
The Intricate Connection between Bacterial α-Diversity and Fungal Engraftment in the Human Gut of Healthy and Impaired Individuals as Studied Using the In Vitro SHIME® Model


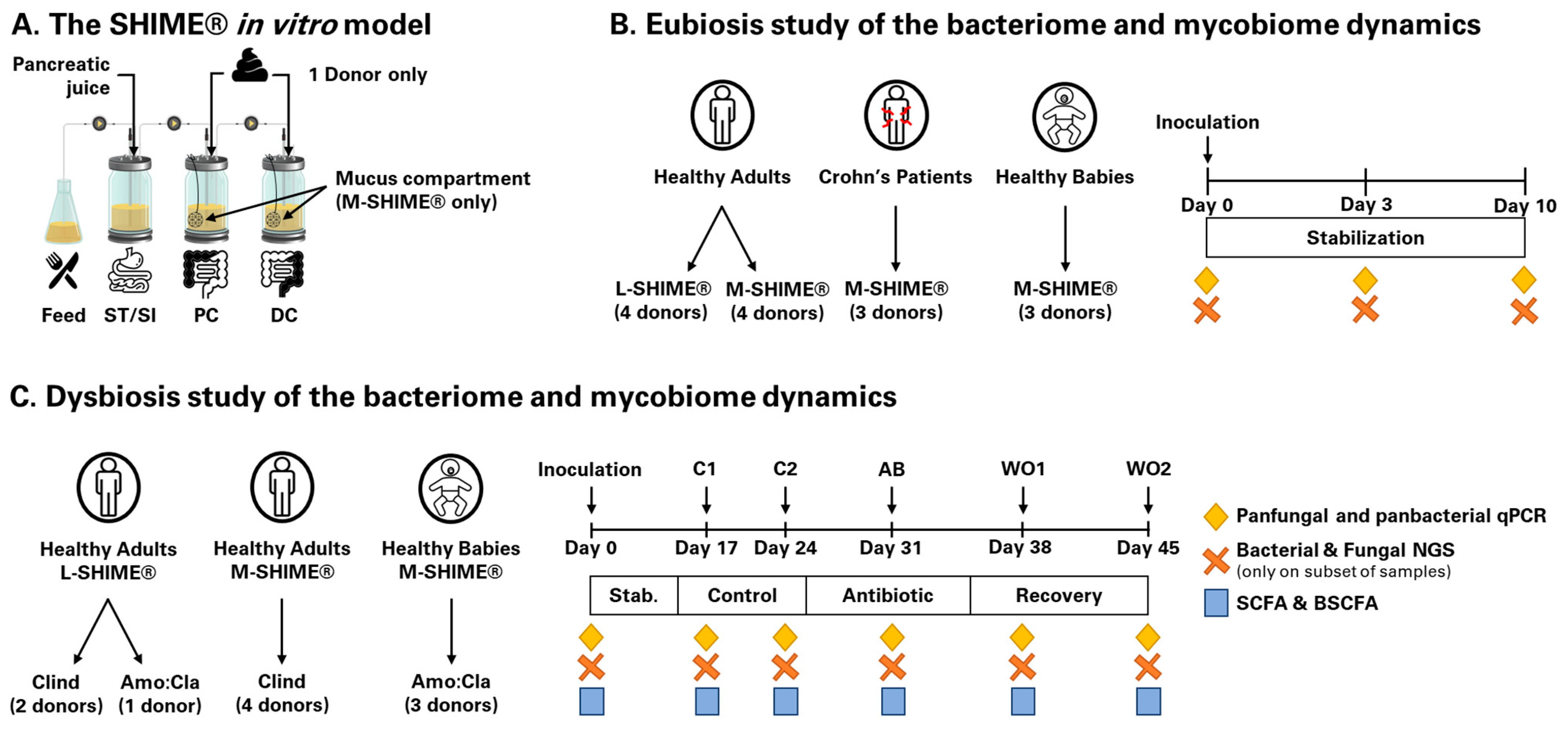{kind=link}
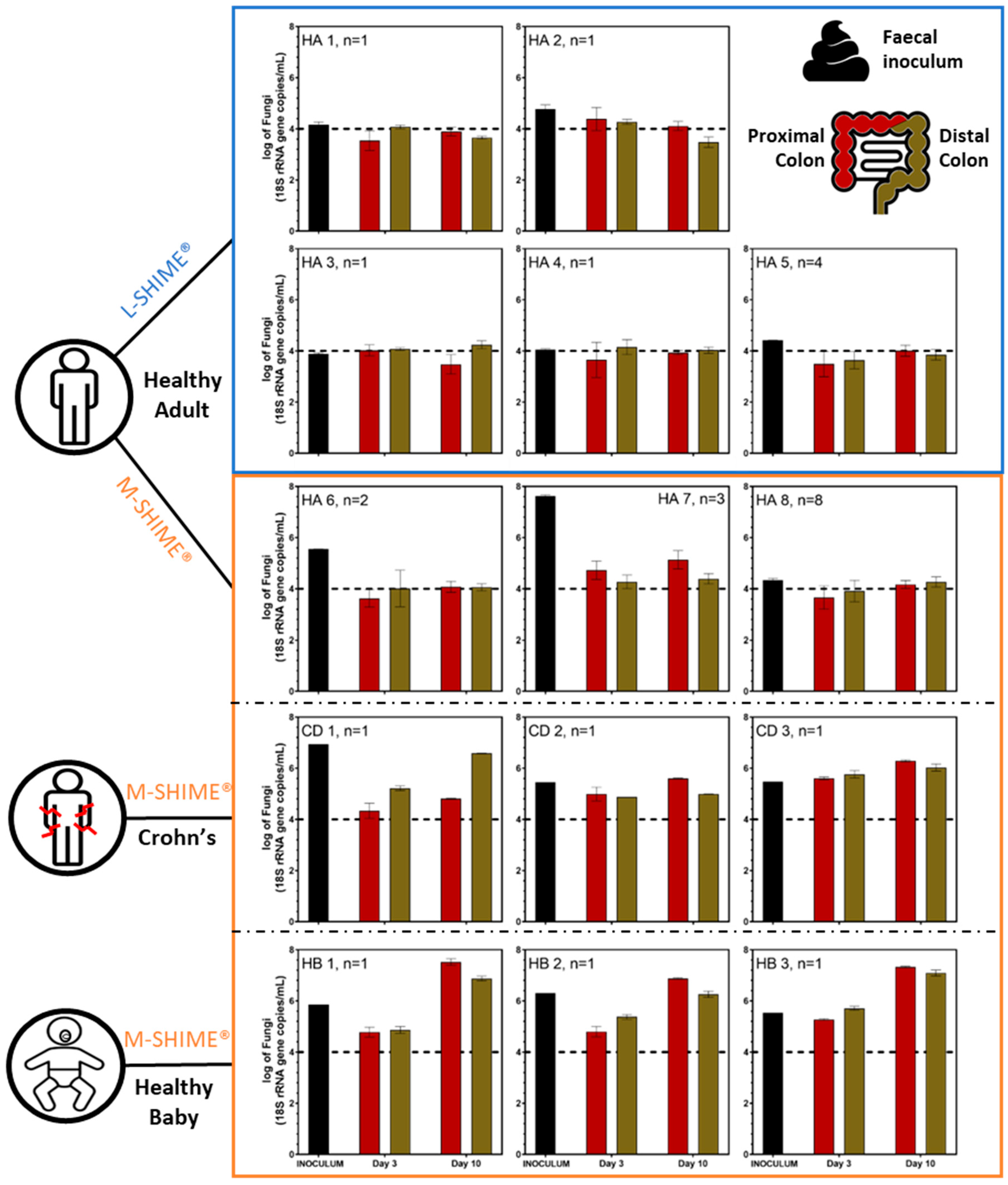{kind=link}
{kind=link}
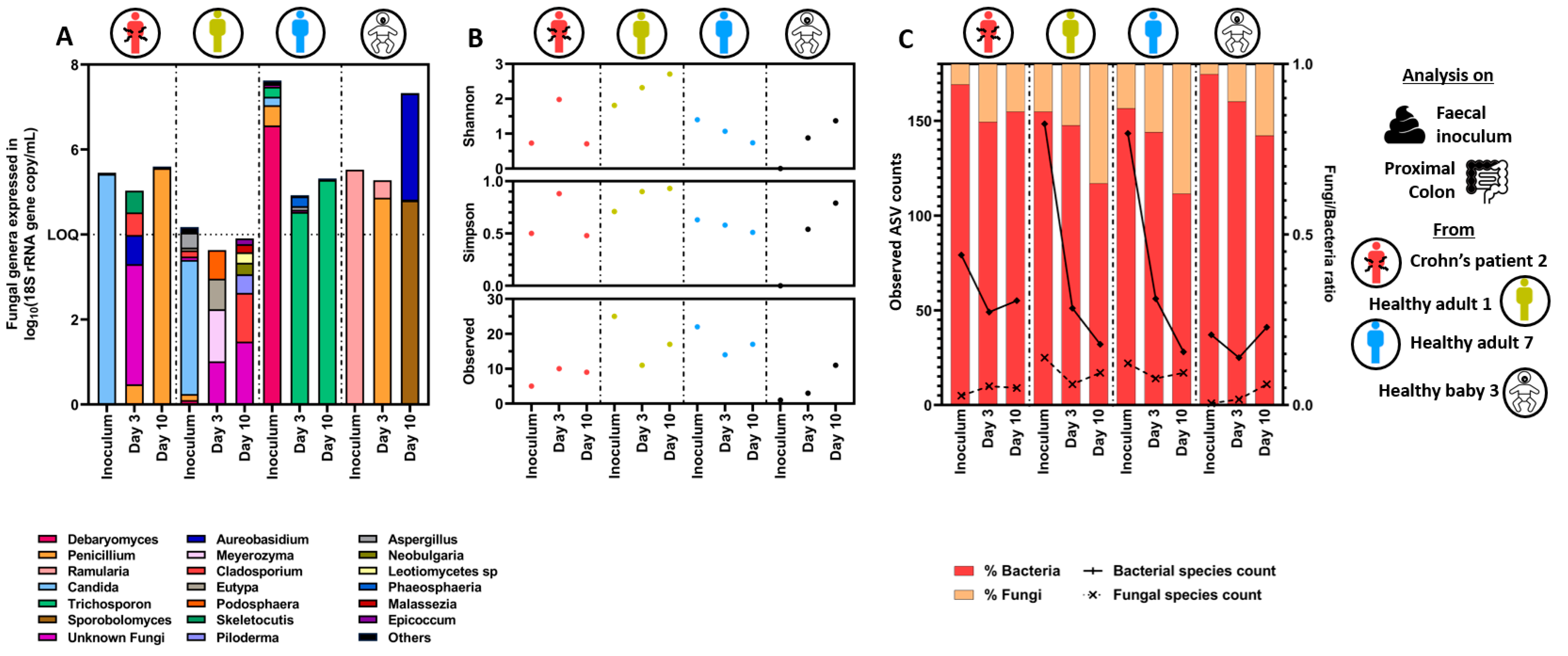
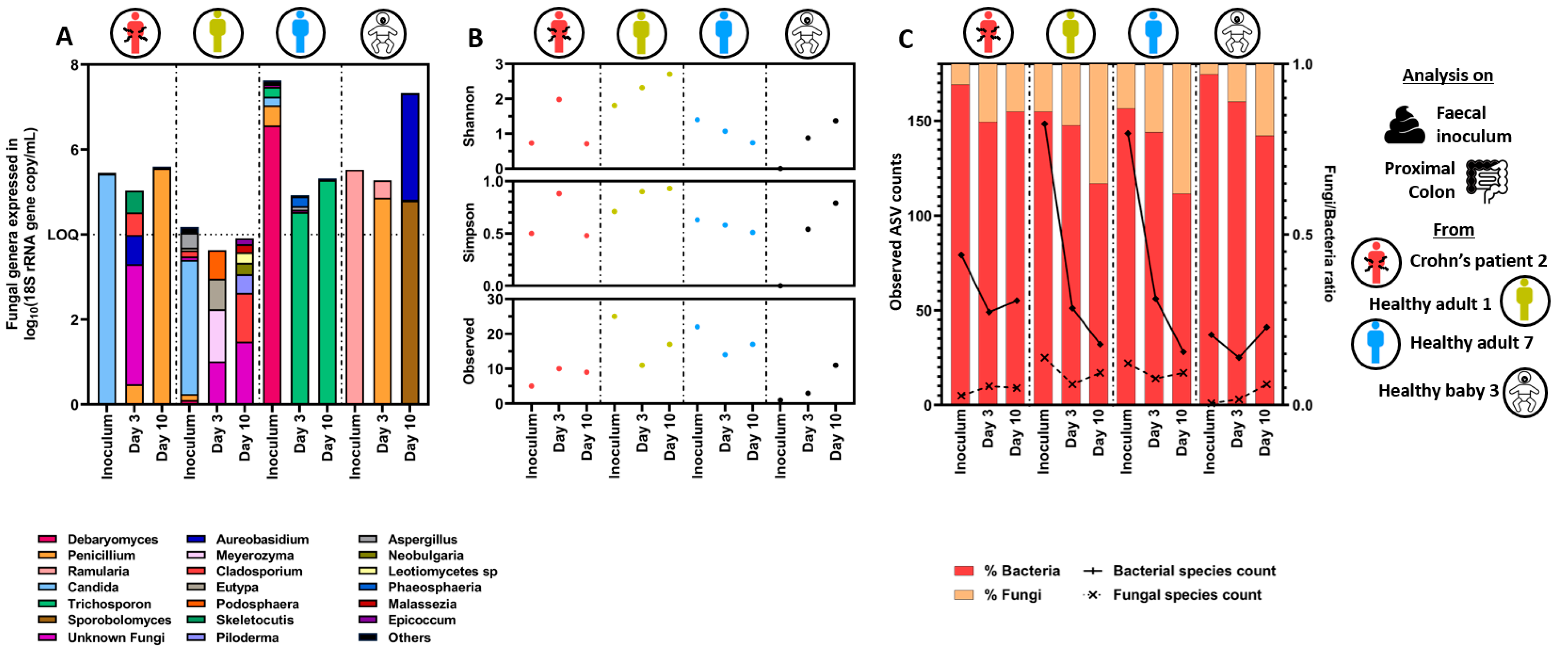{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemical Products
2.2. Sample Collection and Donor Descripion
2.3. Experimental Design of Long-Term L and M-SHIME®
2.3.1. Eubiosis Study
2.3.2. Dysbiosis Study
2.4. Metabolic Analysis
2.5. Microbial Community Analysis by qPCR
2.6. Microbial Community Analysis by 16S rRNA and ITS1 rRNA Gene-Targeted Sequencing
2.7. Data Analysis
3. Results
3.1. Wash-Out of Microbes during Stabilization Period in Eubiosis Conditions
3.1.1. Microbial Concentration in Eubiosis
3.1.2. Quantitative Microbiome Profiling and Microbial Diversity Dynamics in Eubiosis
3.2. Fungal Overgrowth Following Bacterial Diversity Reduction during Dysbiosis Induced with Different Antibiotic Treatments
3.2.1. Microbial Concentration during Eubiosis and the Antibiotic Treatment Period
3.2.2. Short-Chain Fatty Acid Production during Eubiosis and the Antibiotic Treatment Period
3.2.3. Quantitative Microbiome Profiling and Microbial Diversity Dynamics during Eubiosis and the Antibiotic Treatment Period
3.3. Specific Bacterial Signatures Are Associated with Changes in Fungal Concentration
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hawksworth, D.L.; Lucking, R. Fungal Diversity Revisited: 2.2 to 3.8 Million Species. Microbiol. Spectr. 2017, 5, 4. [Google Scholar] [CrossRef]
- Whitman, W.B.; Coleman, D.C.; Wiebe, W.J. Prokaryotes: The unseen majority. Proc. Natl. Acad. Sci. USA 1998, 95, 6578–6583. [Google Scholar] [CrossRef]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef]
- Underhill, D.M.; Iliev, I.D. The mycobiota: Interactions between commensal fungi and the host immune system. Nat. Rev. Immunol. 2014, 14, 405–416. [Google Scholar] [CrossRef]
- Rinninella, E.; Raoul, P.; Cintoni, M.; Franceschi, F.; Miggiano, G.A.D.; Gasbarrini, A.; Mele, M.C. What is the Healthy Gut Microbiota Composition? A Changing Ecosystem across Age, Environment, Diet, and Diseases. Microorganisms 2019, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Faust, K.; Raes, J. Microbial interactions: From networks to models. Nat. Rev. Microbiol. 2012, 10, 538–550. [Google Scholar] [CrossRef]
- Zheng, D.; Liwinski, T.; Elinav, E. Interaction between microbiota and immunity in health and disease. Cell Res. 2020, 30, 492–506. [Google Scholar] [CrossRef]
