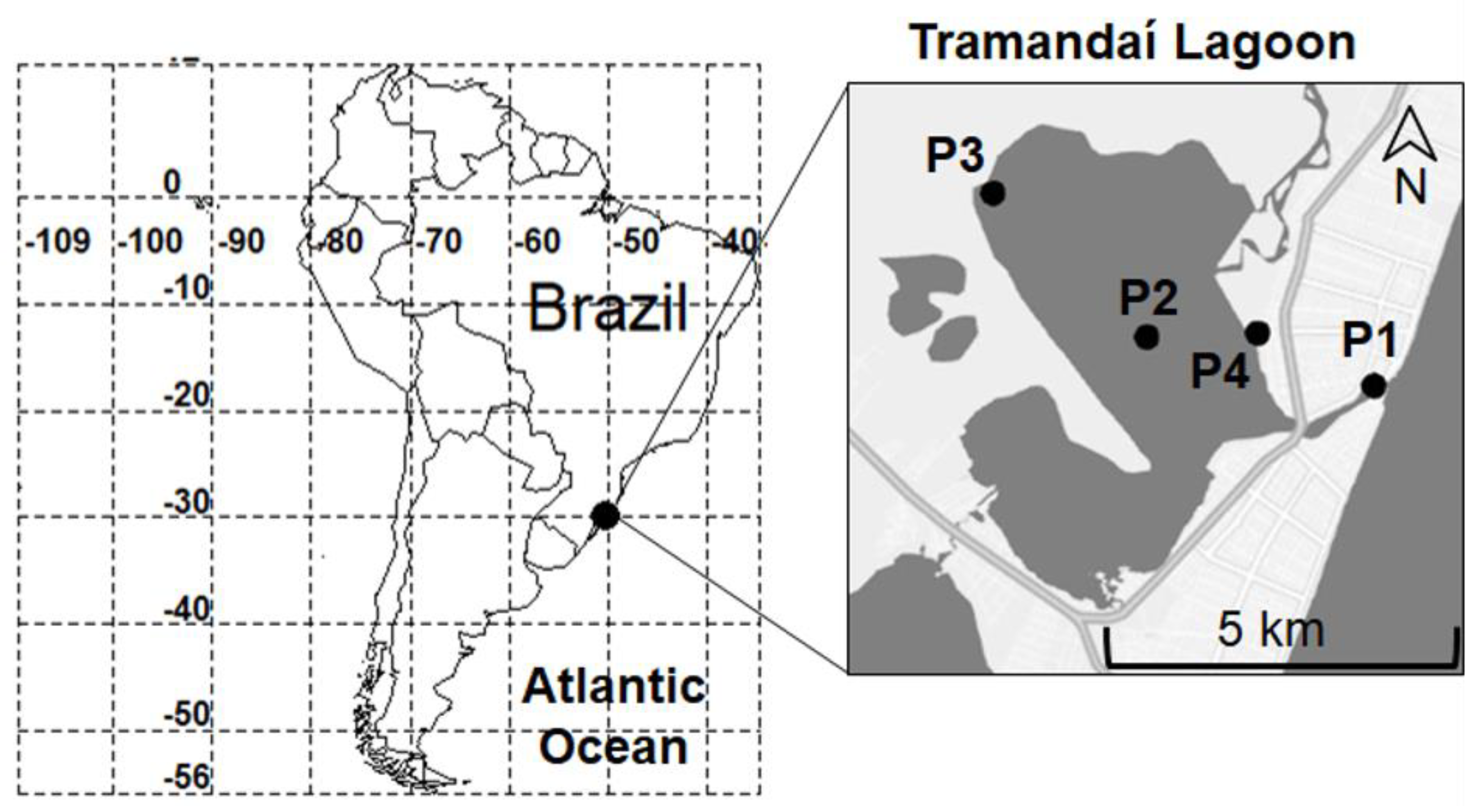
2.1. Study Area
2.2. Sampling Design and Collection
2.3. eDNA Extraction, Amplification, and High-Throughput Sequencing
2.4. Bioinformatic Analyses
2.5. Phylogenetic Analyses
2.6. Community Ecology Analyses
2.6.1. Alpha Diversity
2.6.2. Beta Diversity
2.6.3. Complex Network Analyses
2.6.4. Canonical Correspondence Analyses
3. Results
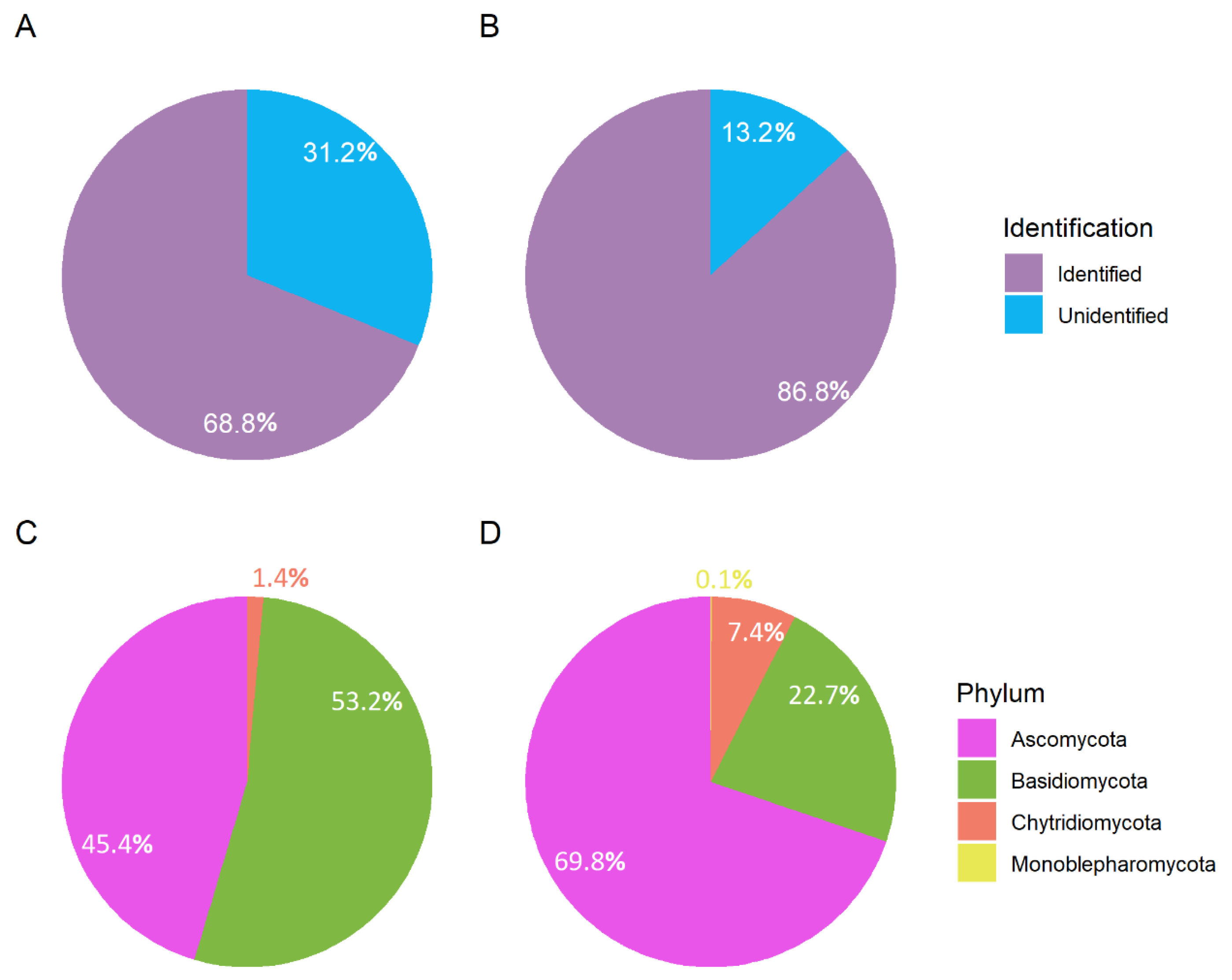
3.1. Taxonomic Composition
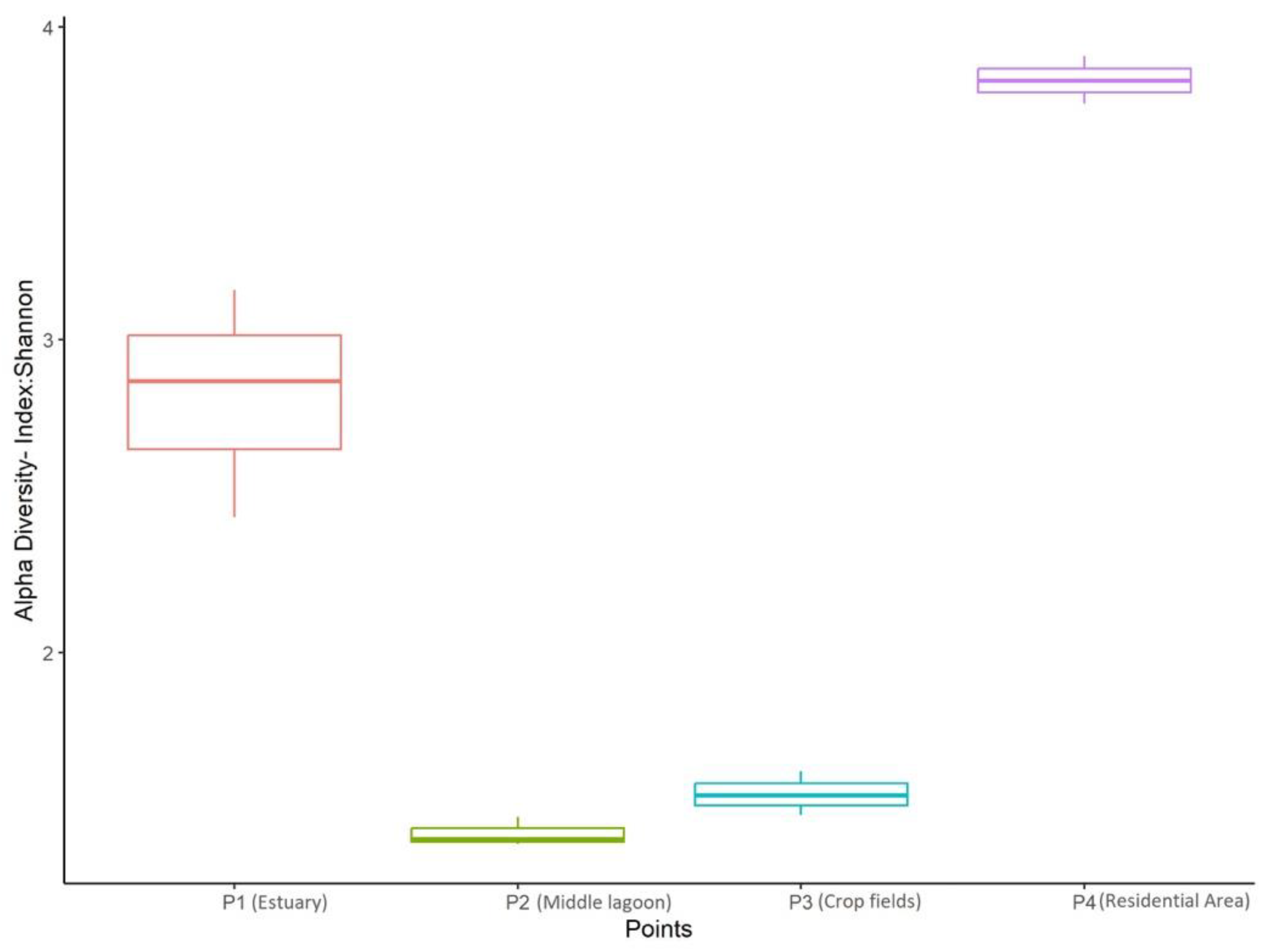
3.2. Alpha Diversity
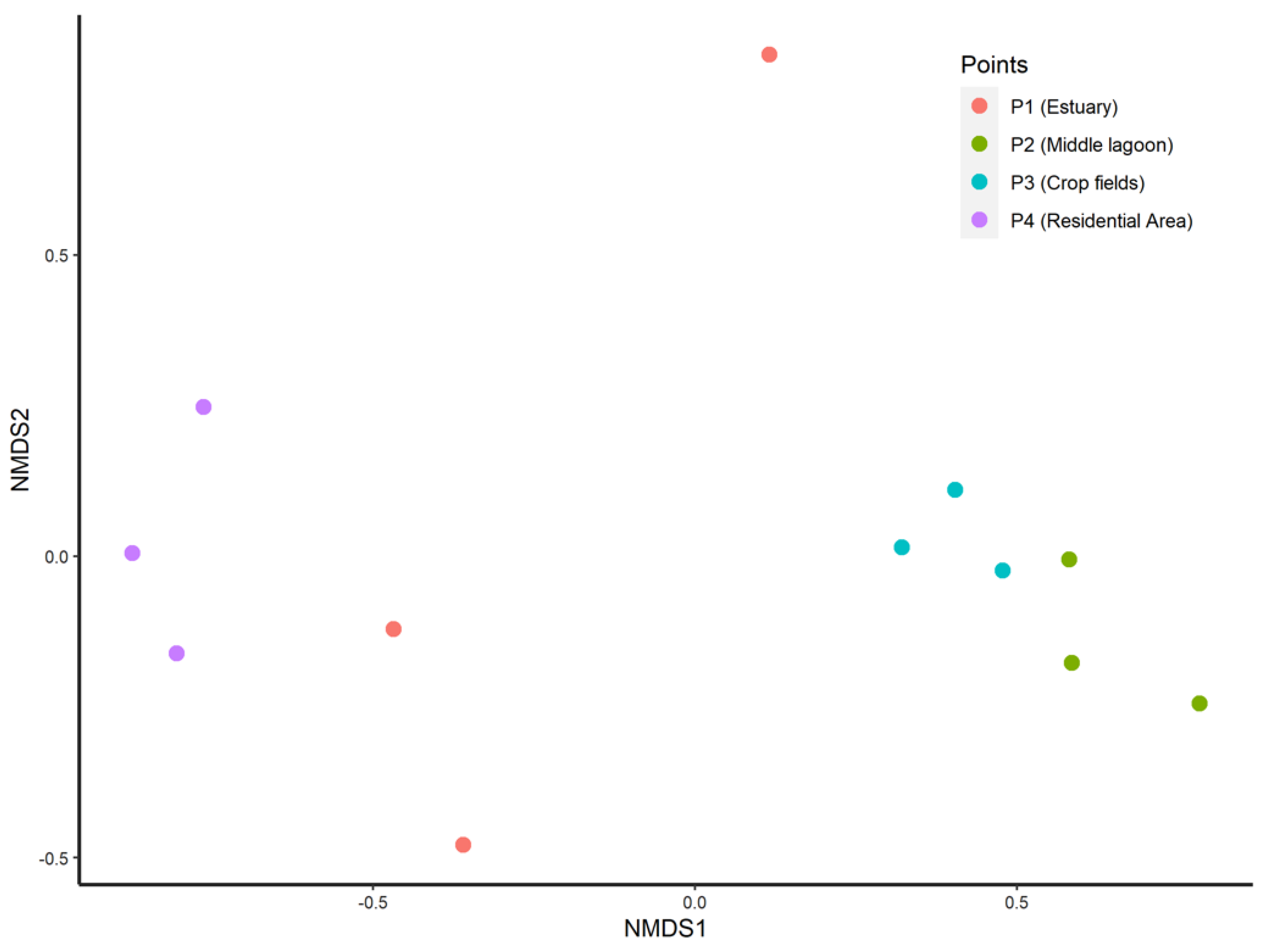
3.3. Beta Diversity
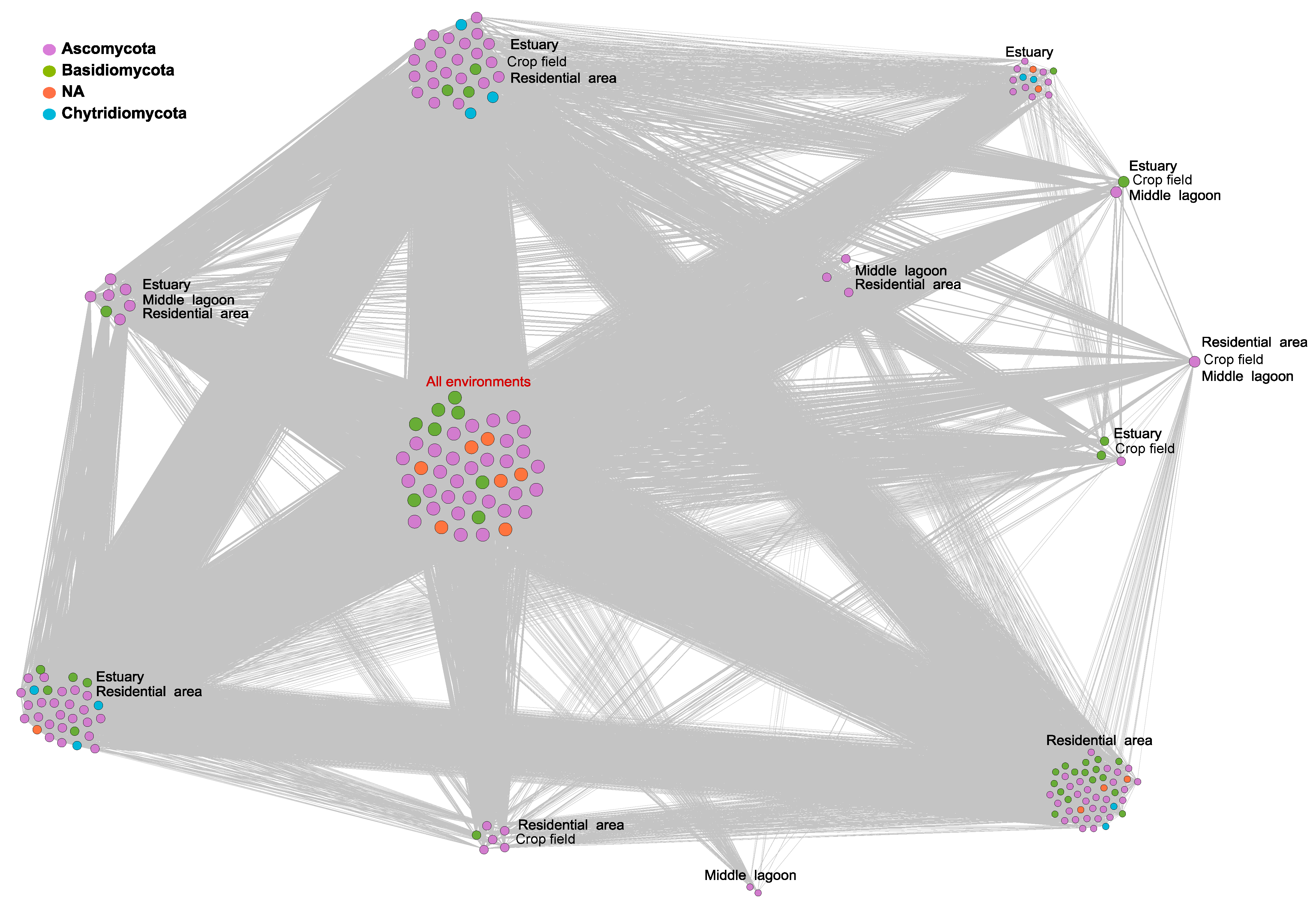
3.4. Co-Occurrence Networks
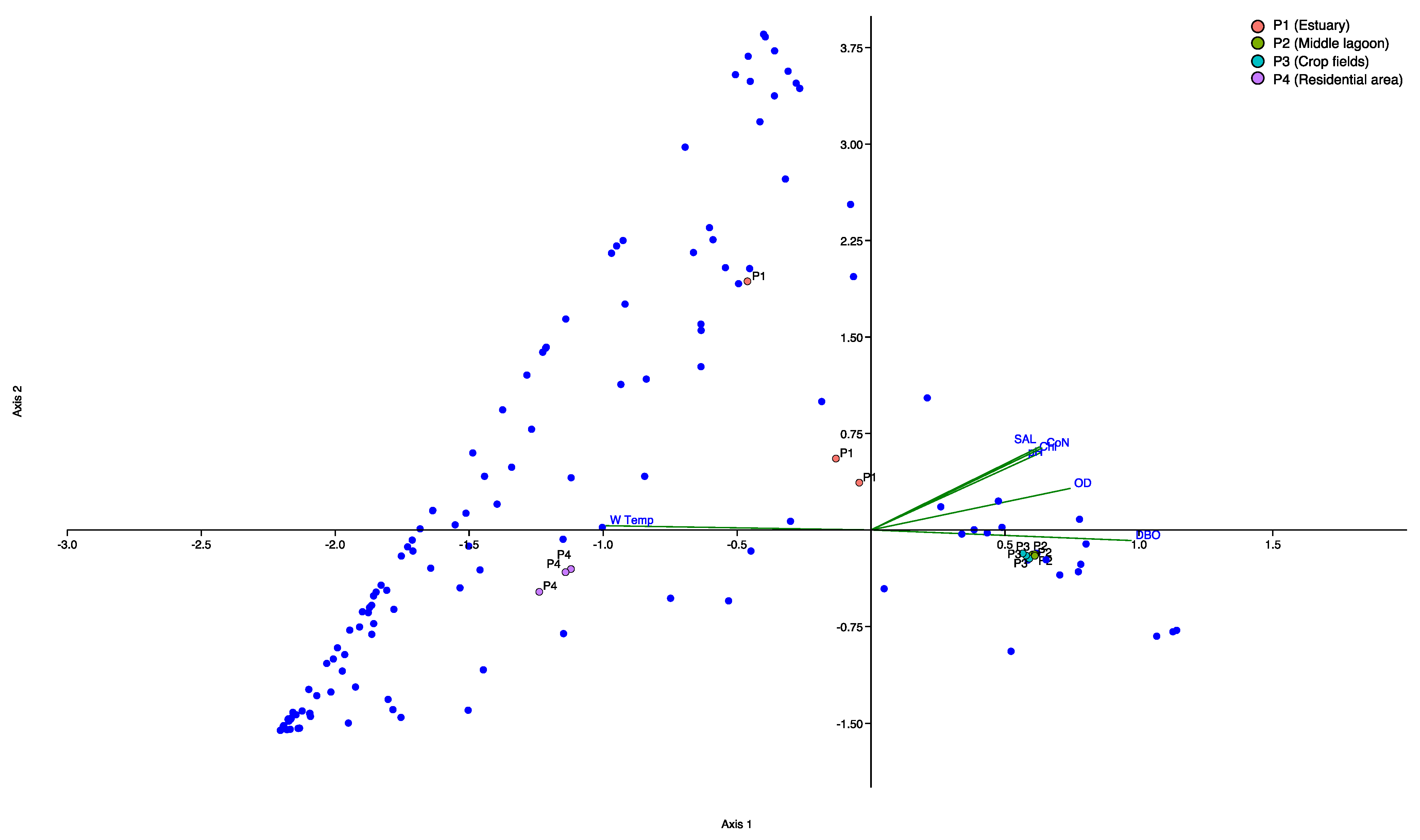
3.5. Canonical Correspondence Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Balian, E.V.; Segers, H.; Martens, K.; Lévéque, C. The Freshwater Animal Diversity Assessment: An Overview of the Results. In Freshwater Animal Diversity Assessment; Balian, E.V., Lévêque, C., Segers, H., Martens, K., Eds.; Developments in Hydrobiology; Springer: Dordrecht, The Netherlands, 2008; Volume 198, pp. 627–637. ISBN 978-1-4020-8258-0. [Google Scholar]
- Strayer, D.L.; Dudgeon, D. Freshwater Biodiversity Conservation: Recent Progress and Future Challenges. J. N. Am. Benthol. Soc. 2010, 29, 344–358. [Google Scholar] [CrossRef]
- Dudgeon, D.; Arthington, A.H.; Gessner, M.O.; Kawabata, Z.-I.; Knowler, D.J.; Lévêque, C.; Naiman, R.J.; Prieur-Richard, A.-H.; Soto, D.; Stiassny, M.L.J.; et al. Freshwater biodiversity: Importance, threats, status and conservation challenges. Biol. Rev. 2006, 81, 163. [Google Scholar] [CrossRef] [PubMed]
- Shearer, C.A.; Descals, E.; Kohlmeyer, B.; Kohlmeyer, J.; Marvanová, L.; Padgett, D.; Porter, D.; Raja, H.A.; Schmit, J.P.; Thorton, H.A.; et al. Fungal biodiversity in aquatic habitats. Biodivers. Conserv. 2007, 16, 49–67. [Google Scholar] [CrossRef]
- Grossart, H.-P.; Van Den Wyngaert, S.; Kagami, M.; Wurzbacher, C.; Cunliffe, M.; Rojas-Jimenez, K. Fungi in aquatic ecosystems. Nat. Rev. Microbiol. 2019, 17, 339–354. [Google Scholar] [CrossRef] [PubMed]
- Lepère, C.; Domaizon, I.; Humbert, J.-F.; Jardillier, L.; Hugoni, M.; Debroas, D. Diversity, spatial distribution and activity of fungi in freshwater ecosystems. PeerJ 2019, 7, e6247. [Google Scholar] [CrossRef]
- Gutiérrez, M.; Vera, J.; Srain, B.; Quiñones, R.; Wörmer, L.; Hinrichs, K.; Pantoja-Gutiérrez, S. Biochemical fingerprints of marine fungi: Implications for trophic and biogeochemical studies. Aquat. Microb. Ecol. 2020, 84, 75–90. [Google Scholar] [CrossRef]
- Gutiérrez, M.H.; Pantoja, S.; Tejos, E.; Quiñones, R.A. The role of fungi in processing marine organic matter in the upwelling ecosystem off Chile. Mar. Biol. 2011, 158, 205–219. [Google Scholar] [CrossRef]
