1. Introduction
Metschnikowia bicuspidata was initially isolated from diseased
Daphnia magna in 1884 [
1], and there are currently three reported variants:
M. bicuspidata var. Bicuspidata,
M. bicuspidata var. California, and
M. bicuspidata var. Chathamia [
2,
3].
Metschnikowia bicuspidata is an opportunistic pathogen that can infect various economically important aquatic animals, such as
Eriocheir sinensis,
Macrobrachium rosenbergii,
Palaemonetes sinensis,
Portunus trituberculatus, and
Oncorhyncus tshawytscha. Their presence can thus seriously hinder healthy development in aquaculture systems [
3,
4,
5,
6,
7,
8,
9,
10].
Chinese mitten crabs,
Eriocheir sinensis, when infected with
M. bicuspidata, exhibit symptoms including swollen joints, white muscles, and milky hemolymph, which is why it is also referred to as “milky disease”. Following infection, the vitality of crabs decreases, and their appendages become prone to detachment, ultimately leading to death due to extensive yeast infections throughout the body [
11]. Since 2019, outbreaks of “milky disease” in many Chinese mitten crab breeding areas have resulted in significant economic losses to the aquaculture industry.
The current commonly employed detection method is conventional PCR technology, with primers designed for the identification of yeast 26S rDNA and 5.8S ITS rDNA gene sequences [
11,
12]. However, these methods exhibit low specificity and sensitivity, which may lead to false positive results owing to cross-reactivity with other similar aquatic animal diseases. However, the accurate diagnosis of crabs with mild initial infection levels is hindered by low sensitivity, thereby impeding disease prevention and treatment efforts. The previously reported nested PCR detection technology, which is highly sensitive and specific, is time-consuming and does not enable the direct quantification of
M. bicuspidata in Chinese mitten crabs [
13].
Real-time quantitative PCR (qPCR) is a modified version of conventional PCR that utilizes fluorescent probes to detect and quantify unknown templates through the accumulation of fluorescence signals [
14]. Commonly employed methods for the quantitative detection of yeast fluorescence include both dye and probe approaches. The TaqMan probe method uses an oligonucleotide segment that can hybridize and complement a target gene. Chen et al. [
15] established a qPCR identification method for
Saccharomyces cerevisiae,
Pichia spartinae,
Zygosaccharomyces rouxii, and
Dekkera bruxellensis by targeting a conserved sequence of the yeast gene. Liu et al. [
16] developed a detection method for
M. bicuspidata utilizing the TaqMan probe method; however, the design of the probe proved challenging, and its synthesis cost exorbitant, resulting in a high level of expense. The SYBR Green I fluorescence method is a cost-effective and user-friendly nonspecific minor groove binder that intercalates with the DNA double helix, making it a widely applicable approach for disease detection.
In this study, we aim to develop a SYBR Green I qPCR method that exhibits robust specificity and high sensitivity in detecting M. bicuspidata, thereby providing valuable assistance for the prevention and treatment of milky disease in Chinese mitten crabs.
4. Discussion
Severe outbreaks of “milky disease” have occurred during the breeding of Chinese mitten crabs in northern China in recent years. The infected crabs exhibited weakened vitality, tissue emulsification, and mortality rates exceeding 20%. Currently, this disease is spreading nationwide, with an increasing annual infection rate. The pathogen responsible for this epidemic is
M. bicuspidata, for which there are no effective treatments currently available. Zhang et al. [
12] discovered that Massoia lactone, produced by
Aureobasidium melanogenum, exhibits potent anti-
M. bicuspidata activity. Ma et al. [
9] also observed the inhibitory effects of various antifungal drugs, including ketoconazole, fluconazole, econazole, clotrimazole, amphotericin B, itraconazole, and nystatin, against
M. bicuspidata. However, the clinical efficacy of these drugs is yet to be demonstrated. Therefore, early interruption of the transmission route is the primary approach for preventing disease occurrence. Diseased crabs, in their initial stages, can only be detected using highly sensitive molecular biology techniques because no apparent abnormalities can be observed with the naked eye. Currently, the PCR method commonly used to quantify
M. bicuspidata lacks accuracy and requires agarose electrophoresis to determine the amplification results, which is a complex and time-consuming process [
11,
12]. Nested PCR has a higher sensitivity and specificity than conventional PCR; however, its operation is complex and time-consuming. Additionally, it cannot quantify the extent of pathogen infection [
13]. qPCR technology possesses not only high sensitivity, good specificity, and accuracy, but also ease of operation and implementation, rendering it widely applicable for the detection of aquatic pathogens [
19].