- Vandeputte, D.; De Commer, L.; Tito, R.Y.; Kathagen, G.; Sabino, J.; Vermeire, S.; Faust, K.; Raes, J. Temporal variability in quantitative human gut microbiome profiles and implications for clinical research. Nat. Commun. 2021, 12, 6740. [Google Scholar] [CrossRef]
- Chehoud, C.; Albenberg, L.G.; Judge, C.; Hoffmann, C.; Grunberg, S.; Bittinger, K.; Baldassano, R.N.; Lewis, J.D.; Bushman, F.D.; Wu, G.D. Fungal Signature in the Gut Microbiota of Pediatric Patients With Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2015, 21, 1948–1956. [Google Scholar] [CrossRef]
- Nash, A.K.; Auchtung, T.A.; Wong, M.C.; Smith, D.P.; Gesell, J.R.; Ross, M.C.; Stewart, C.J.; Metcalf, G.A.; Muzny, D.M.; Gibbs, R.A.; et al. The gut mycobiome of the Human Microbiome Project healthy cohort. Microbiome 2017, 5, 153. [Google Scholar] [CrossRef]
- d’Enfert, C.; Kaune, A.K.; Alaban, L.R.; Chakraborty, S.; Cole, N.; Delavy, M.; Kosmala, D.; Marsaux, B.; Frois-Martins, R.; Morelli, M.; et al. The impact of the Fungus-Host-Microbiota interplay upon Candida albicans infections: Current knowledge and new perspectives. FEMS Microbiol. Rev. 2021, 45, fuaa060. [Google Scholar] [CrossRef] [PubMed]
- Hallen-Adams, H.E.; Suhr, M.J. Fungi in the healthy human gastrointestinal tract. Virulence 2017, 8, 352–358. [Google Scholar] [CrossRef]
- Santus, W.; Devlin, J.R.; Behnsen, J. Crossing Kingdoms: How the Mycobiota and Fungal-Bacterial Interactions Impact Host Health and Disease. Infect. Immun. 2021, 89, 4. [Google Scholar] [CrossRef]
- Lawley, T.D.; Walker, A.W. Intestinal colonization resistance. Immunology 2013, 138, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Pappas, P.G. Opportunistic fungi: A view to the future. Am. J. Med. Sci. 2010, 340, 253–257. [Google Scholar] [CrossRef]
- Bartlett, J.G. Antibiotic-associated diarrhea. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 1992, 15, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Wu, G.D.; Albenberg, L.; Tomov, V.T. Gut microbiota and IBD: Causation or correlation? Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 573–584. [Google Scholar] [CrossRef]
- Lange, K.; Buerger, M.; Stallmach, A.; Bruns, T. Effects of Antibiotics on Gut Microbiota. Dig. Dis. 2016, 34, 260–268. [Google Scholar] [CrossRef]
- Fan, D.; Coughlin, L.A.; Neubauer, M.M.; Kim, J.; Kim, M.S.; Zhan, X.; Simms-Waldrip, T.R.; Xie, Y.; Hooper, L.V.; Koh, A.Y. Activation of HIF-1alpha and LL-37 by commensal bacteria inhibits Candida albicans colonization. Nat. Med. 2015, 21, 808–814. [Google Scholar] [CrossRef]
- Natividad, J.M.; Verdu, E.F. Modulation of intestinal barrier by intestinal microbiota: Pathological and therapeutic implications. Pharmacol. Res. 2013, 69, 42–51. [Google Scholar] [CrossRef]