- Gulis, V.; Ferreira, V.; Graca, M.A.S. Stimulation of leaf litter decomposition and associated fungi and invertebrates by moderate eutrophication: Implications for stream assessment. Freshw. Biol. 2006, 51, 1655–1669. [Google Scholar] [CrossRef]
- Jobard, M.; Rasconi, S.; Sime-Ngando, T. Diversity and functions of microscopic fungi: A missing component in pelagic food webs. Aquat. Sci. 2010, 72, 255–268. [Google Scholar] [CrossRef]
- Valentini, A.; Pompanon, F.; Taberlet, P. DNA barcoding for ecologists. Trends Ecol. Evol. 2009, 24, 110–117. [Google Scholar] [CrossRef]
- Matsuoka, S.; Sugiyama, Y.; Sato, H.; Katano, I.; Harada, K.; Doi, H. Spatial structure of fungal DNA assemblages revealed with eDNA metabarcoding in a forest river network in western Japan. MBMG 2019, 3, e36335. [Google Scholar] [CrossRef]
- Díaz-Ferguson, E.E.; Moyer, G.R. History, applications, methodological issues and perspectives for the use environmental DNA (eDNA) in marine and freshwater environments. RBT 2014, 62, 1273. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wang, M.M.; Wang, X.G.; Cheng, X.L.; Guo, J.J.; Bian, X.M.; Cai, L. Fungal communities in sediments of subtropical Chinese seas as estimated by DNA metabarcoding. Sci. Rep. 2016, 6, 26528. [Google Scholar] [CrossRef] [PubMed]
- Richards, T.A.; Leonard, G.; Mahé, F.; Del Campo, J.; Romac, S.; Jones, M.D.M.; Maguire, F.; Dunthorn, M.; De Vargas, C.; Massana, R.; et al. Molecular diversity and distribution of marine fungi across 130 European environmental samples. Proc. R. Soc. B. 2015, 282, 20152243. [Google Scholar] [CrossRef] [PubMed]
- Khomich, M.; Davey, M.L.; Kauserud, H.; Rasconi, S.; Andersen, T. Fungal communities in Scandinavian lakes along a longitudinal gradient. Fungal Ecol. 2017, 27, 36–46. [Google Scholar] [CrossRef]
- Marchioro, C.A.; Foerster, L.A. Development and Survival of the Diamondback Moth, Plutella xylostella (L.) (Lepidoptera: Yponomeutidae) as a Function of Temperature: Effect on the Number of Generations in Tropical and Subtropical Regions. Neotrop. Entomol. 2011, 40, 533–541. [Google Scholar]
- Houze, R.A.; Rasmussen, K.L.; Zuluaga, M.D.; Brodzik, S.R. The variable nature of convection in the tropics and subtropics: A legacy of 16 years of the Tropical Rainfall Measuring Mission satellite. Rev. Geophys. 2015, 53, 994–1021. [Google Scholar] [CrossRef]
- Jones, E.B.G.; Pang, K.-L. Tropical aquatic fungi. Biodivers. Conserv. 2012, 21, 2403–2423. [Google Scholar] [CrossRef]
- Hawksworth, D.L. The magnitude of fungal diversity: The 1.5 million species estimate revisited. Mycol. Res. 2001, 105, 1422–1432. [Google Scholar] [CrossRef]
