
A Whole-Genome Assembly for Hyaloperonospora parasitica, A Pathogen Causing Downy Mildew in Cabbage (Brassica oleracea var. capitata L.)

 ,
,  ,
, 

Abstract
1. Introduction
2. Materials and Methods
2.1. Purification of H. parasitica
2.2. Library Construction and Sequencing
2.3. De Novo Genome Assembly
2.4. Gene Prediction and Functional Annotation
2.5. Identification of RNAs and Repeated Sequences
2.6. Secretome and Effector Identification
2.7. Genomic Comparison and Phylogenomic Analysis
3. Results
3.1. Genome Assembly of Strain BJ2020
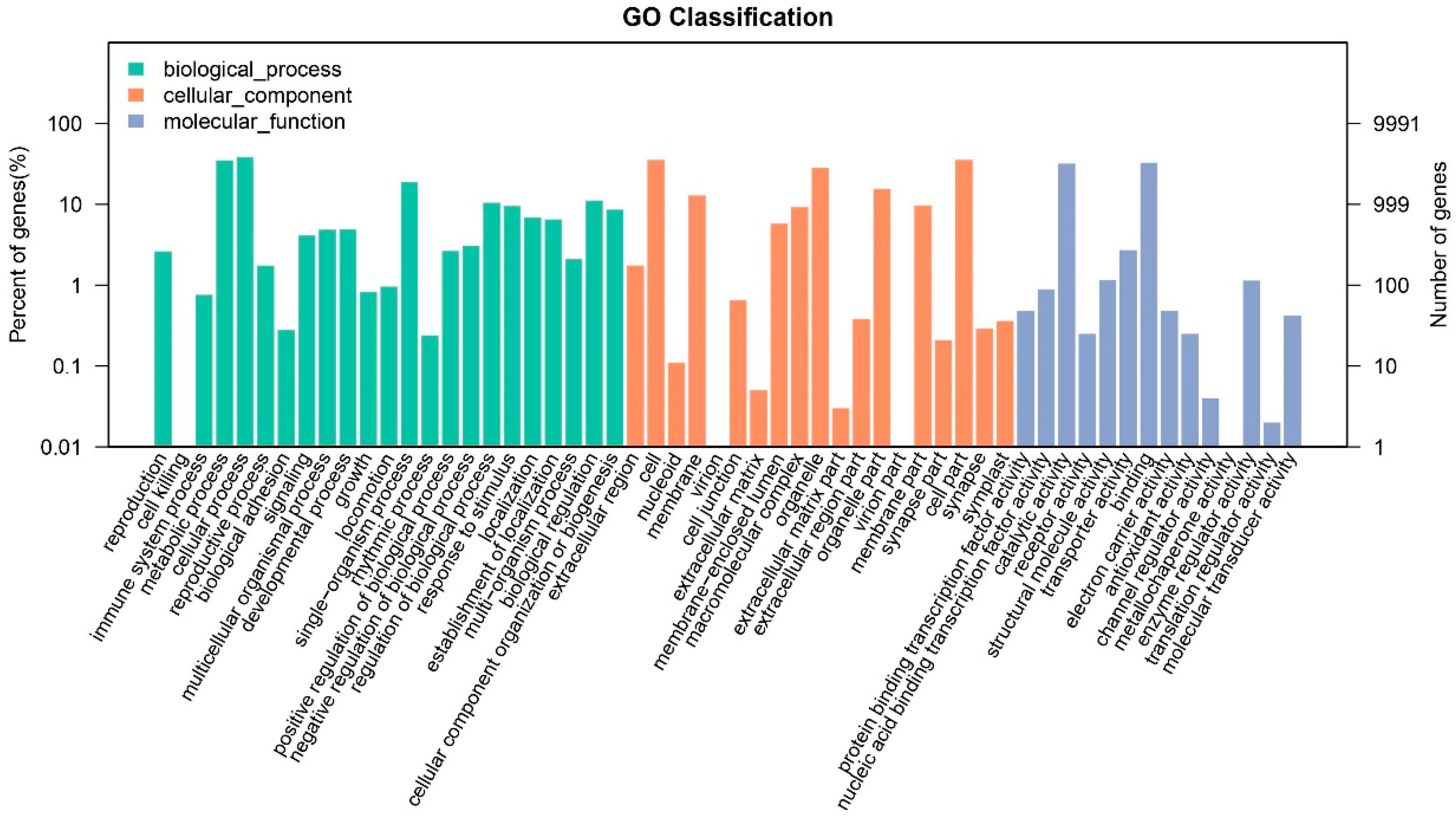
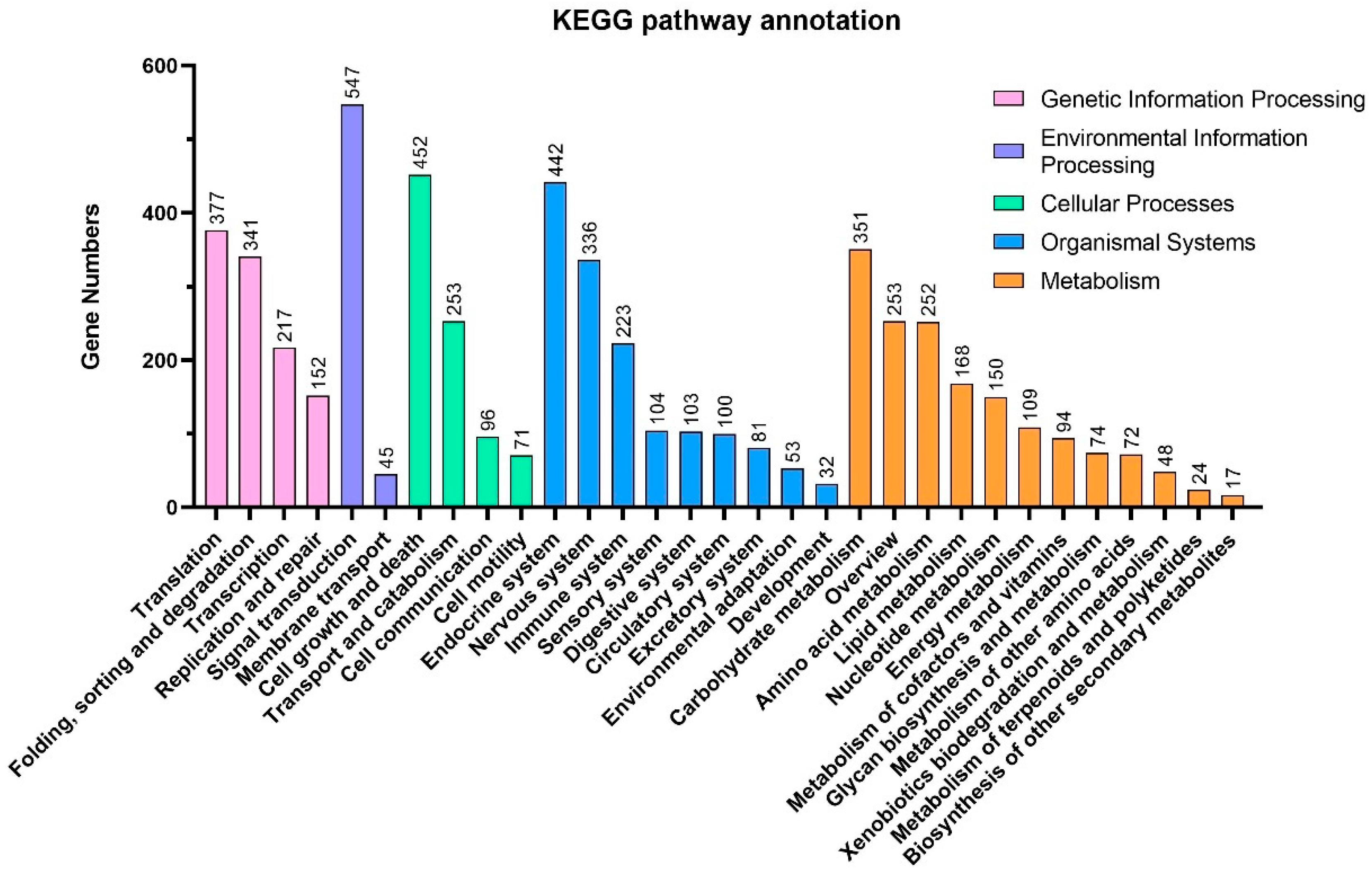
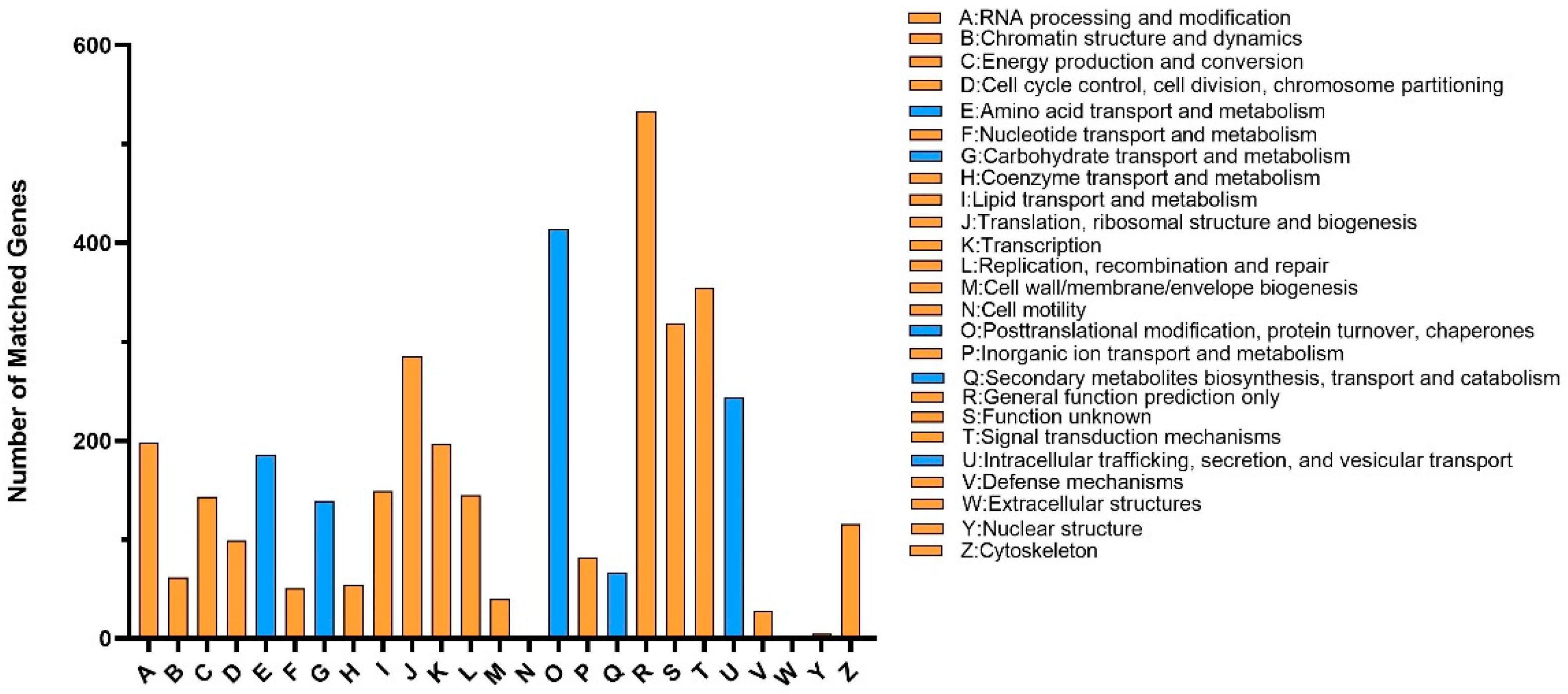
3.2. Gene Functional Annotation
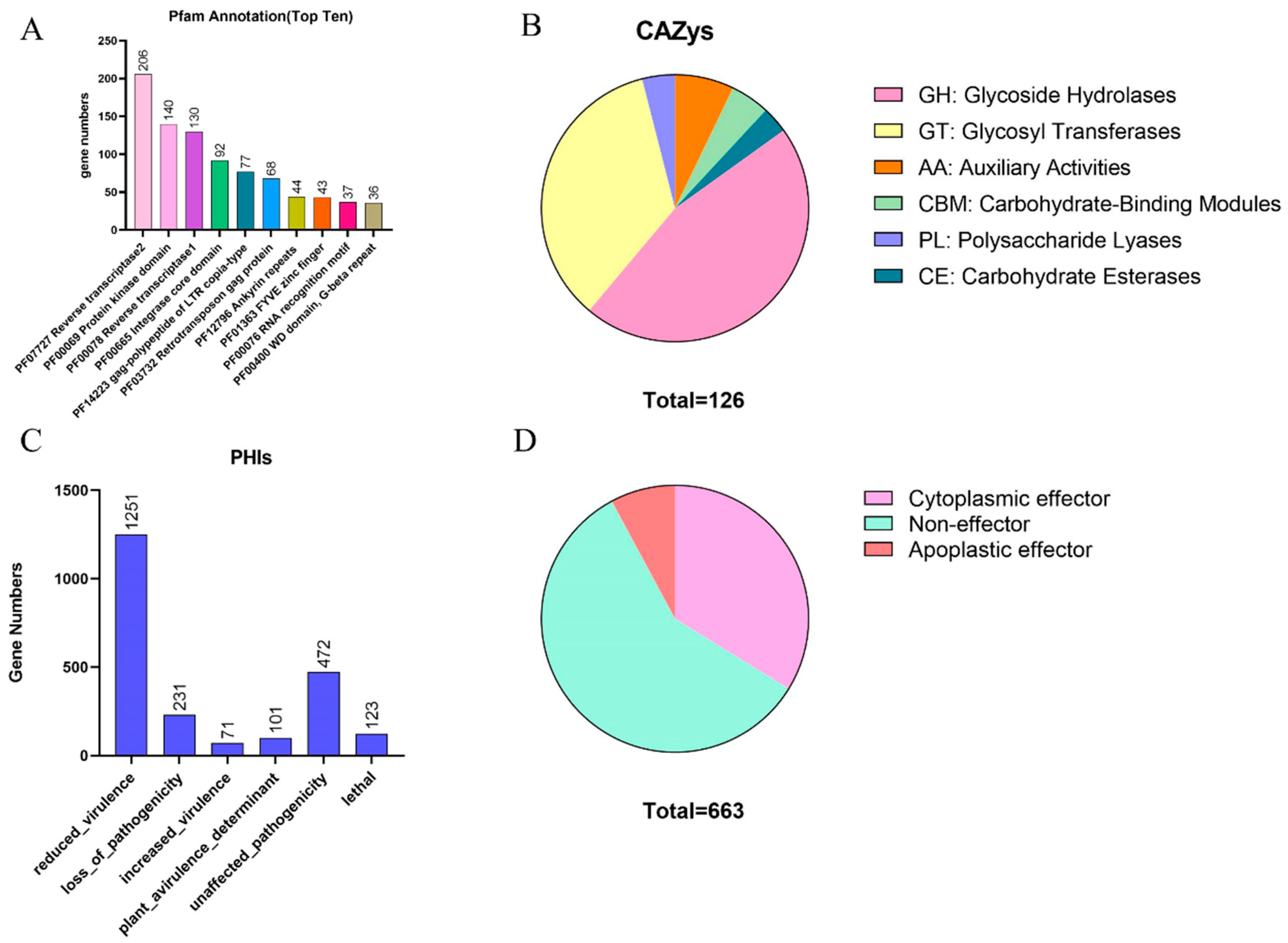
3.3. Identification of Disease-Related Genes
3.4. Comparison between H. parasitica and Other Brassicaceae Crop Downy Mildew Pathogens
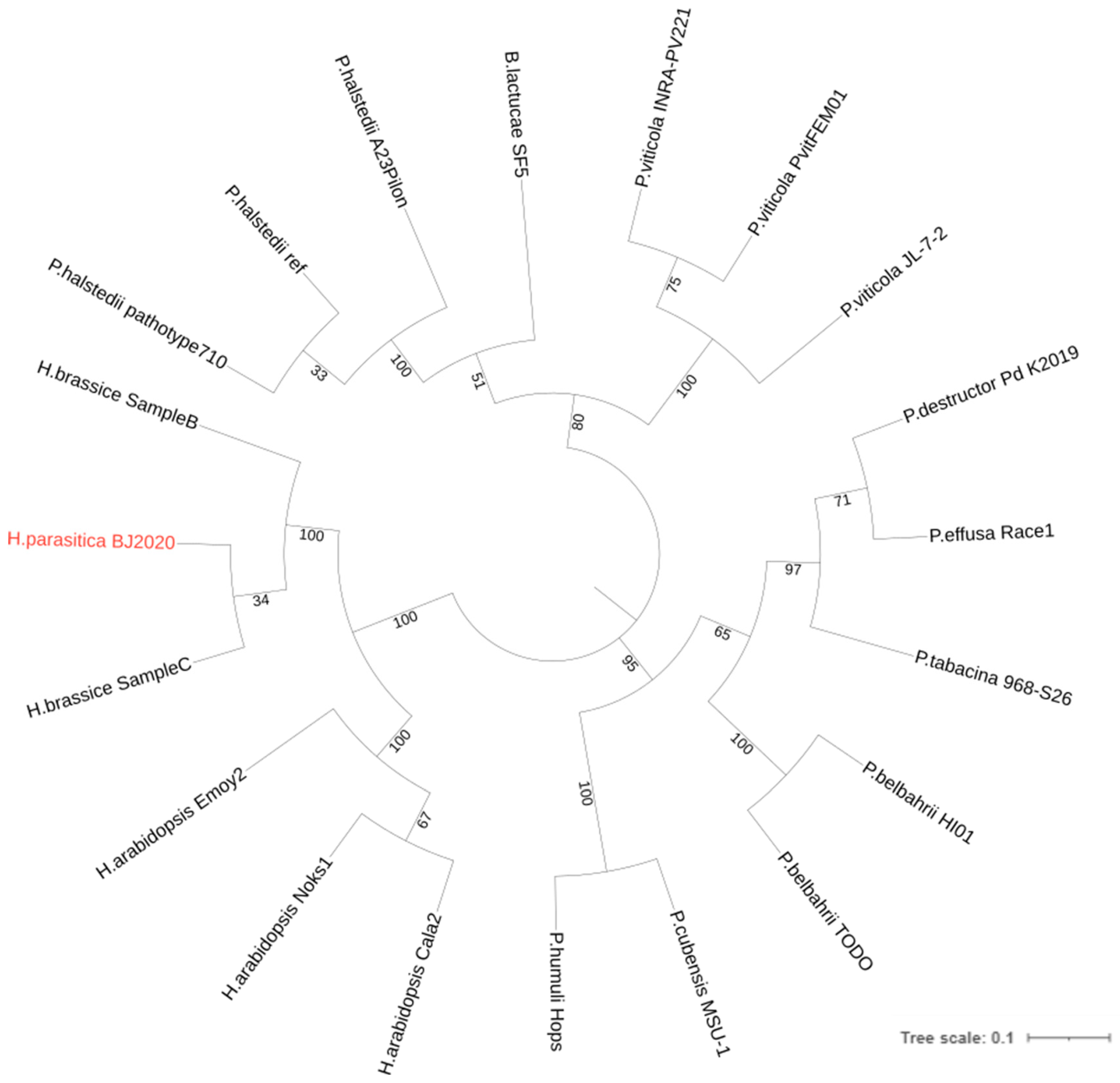
3.5. Phylogenomic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, M.-Y.; Jiao, Y.-T.; Wang, Y.-T.; Zhang, N.; Wang, B.-B.; Liu, R.-Q.; Yin, X.; Xu, Y.; Liu, G.-T. CRISPR/Cas9-mediated VvPR4b editing decreases downy mildew resistance in grapevine (Vitis vinifera L.). Hortic. Res. 2020, 7, 149. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.K.; Shen, Y.; Zhao, Z.; Sheng, X.; Wang, J.; Yu, H.; Gu, H. Molecular Breeding Strategy and Challenges Towards Improvement of Downy Mildew Resistance in Cauliflower (Brassica oleracea var. botrytis L.). Front. Plant Sci. 2021, 12, 667757. [Google Scholar]
- Bhattarai, G.; Olaoye, D.; Mou, B.; Correll, J.C.; Shi, A. Mapping and selection of downy mildew resistance in spinach cv. Whale by low coverage whole genome sequencing. Front. Plant Sci. 2022, 13, 1012923. [Google Scholar]