- den Besten, G.; van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.J.; Bakker, B.M. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 2013, 54, 2325–2340. [Google Scholar] [CrossRef] [PubMed]
- Baumler, A.J.; Sperandio, V. Interactions between the microbiota and pathogenic bacteria in the gut. Nature 2016, 535, 85–93. [Google Scholar] [CrossRef]
- Gensollen, T.; Iyer, S.S.; Kasper, D.L.; Blumberg, R.S. How colonization by microbiota in early life shapes the immune system. Science 2016, 352, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Van de Wiele, T.; Van den Abbeele, P.; Ossieur, W.; Possemiers, S.; Marzorati, M. The Simulator of the Human Intestinal Microbial Ecosystem (SHIME®). In The Impact of Food Bioactives on Health: In Vitro and Ex Vivo Models; Verhoeckx, K., Cotter, P., Lopez-Exposito, I., Kleiveland, C., Lea, T., Mackie, A., Requena, T., Swiatecka, D., Wichers, H., Eds.; Springer: Cham, Switzerland, 2015; pp. 305–317. [Google Scholar]
- Roussel, C.; De Paepe, K.; Galia, W.; de Bodt, J.; Chalancon, S.; Denis, S.; Leriche, F.; Vandekerkove, P.; Ballet, N.; Blanquet-Diot, S.; et al. Multi-targeted properties of the probiotic Saccharomyces cerevisiae CNCM I-3856 against enterotoxigenic Escherichia coli (ETEC) H10407 pathogenesis across human gut models. Gut Microbes 2021, 13, 1953246. [Google Scholar] [CrossRef] [PubMed]
- Natividad, J.M.; Marsaux, B.; Rodenas, C.L.G.; Rytz, A.; Vandevijver, G.; Marzorati, M.; Van den Abbeele, P.; Calatayud, M.; Rochat, F. Human Milk Oligosaccharides and Lactose Differentially Affect Infant Gut Microbiota and Intestinal Barrier In Vitro. Nutrients 2022, 14, 2546. [Google Scholar] [CrossRef]
- Van den Abbeele, P.; Roos, S.; Eeckhaut, V.; MacKenzie, D.A.; Derde, M.; Verstraete, W.; Marzorati, M.; Possemiers, S.; Vanhoecke, B.; Van Immerseel, F.; et al. Incorporating a mucosal environment in a dynamic gut model results in a more representative colonization by lactobacilli. Microb. Biotechnol. 2012, 5, 106–115. [Google Scholar] [CrossRef]
- Van den Abbeele, P.; Belzer, C.; Goossens, M.; Kleerebezem, M.; De Vos, W.M.; Thas, O.; De Weirdt, R.; Kerckhof, F.M.; Van de Wiele, T. Butyrate-producing Clostridium cluster XIVa species specifically colonize mucins in an in vitro gut model. ISME J. 2013, 7, 949–961. [Google Scholar] [CrossRef]
- Calatayud, M.; Duysburgh, C.; Van den Abbeele, P.; Franckenstein, D.; Kuchina-Koch, A.; Marzorati, M. Long-Term Lactulose Administration Improves Dysbiosis Induced by Antibiotic and C. difficile in the PathoGut™ SHIME Model. Antibiotics 2022, 11, 1464. [Google Scholar] [CrossRef]