- Attrill, M.J.; Rundle, S.D. Ecotone or Ecocline: Ecological Boundaries in Estuaries. Estuar. Coast. Shelf Sci. 2002, 55, 929–936. [Google Scholar] [CrossRef]
- Deak, T. Ecology and Biodiversity of Yeasts with Potential Value in Biotechnology. In Yeast Biotechnology: Diversity and Applications; Satyanarayana, T., Kunze, G., Eds.; Springer: Dordrecht, The Netherlands, 2009; pp. 151–168. ISBN 978-1-4020-8291-7. [Google Scholar]
- Pagani, D.M.; Heidrich, D.; Tormente, F.; Milani, G.; Jank, L.; They, N.H.; Valente, P.; Scroferneker, M.L. High MICs for antifungal agents in yeasts from an anthropized lagoon in South America. Microbiol. Res. 2022, 262, 127083. [Google Scholar] [CrossRef] [PubMed]
- Loitzenbauer, E.; Bulhões Mendes, C.A. Salinity dynamics as a tool for water resources management in coastal zones: An application in the Tramandaí River basin, southern Brazil. Ocean Coast. Manag. 2012, 55, 52–62. [Google Scholar] [CrossRef]
- Leite, B.; Chaves, M.A.D.; Nunes, A.A.T.; Jank, L.; Corção, G. Antibiotic resistance in surface waters from a coastal lagoon of Southern Brazil under the impact of anthropogenic activities. Rev. Ambiente Água 2019, 14, 5. [Google Scholar] [CrossRef]
- Rice, E.W.; Baird, R.B.; Eaton, A.D. (Eds.) Standard Methods for the Examination of Water and Wastewater; American Public Health Association, American Water Works Association, Water Environment Federation: Washington, DC, USA, 2017; Volume 22, ISBN 978-0-87553-287-5. [Google Scholar]
- Ihrmark, K.; Bödeker, I.T.M.; Cruz-Martinez, K.; Friberg, H.; Kubartova, A.; Schenck, J.; Strid, Y.; Stenlid, J.; Brandström-Durling, M.; Clemmensen, K.E.; et al. New primers to amplify the fungal ITS2 region-evaluation by 454-sequencing of artificial and natural communities. FEMS Microbiol. Ecol. 2012, 82, 666–677. [Google Scholar] [CrossRef] [PubMed]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In PCR Protocols: A Guide to Methods and Application; Innis, M.A., Gelfand, D.H., Sninsky, J.J., White, T.J., Eds.; Elsevier, Academic Press: New York, NY, USA, 1990; pp. 315–322. ISBN 978-0-12-372180-8. [Google Scholar]
- Reynolds, N.K.; Jusino, M.A.; Stajich, J.E.; Smith, M.E. Understudied, underrepresented, and unknown: Methodological biases that limit detection of early diverging fungi from environmental samples. Mol. Ecol. Resour. 2022, 22, 1065–1085. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- McIlroy, M.D. A Research UNIX Reader: Annotated Excerpts from the Programmer’s Manual, 1971–1986; AT&T Bell Laboratories: Murray Hill, NY, USA, 1987; pp. 1–115. [Google Scholar]
- Martelli, A.; Holden, S.; McGuire, P.; Ravenscroft Martelli, A. Python in a Nutshell: A Desktop Quick Reference, 4th ed.; O’Reilly Media: Sebastopol, CA, USA, 2023; ISBN 978-1-09-811355-1. [Google Scholar]