- Tan, J.; Wang, Y.; Dymerski, R.; Wu, Z.; Weng, Y. Sigma factor binding protein 1 (CsSIB1) is a putative candidate of the major-effect QTL dm5.3 for downy mildew resistance in cucumber (Cucumis sativus). Theor. Appl. Genet. 2022, 135, 4197–4215. [Google Scholar] [PubMed]
- Singh, K.P.; Kumari, P.; Rai, P.K. Current Status of the Disease-Resistant Gene(s)/QTLs, and Strategies for Improvement in Brassica juncea. Plant Sci. 2017, 8, 1788. [Google Scholar]
- Neik, T.X.; Barbetti, M.J.; Batley, J. Current Status and Challenges in Identifying Disease Resistance Genes in Brassica napus. Front. Plant Sci. 2021, 12, 617405. [Google Scholar]
- Lv, H.; Fang, Z.; Yang, L.; Zhang, Y.; Wang, Y. An update on the arsenal: Mining resistance genes for disease management of Brassica crops in the genomic era. Hortic. Res. 2020, 7, 34. [Google Scholar]
- Molinero-Ruiz, L. Sustainable and efficient control of sunflower downy mildew by means of genetic resistance: A review. Theor. Appl. Genet. 2022, 135, 3757–3771. [Google Scholar]
- Coelho, P.S.; Monteiro, A.A. Inheritance of downy mildew resistance in mature broccoli plants. Euphytica 2003, 131, 65–69. [Google Scholar] [CrossRef]
- Constantinescu, O.; Fatehi, J. Peronospora-like fungi (Chromista, Peronosporales) parasitic on Brassicaceae and related hosts. Nova Hedwig. 2002, 74, 291–338. [Google Scholar] [CrossRef]
- Aragona, M.; Haegi, A.; Valente, M.T.; Riccioni, L.; Orzali, L.; Vitale, S.; Luongo, L.; Infantino, A. New-Generation Sequencing Technology in Diagnosis of Fungal Plant Pathogens: A Dream Comes True? J. Fungi 2022, 8, 737. [Google Scholar]
- Bao, J.; Chen, M.; Zhong, Z.; Tang, W.; Lin, L.; Zhang, X.; Jiang, H.; Zhang, D.; Miao, C.; Tang, H.; et al. PacBio Sequencing Reveals Transposable Elements as a Key Contributor to Genomic Plasticity and Virulence Variation in Magnaporthe oryzae. Mol. Plant 2017, 10, 1465–1468. [Google Scholar] [PubMed]