M. bicuspidata is an opportunistic pathogen. The establishment of a qPCR detection method enables rapid and precise quantification of
M. bicuspidata in samples, facilitating accurate diagnosis, even at low levels, during the early stages of the disease outbreak. This is conducive to the prompt implementation of effective prevention and control measures, thereby minimizing economic losses.
In this study, a novel qPCR method was developed to target the mitochondrial cytochrome c oxidase subunit VIA (COX6A) gene in
M. bicuspidata. The COX6A motif, has a circular DNA molecule located outside the nuclear genome that encodes respiratory chain cytochrome oxidase, and is widely conserved in both prokaryotic and eukaryotic organisms [
20]. The utilization of mitochondrial cytochrome C oxidase subunits for species identification has emerged as a new research direction, with many detection methods employing these subunits as target genes. Di Cesare et al. [
21] used a specific mitochondrial COX I gene fragment to establish a PCR assay for
Eucoleus boehmi in canids, whereas Latrofa et al. [
22] developed a multiplex PCR assay for filarioid species infesting dogs using mitochondrial COX I. Echeverry et al. [
23] established a PCR assay based on mitochondrial COX I to detect Plasmodium parasites, which demonstrated superior sensitivity and specificity compared to 18S rRNA gene-based PCR. The mitochondrial COX6A gene utilized in this study, a subunit of the mitochondrial cytochrome C oxidase complex, exhibits conserved characteristics and low amino acid sequence homology with other yeast species (
Table 7). Therefore, mitochondrial COX6A could be used to develop and implement detection technologies for
M. bicuspidata.
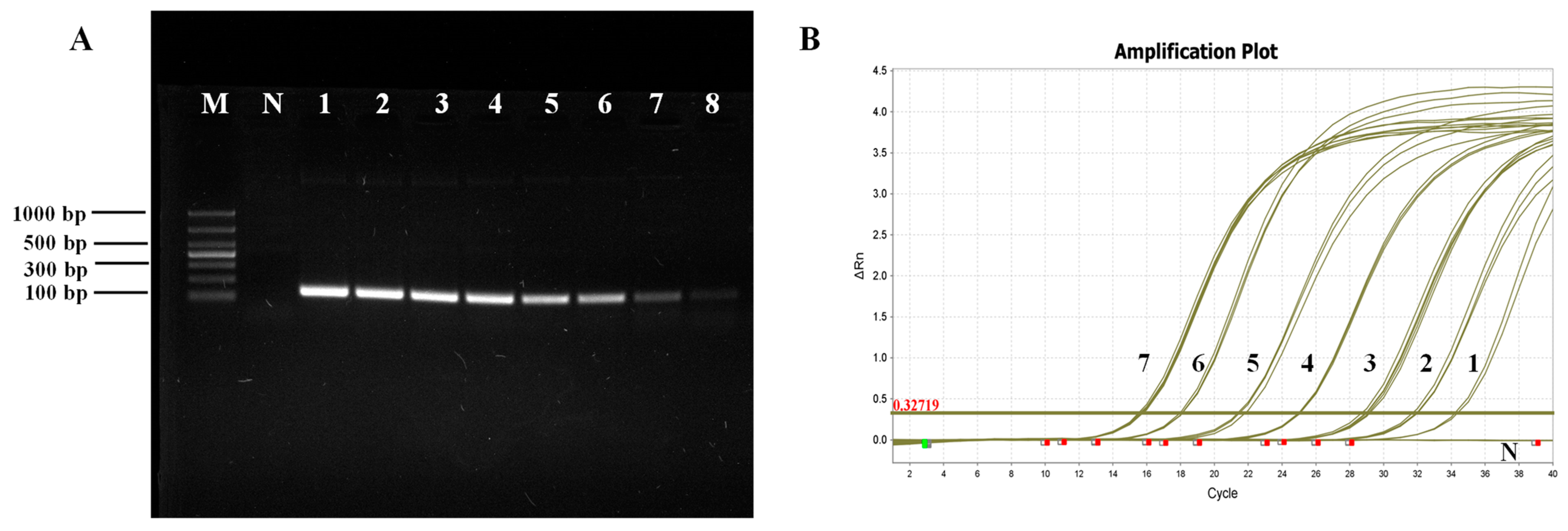
The qPCR technique established in this study exhibited significantly enhanced specificity compared to conventional PCR. The COX6A gene primers demonstrated robust specificity and produced clear bands during sample analysis. Currently, the primers designed for LSU rRNA and its ITS region have the potential to amplify template DNA from other pathogens, resulting in a target band of
Staphylococcus aureus appearing at the same location as
M. bicuspidata [
13]. The presence of other microorganisms in environmental samples or individuals may result in erroneous detection, and thus necessitating the sequencing of the target band to confirm
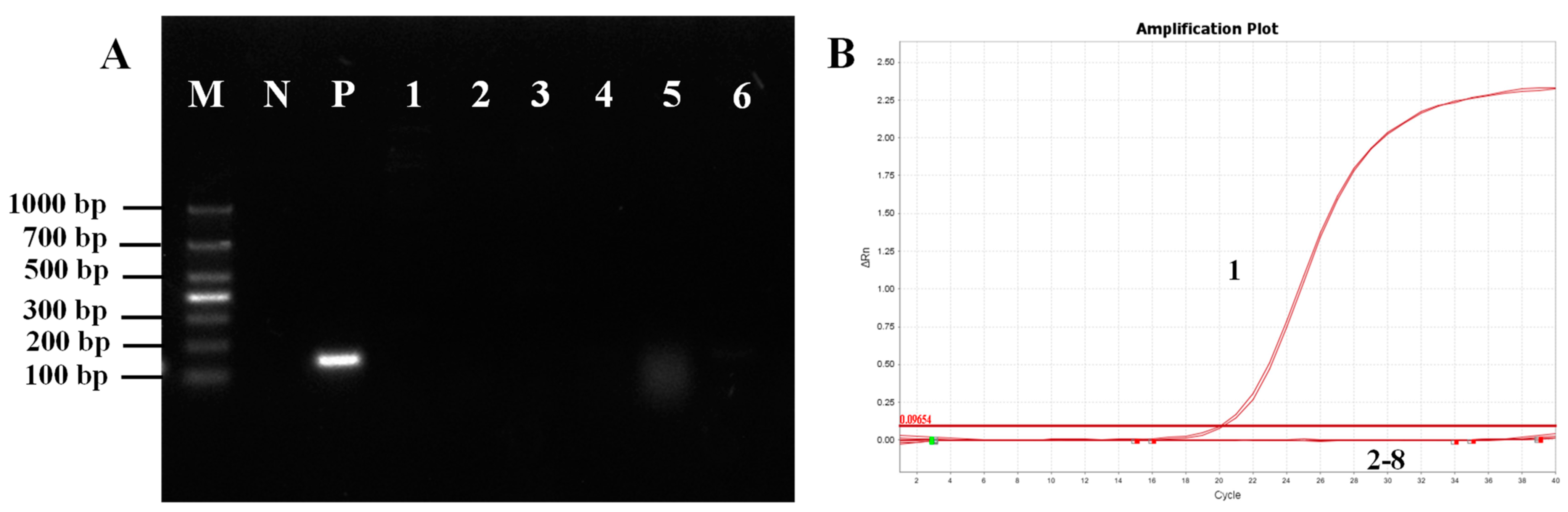
M. bicuspidata identity. The qPCR primers designed for this study exhibited no cross-reactivity with the six other pathogens; thus, any specific amplification confirmed the presence of