- Kõljalg, U.; Nilsson, H.R.; Schigel, D.; Tedersoo, L.; Larsson, K.-H.; May, T.W.; Taylor, A.F.S.; Jeppesen, T.S.; Frøslev, T.G.; Lindahl, B.D.; et al. The Taxon Hypothesis Paradigm—On the Unambiguous Detection and Communication of Taxa. Microorganisms 2020, 8, 1910. [Google Scholar] [CrossRef]
- Edgar, R.C. SINTAX: A Simple Non-Bayesian Taxonomy Classifier for 16S and ITS Sequences. bioRxiv 2016. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- Nguyen, N.H.; Song, Z.; Bates, S.T.; Branco, S.; Tedersoo, L.; Menke, J.; Schilling, J.S.; Kennedy, P.G. FUNGuild: An open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol. 2016, 20, 241–248. [Google Scholar] [CrossRef]
- Geneious Prime. Geneious Prime ®, version 2022.1.1; Geneious: Auckland, New Zealand, 2022.
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Rambaut, A. FigTree: Tree Figure Drawing Tool, version 1.4.3; Andrew Rambaut: Edinburgh, UK, 2016.
- R Core Team. R: A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2023. [Google Scholar]
- Chong, J.; Liu, P.; Zhou, G.; Xia, J. Using MicrobiomeAnalyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nat. Protoc. 2020, 15, 799–821. [Google Scholar] [CrossRef]
- Dhariwal, A.; Chong, J.; Habib, S.; King, I.L.; Agellon, L.B.; Xia, J. MicrobiomeAnalyst: A web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acids Res. 2017, 45, W180–W188. [Google Scholar] [CrossRef]
- Quinn, T.P.; Erb, I.; Gloor, G.; Notredame, C.; Richardson, M.F.; Crowley, T.M. A field guide for the compositional analysis of any-omics data. GigaScience 2019, 8, giz107. [Google Scholar] [CrossRef]
- Bastian, M.; Heymann, S.; Jacomy, M. Gephi: An open source software for exploring and manipulating networks. In Proceedings of the International AAAI Conference on Web and Social Media, Limassol, Cyprus, 5–8 June 2009. [Google Scholar]
- Anderson, M.J. Permutation tests for univariate or multivariate analysis of variance and regression. Can. J. Fish. Aquat. Sci. 2001, 58, 626–639. [Google Scholar] [CrossRef]
- Anderson, M.J. Distance-Based Tests for Homogeneity of Multivariate Dispersions. Biometrics 2006, 62, 245–253. [Google Scholar] [CrossRef]
- Hammer, O.; Harper, D.A.T.; Ryan, P.D. PAST: Paleontological statistics software package for education and data analysis. Palaeontol. Electron. 2001, 4, 9. [Google Scholar]
- Legendre, P.; Legendre, L. Numerical Ecology. In Developments in Environmental Modelling, 2nd ed.; Elsevier: Amsterdam, The Netherlands; New York, NY, USA, 1998; ISBN 978-0-444-89249-2. [Google Scholar]