- Xu, F.; Li, X.; Ren, H.; Zeng, R.; Wang, Z.; Hu, H.; Bao, J.; Que, Y. The First Telomere-to-Telomere Chromosome-Level Genome Assembly of Stagonospora tainanensis Causing Sugarcane Leaf Blight. J. Fungi 2022, 8, 1088. [Google Scholar] [CrossRef]
- Cao, Z.; Zhang, K.; Guo, X.; Turgeon, B.G.; Dong, J. A Genome Resource of Setosphaeria turcica, Causal Agent of Northern Leaf Blight of Maize. Phytopathology® 2020, 110, 2014–2016. [Google Scholar] [CrossRef]
- Winkworth, R.C.; Neal, G.; Ogas, R.A.; Nelson, B.C.W.; McLenachan, P.A.; Bellgard, S.E.; Lockhart, P.J. Comparative Analyses of Complete Peronosporaceae (Oomycota) Mitogenome Sequences-Insights into Structural Evolution and Phylogeny. Genome Biol. Evol. 2022, 14, evac049. [Google Scholar] [PubMed]
- You, M.P.; Akhatar, J.; Mittal, M.; Barbetti, M.J.; Maina, S.; Banga, S.S. Comparative analysis of draft genome assemblies developed from whole genome sequences of two Hyaloperonospora brassicae isolate samples differing in field virulence on Brassica napus. Biotechnol. Rep. 2021, 31, e00653. [Google Scholar]
- Yu, S.; Zhang, F.; Yu, R.; Zou, Y.; Qi, J.; Zhao, X.; Yu, Y.; Zhang, D.; Li, L. Genetic mapping and localization of a major QTL for seedling resistance to downy mildew in Chinese cabbage (Brassica rapa ssp. pekinensis). Mol. Breed. 2009, 23, 573–590. [Google Scholar]
- Zhang, B.; Li, P.; Su, T.; Li, P.; Xin, X.; Wang, W.; Zhao, X.; Yu, Y.; Zhang, D.; Yu, S.; et al. BrRLP48, Encoding a Receptor-Like Protein, Involved in Downy Mildew Resistance in Brassica rapa. Front. Plant Sci. 2018, 9, 1708. [Google Scholar] [CrossRef]