M. bicuspidata, which was validated through clinical sample testing.
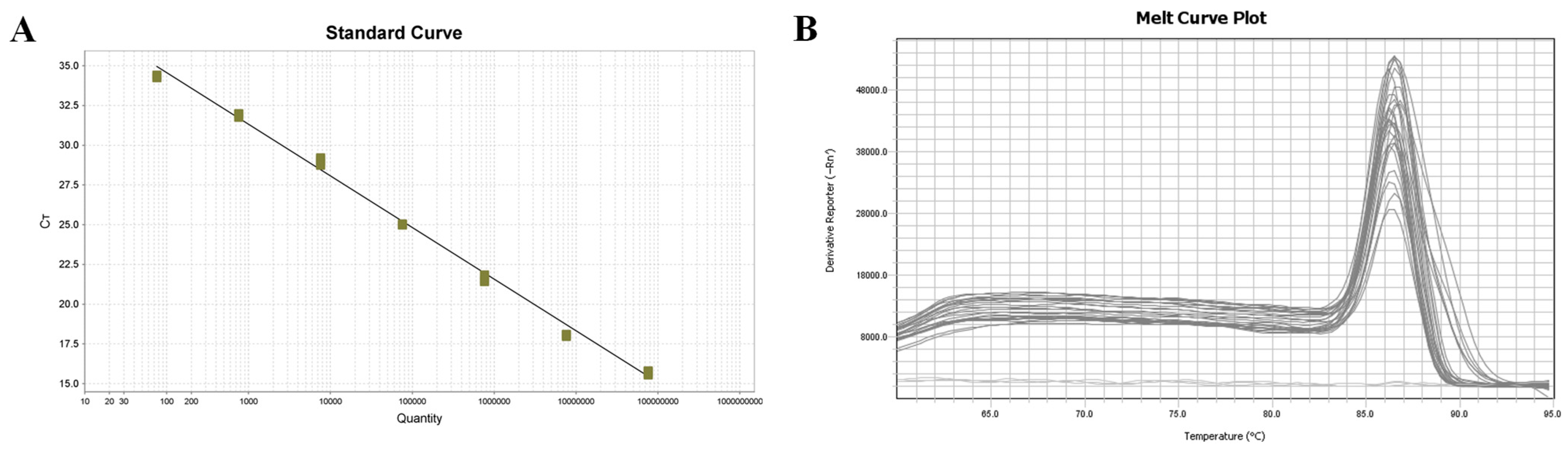
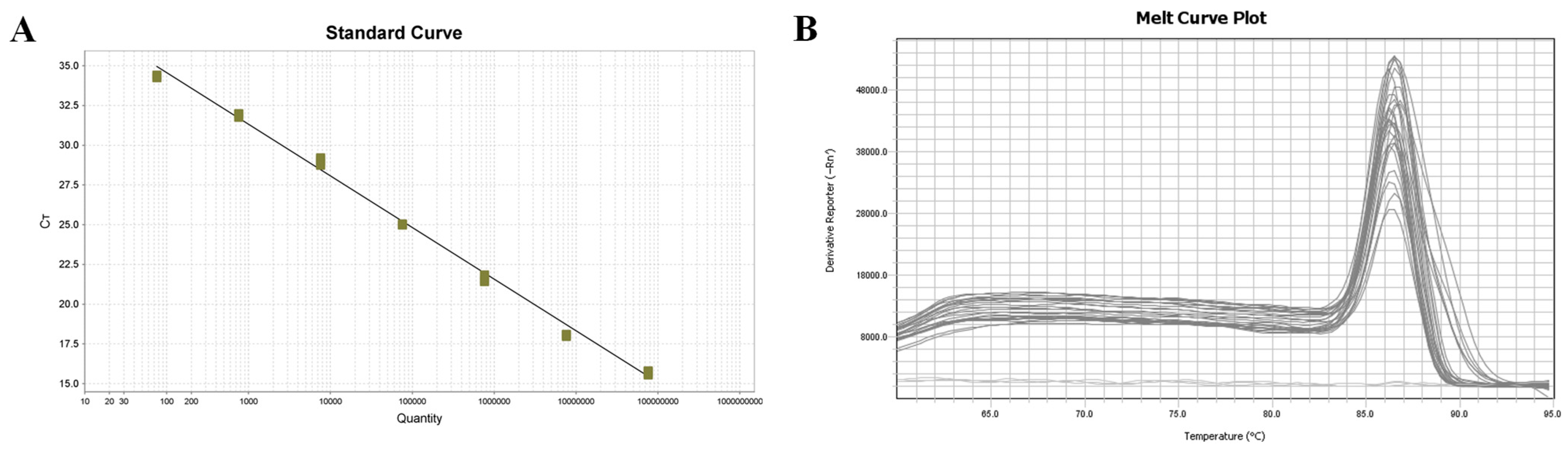
The sensitivity of COX6A-qPCR (7.6 × 101 copies/μL) was notably higher than that of the LSU rRNA group (6.03 × 104 copies/μL) and ITS (6.74 × 105 copies/μL) primers (Bao et al., 2022). COX6A-qPCR detected positive results in 46 of 60 clinical samples (76.67%), and sequence alignment analysis confirmed the presence of a COX6A gene fragment specific to M. bicuspidata. In contrast, nested PCR yielded a positivity rate of 65%, with 28 positive samples in the first round and an additional 11 positives in the second round. However, in clinical samples, the LSU rRNA and ITS primers only achieved positive rates of amplification products of 30% and 48.33%, respectively, which were significantly lower than those obtained using qPCR. This further confirmed that qPCR exhibited high specificity and sensitivity.
Compared with the other detection methods, qPCR possesses the unique ability to quantify and elucidate the degree of disease in Chinese mitten crabs. The range of yeast content in positive samples calculated in this study is 1.0 × 101–02.7 × 106 copies/µL, indicating that this detection method has a broad detection range and can accurately measure the extent of infection. In this study, the established COX6A-qPCR method was used to detect M. bicuspidata in various tissues from the infected Chinese mitten crabs, revealing a significant presence of M. bicuspidata across all tissues, with higher yeast content observed in the hemolymph, stomach, hepatopancreas, and muscle than in other tissues. The identification of these tissues could facilitate the detection of positive samples during periods of low M. bicuspidata load in Chinese mitten crabs, thereby enhancing detection accuracy and contributing significantly to the early diagnosis, prevention, and control of M. bicuspidata disease. The hemolymph of E. sinensis was initially tested to