- Ortiz-Vera, M.P.; Olchanheski, L.R.; Da Silva, E.G.; De Lima, F.R.; Martinez, L.R.D.P.R.; Sato, M.I.Z.; Jaffé, R.; Alves, R.; Ichiwaki, S.; Padilla, G.; et al. Influence of water quality on diversity and composition of fungal communities in a tropical river. Sci. Rep. 2018, 8, 14799. [Google Scholar] [CrossRef]
- Song, P.; Tanabe, S.; Yi, R.; Kagami, M.; Liu, X.; Ban, S. Fungal community structure at pelagic and littoral sites in Lake Biwa determined with high-throughput sequencing. Limnology 2018, 19, 241–251. [Google Scholar] [CrossRef]
- Panzer, K.; Yilmaz, P.; Weiß, M.; Reich, L.; Richter, M.; Wiese, J.; Schmaljohann, R.; Labes, A.; Imhoff, J.F.; Glöckner, F.O.; et al. Identification of Habitat-Specific Biomes of Aquatic Fungal Communities Using a Comprehensive Nearly Full-Length 18S rRNA Dataset Enriched with Contextual Data. PLoS ONE 2015, 10, e0134377. [Google Scholar] [CrossRef]
- Park, D. Methods of detecting funig in organic detritus in water. Trans. Br. Mycol. Soc. 1972, 58, 281–290. [Google Scholar] [CrossRef]
- Bell, D.K. In Vitro Antagonism of Trichoderma species Against Six Fungal Plant Pathogens. Phytopathology 1982, 72, 379. [Google Scholar] [CrossRef]
- Dinardo-Miranda, L.L.; Vasconcelos, A.C.M.; Ferreira, J.M.G.; Garcia, C.A., Jr.; Coelho, Á.L.; Gil, M.A. Eficiência de Metarhizium anisopliae (Metsch.) no controle de Mahanarva fimbriolata (Stål) (Hemiptera: Cercopidae) em cana-de-açúcar. Neotrop. Entomol. 2004, 33, 743–749. [Google Scholar] [CrossRef]
- Kubicek, C.P.; Komon-Zelazowska, M.; Druzhinina, I.S. Fungal genus Hypocrea/Trichoderma: From barcodes to biodiversity. J. Zhejiang Univ. Sci. B 2008, 9, 753–763. [Google Scholar] [CrossRef] [PubMed]
- Castro, D.; Rocha, C.M. Qualidade das águas da bacia hidrográfica do Rio Tramandaí; Via Sapiens: Porto Alegre, Brazil, 2016. [Google Scholar]
- Schmidt, O.; Liese, W. Variability of Wood Degrading Enzymes of Schizophyllum commune. Holzforschung 1980, 34, 67–72. [Google Scholar] [CrossRef]
- Ohm, R.A.; De Jong, J.F.; Lugones, L.G.; Aerts, A.; Kothe, E.; Stajich, J.E.; De Vries, R.P.; Record, E.; Levasseur, A.; Baker, S.E.; et al. Genome sequence of the model mushroom Schizophyllum commune. Nat. Biotechnol. 2010, 28, 957–963. [Google Scholar] [CrossRef]
- Barr, D.J.S. Phylum Chytridiomycota. In Handbook of Protoctis; Margulis, L., Corliss, J.O., Melkonian, M., Chapman, D.J., Eds.; Jones and Bartlett: Boston, MA, USA, 1990; pp. 454–466. [Google Scholar]
- James, T.Y.; Letcher, P.M.; Longcore, J.E.; Mozley-Standridge, S.E.; Porter, D.; Powell, M.J.; Griffith, G.W.; Vilgalys, R. A molecular phylogeny of the flagellated fungi (Chytridiomycota) and description of a new phylum (Blastocladiomycota). Mycologia 2006, 98, 860–871. [Google Scholar] [CrossRef]
- Alexopoulos, C.J.; Mims, C.W.; Blackwell, M. Introductory Mycology, 4th ed.; John Wiley and Sons: Hoboken, NJ, USA, 1996; ISBN 978-0-471-52229-4. [Google Scholar]