- Yu, S.; Zhang, F.; Zhao, X.; Yu, Y.; Zhang, D. Sequence-characterized amplified region and simple sequence repeat markers for identifying the major quantitative trait locus responsible for seedling resistance to downy mildew in Chinese cabbage (Brassica rapa ssp. pekinensis). Plant Breed. 2011, 130, 580–583. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar]
- Boetzer, M.; Pirovano, W. Toward almost closed genomes with GapFiller. Genome Biol. 2012, 13, R56. [Google Scholar] [CrossRef] [PubMed]
- Massouras, A.; Hens, K.; Gubelmann, C.; Uplekar, S.; Decouttere, F.; Rougemont, J.; Cole, S.T.; Deplancke, B. Primer-initiated sequence synthesis to detect and assemble structural variants. Nat. Methods 2010, 7, 485–486. [Google Scholar] [PubMed]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Zdobnov, E.M. BUSCO: Assessing Genomic Data Quality and Beyond. Curr. Protoc. 2021, 1, e323. [Google Scholar]
- Besemer, J.; Borodovsky, M. GeneMark: Web software for gene finding in prokaryotes, eukaryotes and viruses. Nucleic Acids Res. 2005, 33 (Suppl. 2), W451–W454. [Google Scholar] [CrossRef]
- Urban, M.; Cuzick, A.; Seager, J.; Wood, V.; Rutherford, K.; Venkatesh, S.Y.; Sahu, J.; Iyer, S.V.; Khamari, L.; De Silva, N.; et al. PHI-base in 2022: A multi-species phenotype database for Pathogen–Host Interactions. Nucleic Acids Res. 2022, 50, D837–D847. [Google Scholar] [PubMed]
- Drula, E.; Garron, M.-L.; Dogan, S.; Lombard, V.; Henrissat, B.; Terrapon, N. The carbohydrate-active enzyme database: Functions and literature. Nucleic Acids Res. 2022, 50, D571–D577. [Google Scholar]
- Saier, M.H., Jr.; Reddy, V.S.; Moreno-Hagelsieb, G.; Hendargo, K.J.; Zhang, Y.; Iddamsetty, V.; Lam, K.J.K.; Tian, N.; Russum, S.; Wang, J.; et al. The Transporter Classification Database (TCDB): 2021 update. Nucleic Acids Res. 2021, 49, D461–D467. [Google Scholar]