- Ghyselinck, J.; Verstrepen, L.; Moens, F.; Van den Abbeele, P.; Said, J.; Smith, B.; Bjarnason, I.; Basit, A.W.; Gaisford, S. A 4-strain probiotic supplement influences gut microbiota composition and gut wall function in patients with ulcerative colitis. Int. J. Pharm. 2020, 587, 119648. [Google Scholar] [CrossRef]
- Boon, N.; Top, E.M.; Verstraete, W.; Siciliano, S.D. Bioaugmentation as a tool to protect the structure and function of an activated-sludge microbial community against a 3-chloroaniline shock load. Appl. Environ. Microbiol. 2003, 69, 1511–1520. [Google Scholar] [CrossRef]
- Duysburgh, C.; Van den Abbeele, P.; Krishnan, K.; Bayne, T.F.; Marzorati, M. A synbiotic concept containing spore-forming Bacillus strains and a prebiotic fiber blend consistently enhanced metabolic activity by modulation of the gut microbiome in vitro. Int. J. Pharm. X 2019, 1, 100021. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Fujimoto, C.; Haruki, Y.; Maeda, T.; Kokeguchi, S.; Petelin, M.; Arai, H.; Tanimoto, I.; Nishimura, F.; Takashiba, S. Quantitative real-time PCR using TaqMan and SYBR Green for Actinobacillus actinomycetemcomitans, Porphyromonas gingivalis, Prevotella intermedia, tetQ gene and total bacteria. FEMS Immunol. Med. Microbiol. 2003, 39, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Chemidlin Prevost-Boure, N.; Christen, R.; Dequiedt, S.; Mougel, C.; Lelievre, M.; Jolivet, C.; Shahbazkia, H.R.; Guillou, L.; Arrouays, D.; Ranjard, L. Validation and application of a PCR primer set to quantify fungal communities in the soil environment by real-time quantitative PCR. PLoS ONE 2011, 6, e24166. [Google Scholar] [CrossRef] [PubMed]
- Herlemann, D.P.; Labrenz, M.; Jurgens, K.; Bertilsson, S.; Waniek, J.J.; Andersson, A.F. Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J. 2011, 5, 1571–1579. [Google Scholar] [CrossRef]
- Gardes, M.; Bruns, T.D. ITS primers with enhanced specificity for basidiomycetes--application to the identification of mycorrhizae and rusts. Mol. Ecol. 1993, 2, 113–118. [Google Scholar] [CrossRef]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In PCR Protocols a Guide to Methods and Applications; Academic Press: Cambridge, MA, USA, 1990; pp. 315–322. [Google Scholar]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Abarenkov, K.; Kristiansson, E.; Ryberg, M.; Nogal-Prata, S.; Gomez-Martinez, D.; Stuer-Patowsky, K.; Jansson, T.; Polme, S.; Ghobad-Nejhad, M.; Corcoll, N.; et al. The curse of the uncultured fungus. MycoKeys 2022, 86, 177–194. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef]
- Robert, V.; Vu, D.; Amor, A.B.; van de Wiele, N.; Brouwer, C.; Jabas, B.; Szoke, S.; Dridi, A.; Triki, M.; Ben Daoud, S.; et al. MycoBank gearing up for new horizons. IMA Fungus 2013, 4, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Van den Abbeele, P.; Sprenger, N.; Ghyselinck, J.; Marsaux, B.; Marzorati, M.; Rochat, F. A Comparison of the In Vitro Effects of 2’Fucosyllactose and Lactose on the Composition and Activity of Gut Microbiota from Infants and Toddlers. Nutrients 2021, 13, 726. [Google Scholar] [CrossRef]