determine its infection status. This approach also minimized tissue loss and mortality in the sampled individuals. By utilizing this method for detecting biological samples in the environment, it has been determined that M. bicuspidata can be detected in various organisms such as fish, shrimp, copepods, and cladocera, with a higher concentration found within shrimp and cladocera. This suggests that the disinfection of the breeding environment should be conducted beforehand, particularly for environmental organisms such as cladocerans and shrimp, to minimize potential carriers and reduce infection risks.
The qPCR method established in this study exhibited an intragroup coefficient of variation ranging from 0.12–0.51% and an intergroup coefficient of variation ranging from 0.51–1.36%. These results demonstrate that the established method exhibits excellent repeatability. Furthermore, the absence of agarose gel electrophoresis enables the entire detection reaction to be completed within 1.3 h, and real-time fluorescence quantitative amplification curve analysis allows for the interpretation of results in less than half of the time required for conventional and nested PCR. The gene sequences used in this study were derived from a strain of M. bicuspidata isolated in the United States, whereas the test samples were obtained from a Chinese mitten crab strain. Despite their disparate origins, these strains exhibited excellent specificity, underscoring the practicality of this detection method.
The qPCR method for M. bicuspidata established in this study exhibited high sensitivity, strong specificity, and good repeatability. It could thus be utilized for the rapid and quantitative detection of M. bicuspidata in infected individuals as well as for the precise detection of the pathogen in the environment. This is important for elucidating the transmission route of the disease and providing a foundation for accurate prevention and control measures.





{kind=link}
{kind=link}
{kind=link}