- Letcher, P.M.; Powell, M.J.; Barr, D.J.S.; Churchill, P.F.; Wakefield, W.S.; Picard, K.T. Rhizophlyctidales—A new order in Chytridiomycota. Mycol. Res. 2008, 112, 1031–1048. [Google Scholar] [CrossRef]
- Vandenkoornhuyse, P.; Baldauf, S.L.; Leyval, C.; Straczek, J.; Young, J.P.W. Extensive Fungal Diversity in Plant Roots. Science 2002, 295, 2051. [Google Scholar] [CrossRef]
- Rayner, A.D.M.; Boddy, L. Amoebae and Myxomycetes. In Fungal Decomposition of Wood: Its Biology and Ecology; Wiley & Sons: Chichester, UK, 1988; pp. 132–134. [Google Scholar]
- Yelle, D.J.; Ralph, J.; Lu, F.; Hammel, K.E. Evidence for cleavage of lignin by a brown rot basidiomycete. Environ. Microbiol. 2008, 10, 1844–1849. [Google Scholar] [CrossRef]
- Calliari, L.J.; Winterwerp, J.C.; Fernandes, E.; Cuchiara, D.; Vinzon, S.B.; Sperle, M.; Holland, K.T. Fine grain sediment transport and deposition in the Patos Lagoon–Cassino beach sedimentary system. Cont. Shelf Res. 2009, 29, 515–529. [Google Scholar] [CrossRef]
- Mizwar, A.; Surapati, H.U. Biological Oxygent Demand (BOD5) as Bio Indicator of Phytoplankton Diversity Index in the Mangrove Area of Kintap Estuary—South Kalimantan. IOP Conf. Ser. Earth Environ. Sci. 2020, 448, 012126. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genus | Trophic Mode | Ecological Guild | Body Morphology |
|---|---|---|---|
| ASCOMYCOTA | |||
| Alternaria | Pathotroph-Saprotroph-Symbiotroph | Animal Pathogen-Endophyte-Plant Pathogen-Wood Saprotroph | Mycelial |
| Apiospora | Pathotroph-Saprotroph | Plant Pathogen | Mycelial |
| Aureobasidium | Pathotroph-Saprotroph-Symbiotroph | Animal Pathogen-Endophyte-Epiphyte-Plant Pathogen-Undefined Saprotroph | Yeast |
| Cercospora | Pathotroph | Plant Pathogen | Mycelial |
| Clonostachys | Pathotroph | Plant Pathogen | Mycelial |
| Colletotrichum | Pathotroph-Symbiotroph | Endophyte-Plant Pathogen | Mycelial |
| Coniella | Pathotroph | Plant Pathogen | Mycelial |
| Curvularia | Pathotroph | Plant Pathogen | Mycelial |
| Diaporthe | Pathotroph-Symbiotroph | Endophyte-Plant Pathogen | Mycelial |
| Erysiphe | Pathotroph | Plant Pathogen | Mycelial |
| Exserohilum | Pathotroph | Plant Pathogen | Mycelial |
| Fusarium | Pathotroph-Saprotroph-Symbiotroph | Animal Pathogen-Endophyte-Lichen Parasite-Plant Pathogen-Soil Saprotroph-Wood Saprotroph | Mycelial |
| Gliomastix | Saprotroph | Undefined Saprotroph | Mycelial |
| Lophiostoma | Saprotroph | Animal Pathogen | Mycelial |
| Metarhizium | Pathotroph | na | Mycelial |
| Meyerozyma | na | na | Yeast |
| Microdochium | na | na | Mycelial |
| Nannizzia | na | na | Mycelial |
| Neodevriesia | na | na | Mycelial |