- Chan, P.P.; Lin, B.Y.; Mak, A.J.; Lowe, T.M. tRNAscan-SE 2.0: Improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 2021, 49, 9077–9096. [Google Scholar]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Stærfeldt, H.-H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar]
- Kalvari, I.; Nawrocki, E.P.; Ontiveros-Palacios, N.; Argasinska, J.; Lamkiewicz, K.; Marz, M.; Griffiths-Jones, S.; Toffano-Nioche, C.; Gautheret, D.; Weinberg, Z.; et al. Rfam 14: Expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Res. 2021, 49, D192–D200. [Google Scholar]
- Flynn, J.M.; Hubley, R.; Goubert, C.; Rosen, J.; Clark, A.G.; Feschotte, C.; Smit, A.F. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 2020, 117, 9451–9457. [Google Scholar] [CrossRef] [PubMed]
- Almagro Armenteros, J.J.; Tsirigos, K.D.; Sønderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef] [PubMed]
- Möller, S.; Croning, M.D.R.; Apweiler, R. Evaluation of methods for the prediction of membrane spanning regions. Bioinformatics 2001, 17, 646–653. [Google Scholar] [CrossRef]
- Sperschneider, J.; Dodds, P.N. EffectorP 3.0: Prediction of Apoplastic and Cytoplasmic Effectors in Fungi and Oomycetes. Mol. Plant-Microbe Interact. 2021, 35, 146–156. [Google Scholar]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.I.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing Large Minimum Evolution Trees with Profiles instead of a Distance Matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar]
- Baxter, L.; Tripathy, S.; Ishaque, N.; Boot, N.; Cabral, A.; Kemen, E.; Thines, M.; Ah-Fong, A.; Anderson, R.; Badejoko, W.; et al. Signatures of Adaptation to Obligate Biotrophy in the Hyaloperonospora arabidopsidis Genome. Science 2010, 330, 1549–1551. [Google Scholar] [CrossRef]