- Schei, K.; Avershina, E.; Oien, T.; Rudi, K.; Follestad, T.; Salamati, S.; Odegard, R.A. Early gut mycobiota and mother-offspring transfer. Microbiome 2017, 5, 107. [Google Scholar] [CrossRef] [PubMed]
- Breuer, U.; Harms, H. Debaryomyces hansenii—an extremophilic yeast with biotechnological potential. Yeast 2006, 23, 415–437. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.C.; Perfect, J.; Colombo, A.L.; Cornely, O.A.; Groll, A.H.; Seidel, D.; Albus, K.; de Almedia, J.N., Jr.; Garcia-Effron, G.; Gilroy, N.; et al. Global guideline for the diagnosis and management of rare yeast infections: An initiative of the ECMM in cooperation with ISHAM and ASM. Lancet Infect. Dis. 2021, 21, e375–e386. [Google Scholar] [CrossRef]
- Mehta, V.; Nayyar, C.; Gulati, N.; Singla, N.; Rai, S.; Chandar, J. A Comprehensive Review of Trichosporon spp.: An Invasive and Emerging Fungus. Cureus 2021, 13, e17345. [Google Scholar] [CrossRef]
- Sandai, D.; Yin, Z.; Selway, L.; Stead, D.; Walker, J.; Leach, M.D.; Bohovych, I.; Ene, I.V.; Kastora, S.; Budge, S.; et al. The evolutionary rewiring of ubiquitination targets has reprogrammed the regulation of carbon assimilation in the pathogenic yeast Candida albicans. mBio 2012, 3, 6. [Google Scholar] [CrossRef]
- Sokol, H.; Leducq, V.; Aschard, H.; Pham, H.P.; Jegou, S.; Landman, C.; Cohen, D.; Liguori, G.; Bourrier, A.; Nion-Larmurier, I.; et al. Fungal microbiota dysbiosis in IBD. Gut 2017, 66, 1039–1048. [Google Scholar] [CrossRef]
- Verdecia, J.; Jankowski, C.A.; Reynolds, M.L.; McCarter, Y.; Ravi, M. Fungemia due to Aureobasidium pullulans. Med. Mycol. Case Rep. 2022, 37, 26–28. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Ikeda, M.; Ohama, Y.; Sunouchi, T.; Hoshino, Y.; Ito, H.; Yamashita, M.; Kanno, Y.; Okamoto, K.; Yamagoe, S.; et al. Aureobasidium melanigenum catheter-related bloodstream infection: A case report. BMC Infect. Dis. 2022, 22, 335. [Google Scholar] [CrossRef]
- Lin, S.Y.; Lu, P.L.; Tan, B.H.; Chakrabarti, A.; Wu, U.I.; Yang, J.H.; Patel, A.K.; Li, R.Y.; Watcharananan, S.P.; Liu, Z.; et al. The epidemiology of non-Candida yeast isolated from blood: The Asia Surveillance Study. Mycoses 2019, 62, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Backhed, F.; Roswall, J.; Peng, Y.; Feng, Q.; Jia, H.; Kovatcheva-Datchary, P.; Li, Y.; Xia, Y.; Xie, H.; Zhong, H.; et al. Dynamics and Stabilization of the Human Gut Microbiome during the First Year of Life. Cell Host Microbe 2015, 17, 852. [Google Scholar] [CrossRef]