| Neopyrenochaeta | na | Undefined Saprotroph | Mycelial |
| Neurospora | Saprotroph | Undefined Saprotroph | Mycelial |
| Niesslia | Saprotroph | Animal Pathogen | Mycelial |
| Nigrograna | Pathotroph | Undefined Saprotroph | Mycelial |
| Nigrospora | Saprotroph | na | Mycelial |
| Omnidemptus | na | na | Mycelial |
| Paracylindrocarpon | na | Dung Saprotroph-Undefined Saprotroph-Wood Saprotroph | Mycelial |
| Penicillium | Saprotroph | Endophyte-Plant Pathogen-Wood Saprotroph | Mycelial |
| Periconia | Pathotroph-Saprotroph-Symbiotroph | Plant Pathogen | Mycelial |
| Phaeoacremonium | Pathotroph | Endophyte-Plant Pathogen | Mycelial |
| Plectosphaerella | Pathotroph-Symbiotroph | Plant Pathogen | Mycelial |
| Pseudocercospora | Pathotroph | na | Mycelial |
| Pseudopyricularia | na | Endophyte-Lichen Parasite-Undefined Saprotroph | Mycelial |
| Pyrenochaetopsis | Pathotroph-Saprotroph-Symbiotroph | na | Mycelial |
| Ramichloridium | na | Undefined Saprotroph | Mycelial |
| Saccharomyces | Saprotroph | na | Yeast |
| Striaticonidium | na | Undefined Saprotroph | Mycelial |
| Talaromyces | Saprotroph | Undefined Saprotroph | Mycelial |
| Teichospora | Saprotroph | Endophyte-Plant Pathogen | Mycelial |
| Toxicocladosporium | Pathotroph-Symbiotroph | Animal Pathogen-Endophyte-Epiphyte-Fungal Parasite-Plant Pathogen-Wood Saprotroph | Mycelial |
| Trichoderma | Pathotroph-Saprotroph-Symbiotroph | Endophyte | Mycelial |
| Trichomerium | Symbiotroph | Undefined Saprotroph | Mycelial |
| Uwebraunia | Saprotroph | na | Mycelial |
| Zymoseptoria | na | Mycelial | |
| BASIDIOMYCOTA | |||
| Agaricus | na | na | Mycelial |
| Amanita | Saprotroph-Symbiotroph | Ectomycorrhizal-Undefined Saprotroph | Mycelial |
| Atractidochium | na | na | Mycelial |
| Chaetospermum | na | na | Mycelial |
| Cintractia | Pathotroph | Plant Parasite-Plant Pathogen | Mycelial |
| Coprinellus | Saprotroph | Undefined Saprotroph | Mycelial |
| Crustoderma | Saprotroph | Wood Saprotroph | Mycelial |
| Curvibasidium | na | na | Yeast |
| Erythrobasidium | na | na | Yeast |
| Farysia | Pathotroph | Plant Pathogen | Yeast |
| Fellomyces | na | na | Yeast |
| Hannaella | na | na | Yeast |
| Kockovaella | Symbiotroph | Epiphyte | Yeast |
| Limonomyces | Pathotroph | Plant Pathogen | Mycelial |
| Lyomyces | na | na | Mycelial |
| Malassezia | Pathotroph-Saprotroph | Animal Pathogen-Undefined Saprotroph | Yeast |
| Marchandiomyces | Pathotroph | Lichen Parasite | Mycelial |
| Meira | na | na | Yeast |
| Moesziomyces | Pathotroph | Plant Pathogen | Yeast |
| Naganishia | na | na | Yeast |
| Papiliotrema | na | na | Yeast |
| Peniophora | Pathotroph-Saprotroph | Plant Pathogen-Wood Saprotroph | Mycelial |
| Peniophorella | Saprotroph | Undefined Saprotroph | Mycelial |