- Fletcher, K.; Martin, F.; Isakeit, T.; Cavanaugh, K.; Magill, C.; Michelmore, R. The genome of the oomycete Peronosclerospora sorghi, a cosmopolitan pathogen of maize and sorghum, is inflated with dispersed pseudogenes. G3 Genes Genomes Genet. 2023, 13, jkac340. [Google Scholar] [CrossRef]
- Kubicek, C.P.; Starr, T.L.; Glass, N.L. Plant Cell Wall–Degrading Enzymes and Their Secretion in Plant-Pathogenic Fungi. Annu. Rev. Phytopathol. 2014, 52, 427–451. [Google Scholar]
- García-Maceira Fé, I.; Di Pietro, A.; Huertas-González, M.D.; Ruiz-Roldán, M.C.; Roncero, M.I.G. Molecular Characterization of an Endopolygalacturonase from Fusarium oxysporum Expressed during Early Stages of Infection. Appl. Environ. Microbiol. 2001, 67, 2191–2196. [Google Scholar]
- Ospina-Giraldo, M.D.; Griffith, J.G.; Laird, E.W.; Mingora, C. The CAZyome of Phytophthora spp.: A comprehensive analysis of the gene complement coding for carbohydrate-active enzymes in species of the genus Phytophthora. BMC Genom. 2010, 11, 525. [Google Scholar] [CrossRef]
- Solomon, K.V.; Haitjema, C.H.; Henske, J.K.; Gilmore, S.P.; Borges-Rivera, D.; Lipzen, A.; Brewer, H.M.; Purvine, S.O.; Wright, A.T.; Theodorou, M.K.; et al. Early-branching gut fungi possess a large, comprehensive array of biomass-degrading enzymes. Science 2016, 351, 1192–1195. [Google Scholar] [CrossRef]
- Zhou, J.; Qi, Y.; Nie, J.; Guo, L.; Luo, M.; McLellan, H.; Boevink, P.C.; Birch, P.R.J.; Tian, Z. A Phytophthora effector promotes homodimerization of host transcription factor StKNOX3 to enhance susceptibility. J. Exp. Bot. 2022, 73, 6902–6915. [Google Scholar] [CrossRef]