- Klainer, A.S. Clindamycin. Med. Clin. N. Am. 1987, 71, 1169–1175. [Google Scholar] [CrossRef] [PubMed]
- Huttner, A.; Bielicki, J.; Clements, M.N.; Frimodt-Moller, N.; Muller, A.E.; Paccaud, J.P.; Mouton, J.W. Oral amoxicillin and amoxicillin-clavulanic acid: Properties, indications and usage. Clin. Microbiol. Infect. Off. Publ. Eur. Soc. Clin. Microbiol. Infect. Dis. 2020, 26, 871–879. [Google Scholar] [CrossRef]
- Espinosa-Gongora, C.; Jessen, L.R.; Kieler, I.N.; Damborg, P.; Bjornvad, C.R.; Gudeta, D.D.; Pires Dos Santos, T.; Sablier-Gallis, F.; Sayah-Jeanne, S.; Corbel, T.; et al. Impact of oral amoxicillin and amoxicillin/clavulanic acid treatment on bacterial diversity and beta-lactam resistance in the canine faecal microbiota. J. Antimicrob. Chemother. 2020, 75, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Spatz, M.; Da Costa, G.; Ventin-Holmberg, R.; Planchais, J.; Michaudel, C.; Wang, Y.; Danne, C.; Lapiere, A.; Michel, M.L.; Kolho, K.L.; et al. Antibiotic treatment using amoxicillin-clavulanic acid impairs gut mycobiota development through modification of the bacterial ecosystem. Microbiome 2023, 11, 73. [Google Scholar] [CrossRef]
- Mancabelli, L.; Mancino, W.; Lugli, G.A.; Argentini, C.; Longhi, G.; Milani, C.; Viappiani, A.; Anzalone, R.; Bernasconi, S.; van Sinderen, D.; et al. Amoxicillin-Clavulanic Acid Resistance in the Genus Bifidobacterium. Appl. Environ. Microbiol. 2021, 87, e03137-20. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, G.P.; Lee, S.M.; Mazmanian, S.K. Gut biogeography of the bacterial microbiota. Nat. Rev. Microbiol. 2016, 14, 20–32. [Google Scholar] [CrossRef]
- Alsina, A.; Mason, M.; Uphoff, R.A.; Riggsby, W.S.; Becker, J.M.; Murphy, D. Catheter-associated Candida utilis fungemia in a patient with acquired immunodeficiency syndrome: Species verification with a molecular probe. J. Clin. Microbiol. 1988, 26, 621–624. [Google Scholar] [CrossRef]
- Dorko, E.; Kmetova, M.; Pilipcinec, E.; Bracokova, I.; Dorko, F.; Danko, J.; Svicky, E.; Tkacikova, L. Rare non-albicans Candida species detected in different clinical diagnoses. Folia Microbiol. 2000, 45, 364–368. [Google Scholar] [CrossRef]
- Hazen, K.C.; Theisz, G.W.; Howell, S.A. Chronic urinary tract infection due to Candida utilis. J. Clin. Microbiol. 1999, 37, 824–827. [Google Scholar] [CrossRef]
- Kaur, H.; Singh, S.; Rudramurthy, S.M.; Jayashree, M.; Peters, N.J.; Ray, P.; Samujh, R.; Ghosh, A.; Chakrabarti, A. Fungaemia due to rare yeasts in paediatric intensive care units: A prospective study. Mycoses 2021, 64, 1387–1395. [Google Scholar] [CrossRef] [PubMed]
- Chavapradit, N.; Angkasekwinai, N. Disseminated cryptococcosis in Crohn’s disease: A case report. BMC Infect. Dis. 2018, 18, 620. [Google Scholar] [CrossRef]