| Phellinus | Pathotroph-Saprotroph | Plant Pathogen-Wood Saprotroph | Mycelial |
| Phlebiopsis | Saprotroph | Undefined Saprotroph | Mycelial |
| Pseudohyphozyma | na | na | Yeast |
| Pseudomicrostroma | na | na | Yeast |
| Resinicium | Saprotroph | Undefined Saprotroph | Mycelial |
| Rhizopogon | Symbiotroph | Ectomycorrhizal | Mycelial |
| Rhodotorula | Pathotroph-Saprotroph | Animal Endosymbiont-Animal Pathogen-Endophyte-Plant Pathogen-Undefined Saprotroph | Yeast |
| Saitozyma | na | na | Yeast |
| Schizophyllum | Saprotroph | Wood Saprotroph | Mycelial |
| Sterigmatomyces | na | na | Yeast |
| Suillus | Symbiotroph | Ectomycorrhizal | Mycelial |
| Trechispora | Saprotroph | Wood Saprotroph | Mycelial |
| Tritirachium | Saprotroph | Undefined Saprotroph | Mycelial |
| Vishniacozyma | na | na | Yeast |
| Wallemia | Saprotroph | Undefined Saprotroph | Yeast |
| Xylodon | Saprotroph | Undefined Saprotroph | Mycelial |
| CHYTRIDIOMYCOTA | |||
| Entoplyctis | na | na | Rhizomycelial |
| Rhizoplyctis | na | na | Rhizomycelial |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pagani, D.M.; Ventura, S.P.R.; Vu, D.; Mendes-Pereira, T.; Ribeiro Tomé, L.M.; Carvalho, D.S.d.; Costa-Rezende, D.H.; Kato, R.B.; García, G.J.Y.; Geml, J.; et al. Unveiling Fungal Community Structure along Different Levels of Anthropic Disturbance in a South American Subtropical Lagoon. J. Fungi 2023, 9, 890. https://doi.org/10.3390/jof9090890
Pagani DM, Ventura SPR, Vu D, Mendes-Pereira T, Ribeiro Tomé LM, Carvalho DSd, Costa-Rezende DH, Kato RB, García GJY, Geml J, et al. Unveiling Fungal Community Structure along Different Levels of Anthropic Disturbance in a South American Subtropical Lagoon. Journal of Fungi. 2023; 9(9):890. https://doi.org/10.3390/jof9090890
Chicago/Turabian StylePagani, Danielle Machado, Stefânia P. R. Ventura, Duong Vu, Thairine Mendes-Pereira, Luiz Marcelo Ribeiro Tomé, Daniel Santana de Carvalho, Diogo Henrique Costa-Rezende, Rodrigo Bentes Kato, Glen Jasper Yupanqui García, József Geml, and et al. 2023. "Unveiling Fungal Community Structure along Different Levels of Anthropic Disturbance in a South American Subtropical Lagoon" Journal of Fungi 9, no. 9: 890. https://doi.org/10.3390/jof9090890
APA StylePagani, D. M., Ventura, S. P. R., Vu, D., Mendes-Pereira, T., Ribeiro Tomé, L. M., Carvalho, D. S. d., Costa-Rezende, D. H., Kato, R. B., García, G. J. Y., Geml, J., Robert, V., They, N. H., Brenig, B., Azevedo, V., Scroferneker, M. L., Valente, P., & Góes-Neto, A. (2023). Unveiling Fungal Community Structure along Different Levels of Anthropic Disturbance in a South American Subtropical Lagoon. Journal of Fungi, 9(9), 890. https://doi.org/10.3390/jof9090890