- Yin, Z.; Liu, H.; Li, Z.; Ke, X.; Dou, D.; Gao, X.; Song, N.; Dai, Q.; Wu, Y.; Xu, J.-R.; et al. Genome sequence of Valsa canker pathogens uncovers a potential adaptation of colonization of woody bark. New Phytol. 2015, 208, 1202–1216. [Google Scholar] [CrossRef]
- Lo Presti, L.; Lanver, D.; Schweizer, G.; Tanaka, S.; Liang, L.; Tollot, M.; Zuccaro, A.; Reissmann, S.; Kahmann, R. Fungal Effectors and Plant Susceptibility. Annu. Rev. Plant Biol. 2015, 66, 513–545. [Google Scholar] [CrossRef]
- Zhang, S.; Xu, J.-R. Effectors and Effector Delivery in Magnaporthe oryzae. PLoS Pathog. 2014, 10, e1003826. [Google Scholar] [CrossRef]
- Wang, S.; Boevink, P.C.; Welsh, L.; Zhang, R.; Whisson, S.C.; Birch, P.R.J. Delivery of cytoplasmic and apoplastic effectors from Phytophthora infestans haustoria by distinct secretion pathways. New Phytol. 2017, 216, 205–215. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Features | BJ2020 |
|---|---|
| Genome size (bp) | 37,102,749 |
| Number of contigs | 4631 |
| N50 (bp) | 20,542 |
| GC content (%) | 51% |
| Protein-coding genes | 9991 |
| Gene density (number of genes per Mb) | 269 |
| Min length (bp) | 118 |
| Max length (bp) | 18,240 |
| Average length (bp) | 1191.66 |
| Total coding gene length (bp) | 11,905,868 |
| tRNA | 237 |
| rRNA | 13 |
| Repeat regions (bases) | 11,653,830 |
| Repeat ratio (%) | 31.41% |
| Simple repeats | 8712 |
| Database | Number of Genes | Percentage |
|---|---|---|
| CDD | 5447 | 54.52% |
| KOG | 3915 | 39.19% |
| NR | 5280 | 52.85% |
| PFAM | 4829 | 48.33% |
| SwissProt | 5181 | 51.86% |
| TrEMBL | 5223 | 52.28% |
| GO | 5051 | 50.56% |
| KEGG | 2254 | 22.56% |
| Annotated in at least one database | 6128 | 61.34% |
| Annotated in all databases | 1955 | 19.57% |