- Quincho-Lopez, A.; Kojima, N.; Nesemann, J.M.; Verona-Rubio, R.; Carayhua-Perez, D. Cryptococcal infection of the colon in a patient without concurrent human immunodeficiency infection: A case report and literature review. Eur. J. Clin. Microbiol. Infect. Dis. Off. Publ. Eur. Soc. Clin. Microbiol. 2021, 40, 2623–2626. [Google Scholar] [CrossRef] [PubMed]
- Ricci, L.; Mackie, J.; Donachie, G.E.; Chapuis, A.; Mezerova, K.; Lenardon, M.D.; Brown, A.J.P.; Duncan, S.H.; Walker, A.W. Human gut bifidobacteria inhibit the growth of the opportunistic fungal pathogen Candida albicans. FEMS Microbiol. Ecol. 2022, 98, fiac095. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, K.E.; Sitarik, A.R.; Havstad, S.; Lin, D.L.; Levan, S.; Fadrosh, D.; Panzer, A.R.; LaMere, B.; Rackaityte, E.; Lukacs, N.W.; et al. Neonatal gut microbiota associates with childhood multisensitized atopy and T cell differentiation. Nat. Med. 2016, 22, 1187–1191. [Google Scholar] [CrossRef] [PubMed]
- Panpetch, W.; Somboonna, N.; Palasuk, M.; Hiengrach, P.; Finkelman, M.; Tumwasorn, S.; Leelahavanichkul, A. Oral Candida administration in a Clostridium difficile mouse model worsens disease severity but is attenuated by Bifidobacterium. PLoS ONE 2019, 14, e0210798. [Google Scholar] [CrossRef]
- Charlet, R.; Bortolus, C.; Sendid, B.; Jawhara, S. Bacteroides thetaiotaomicron and Lactobacillus johnsonii modulate intestinal inflammation and eliminate fungi via enzymatic hydrolysis of the fungal cell wall. Sci. Rep. 2020, 10, 11510. [Google Scholar] [CrossRef]
- Kalyana Chakravarthy, S.; Jayasudha, R.; Ranjith, K.; Dutta, A.; Pinna, N.K.; Mande, S.S.; Sharma, S.; Garg, P.; Murthy, S.I.; Shivaji, S. Alterations in the gut bacterial microbiome in fungal Keratitis patients. PLoS ONE 2018, 13, e0199640. [Google Scholar] [CrossRef]
- Guinan, J.; Thangamani, S. Antibiotic-induced alterations in taurocholic acid levels promote gastrointestinal colonization of Candida albicans. FEMS Microbiol. Lett. 2018, 365, fny196. [Google Scholar] [CrossRef]
- Gutierrez, D.; Weinstock, A.; Antharam, V.C.; Gu, H.; Jasbi, P.; Shi, X.; Dirks, B.; Krajmalnik-Brown, R.; Maldonado, J.; Guinan, J.; et al. Antibiotic-induced gut metabolome and microbiome alterations increase the susceptibility to Candida albicans colonization in the gastrointestinal tract. FEMS Microbiol. Ecol. 2020, 96, fiz187. [Google Scholar] [CrossRef]
- McDonough, L.D.; Mishra, A.A.; Tosini, N.; Kakade, P.; Penumutchu, S.; Liang, S.H.; Maufrais, C.; Zhai, B.; Taur, Y.; Belenky, P.; et al. Candida albicans Isolates 529L and CHN1 Exhibit Stable Colonization of the Murine Gastrointestinal Tract. mBio 2021, 12, e0287821. [Google Scholar] [CrossRef] [PubMed]