| Total Unigenes | 9991 | 100.00% |
| H. parasitica | H. brassicae | H. arabidopsidis | ||
|---|---|---|---|---|
| Strain | BJ2020 | Sample B | Sample C | Emoy2 |
| Total size | 37.10 Mb | 79.39 Mb | 92.19 Mb | 78.38 Mb |
| Protein coding genes (≥250 bp) | 9991 | 36,819 | 40,346 | 14,321 |
| Number of contigs | 4631 | 6438 | 6470 | 10,486 |
| N50 | 20.5 Kb | 23.5 Kb | 24.5 Kb | 41.9 Kb |
| GC (%) | 51% | 47% | 47% | 47% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Y.; Zhang, B.; Liu, S.; Zhao, Z.; Ren, W.; Chen, L.; Yang, L.; Zhuang, M.; Lv, H.; Wang, Y.; et al. A Whole-Genome Assembly for Hyaloperonospora parasitica, A Pathogen Causing Downy Mildew in Cabbage (Brassica oleracea var. capitata L.). J. Fungi 2023, 9, 819. https://doi.org/10.3390/jof9080819
Wu Y, Zhang B, Liu S, Zhao Z, Ren W, Chen L, Yang L, Zhuang M, Lv H, Wang Y, et al. A Whole-Genome Assembly for Hyaloperonospora parasitica, A Pathogen Causing Downy Mildew in Cabbage (Brassica oleracea var. capitata L.). Journal of Fungi. 2023; 9(8):819. https://doi.org/10.3390/jof9080819
Chicago/Turabian StyleWu, Yuankang, Bin Zhang, Shaobo Liu, Zhiwei Zhao, Wenjing Ren, Li Chen, Limei Yang, Mu Zhuang, Honghao Lv, Yong Wang, and et al. 2023. "A Whole-Genome Assembly for Hyaloperonospora parasitica, A Pathogen Causing Downy Mildew in Cabbage (Brassica oleracea var. capitata L.)" Journal of Fungi 9, no. 8: 819. https://doi.org/10.3390/jof9080819
APA StyleWu, Y., Zhang, B., Liu, S., Zhao, Z., Ren, W., Chen, L., Yang, L., Zhuang, M., Lv, H., Wang, Y., Ji, J., Han, F., & Zhang, Y. (2023). A Whole-Genome Assembly for Hyaloperonospora parasitica, A Pathogen Causing Downy Mildew in Cabbage (Brassica oleracea var. capitata L.). Journal of Fungi, 9(8), 819. https://doi.org/10.3390/jof9080819