- Guinan, J.; Villa, P.; Thangamani, S. Secondary bile acids inhibit Candida albicans growth and morphogenesis. Pathog. Dis. 2018, 76, 3. [Google Scholar] [CrossRef] [PubMed]
- Payne, S.; Gibson, G.; Wynne, A.; Hudspith, B.; Brostoff, J.; Tuohy, K. In vitro studies on colonization resistance of the human gut microbiota to Candida albicans and the effects of tetracycline and Lactobacillus plantarum LPK. Curr. Issues Intest. Microbiol. 2003, 4, 1–8. [Google Scholar]
- Gibson, G.R.; Cummings, J.H.; Macfarlane, G.T. Use of a three-stage continuous culture system to study the effect of mucin on dissimilatory sulfate reduction and methanogenesis by mixed populations of human gut bacteria. Appl. Environ. Microbiol. 1988, 54, 2750–2755. [Google Scholar] [CrossRef] [PubMed]
- Wynne, A.G.; McCartney, A.L.; Brostoff, J.; Hudspith, B.N.; Gibson, G.R. An in vitro assessment of the effects of broad-spectrum antibiotics on the human gut microflora and concomitant isolation of a Lactobacillus plantarum with anti-Candida activities. Anaerobe 2004, 10, 165–169. [Google Scholar] [CrossRef]
- Maas, E.; Penders, J.; Venema, K. Studying Fungal-Bacterial Relationships in the Human Gut Using an In Vitro Model (TIM-2). J. Fungi 2023, 9, 174. [Google Scholar] [CrossRef]
- Maas, E.; Penders, J.; Venema, K. Modelling the Gut Fungal-Community in TIM-2 with a Microbiota from Healthy Individuals. J. Fungi 2023, 9, 104. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marsaux, B.; Moens, F.; Marzorati, M.; Van de Wiele, T. The Intricate Connection between Bacterial α-Diversity and Fungal Engraftment in the Human Gut of Healthy and Impaired Individuals as Studied Using the In Vitro SHIME® Model. J. Fungi 2023, 9, 877. https://doi.org/10.3390/jof9090877
Marsaux B, Moens F, Marzorati M, Van de Wiele T. The Intricate Connection between Bacterial α-Diversity and Fungal Engraftment in the Human Gut of Healthy and Impaired Individuals as Studied Using the In Vitro SHIME® Model. Journal of Fungi. 2023; 9(9):877. https://doi.org/10.3390/jof9090877
Chicago/Turabian StyleMarsaux, Benoît, Frédéric Moens, Massimo Marzorati, and Tom Van de Wiele. 2023. "The Intricate Connection between Bacterial α-Diversity and Fungal Engraftment in the Human Gut of Healthy and Impaired Individuals as Studied Using the In Vitro SHIME® Model" Journal of Fungi 9, no. 9: 877. https://doi.org/10.3390/jof9090877
APA StyleMarsaux, B., Moens, F., Marzorati, M., & Van de Wiele, T. (2023). The Intricate Connection between Bacterial α-Diversity and Fungal Engraftment in the Human Gut of Healthy and Impaired Individuals as Studied Using the In Vitro SHIME® Model. Journal of Fungi, 9(9), 877. https://doi.org/10.3390/jof9090877