
Transcriptomic and Metabolomic Profiles Provide Insights into the Red-Stipe Symptom of Morel Fruiting Bodies

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. RNA Sequencing
2.3. De Novo Assembly, Unigene Annotation, and Functional Classification
2.4. Differentially Expressed Unigene Analysis
2.5. Metabolite Extraction and Parameter Setting
2.6. Metabolite Identification and Quantification
2.7. Identification of Key Pathways and Validation of Gene Expression
3. Results
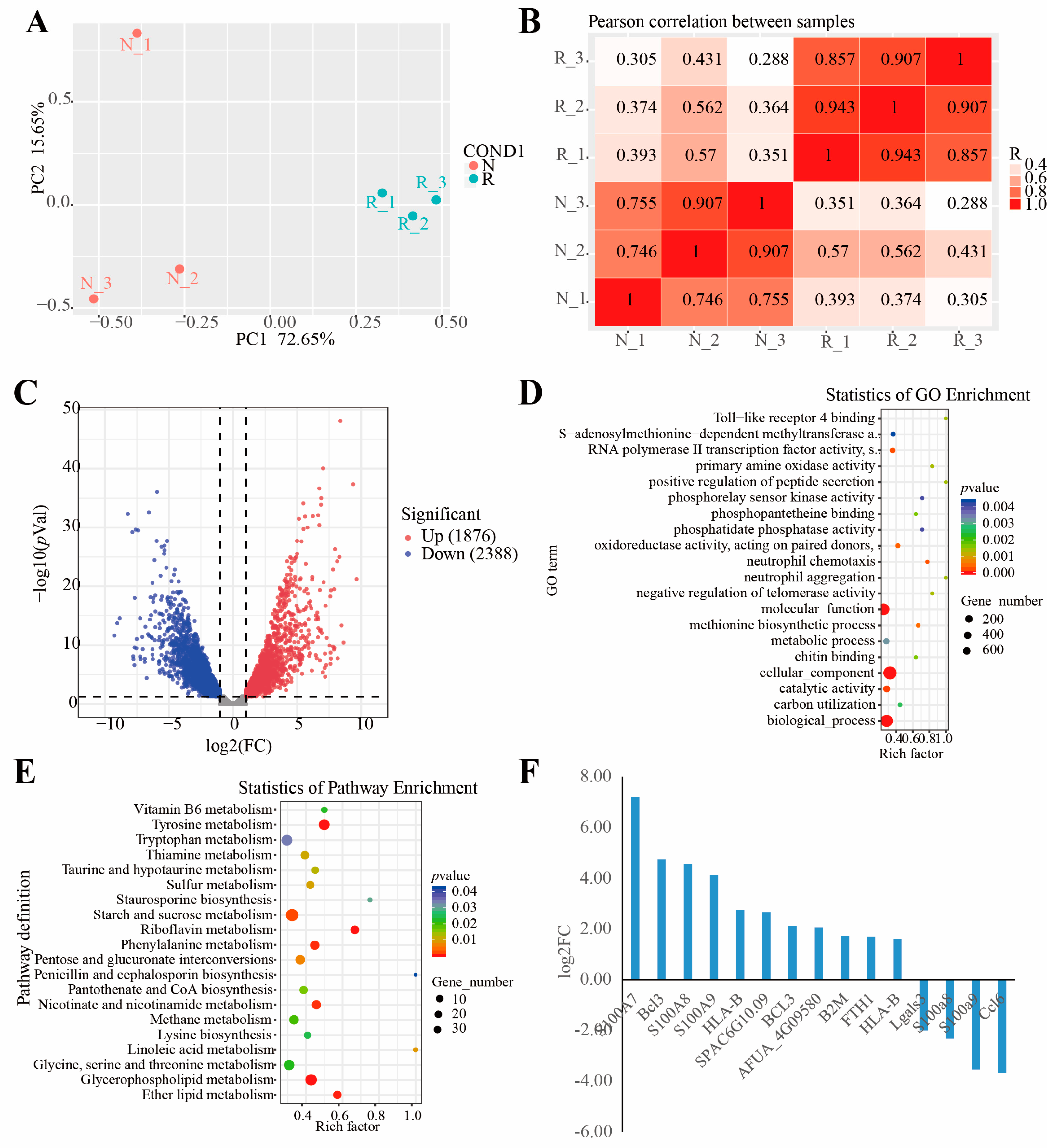
3.1. Transcriptome Sequencing and Functional Annotation
3.2. Global Changes in Gene Expression
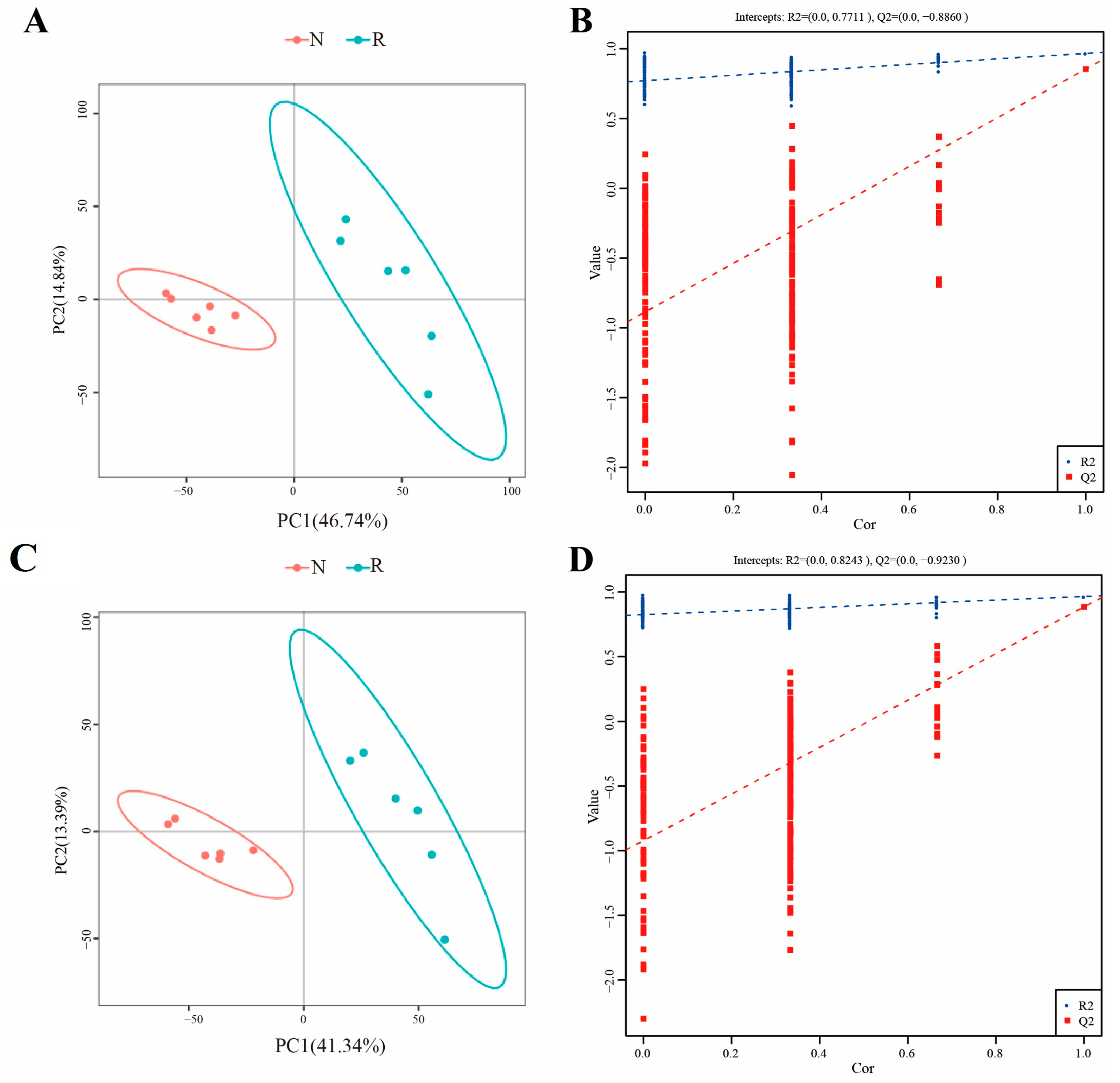
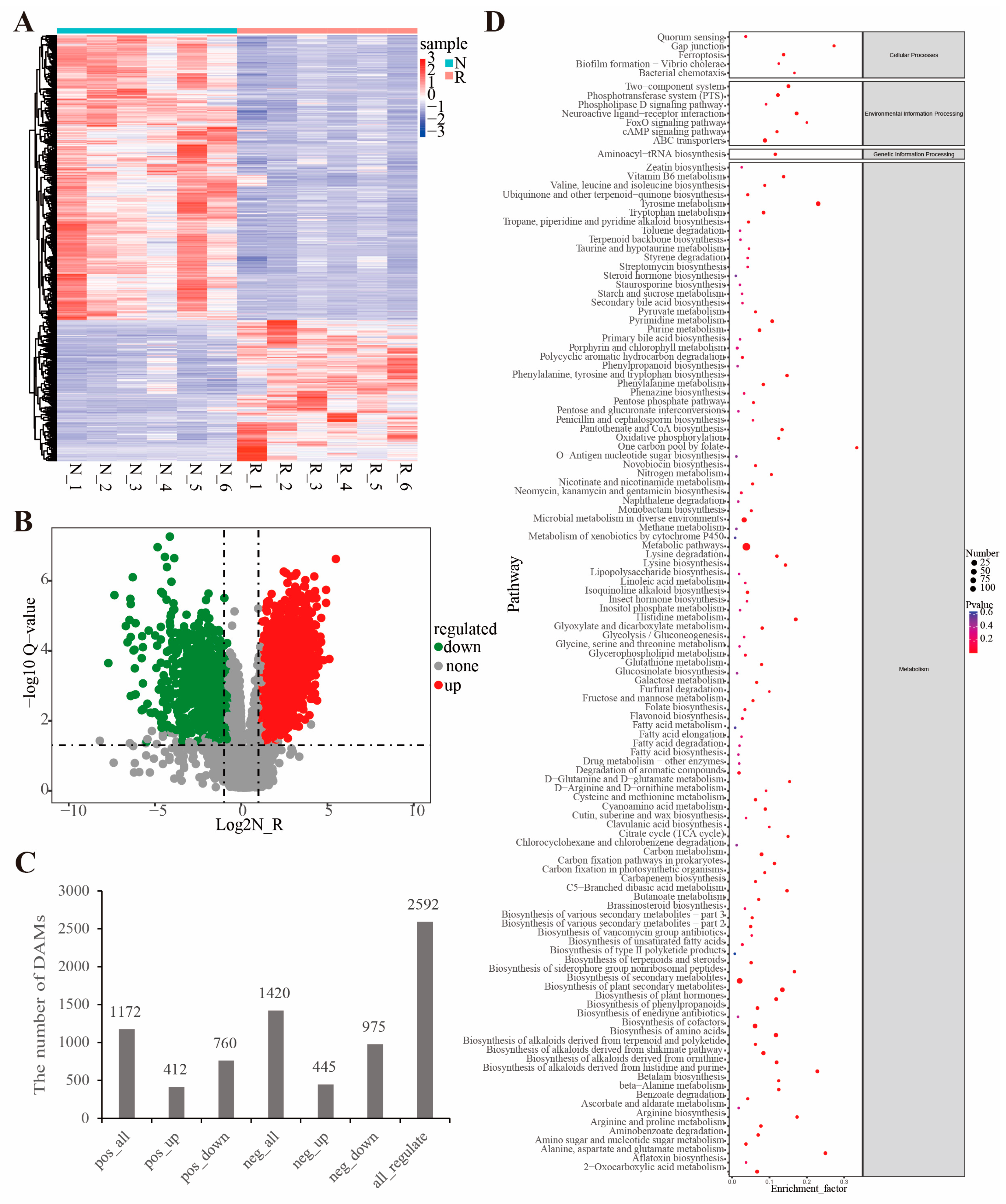
3.3. Metabolome Profiling
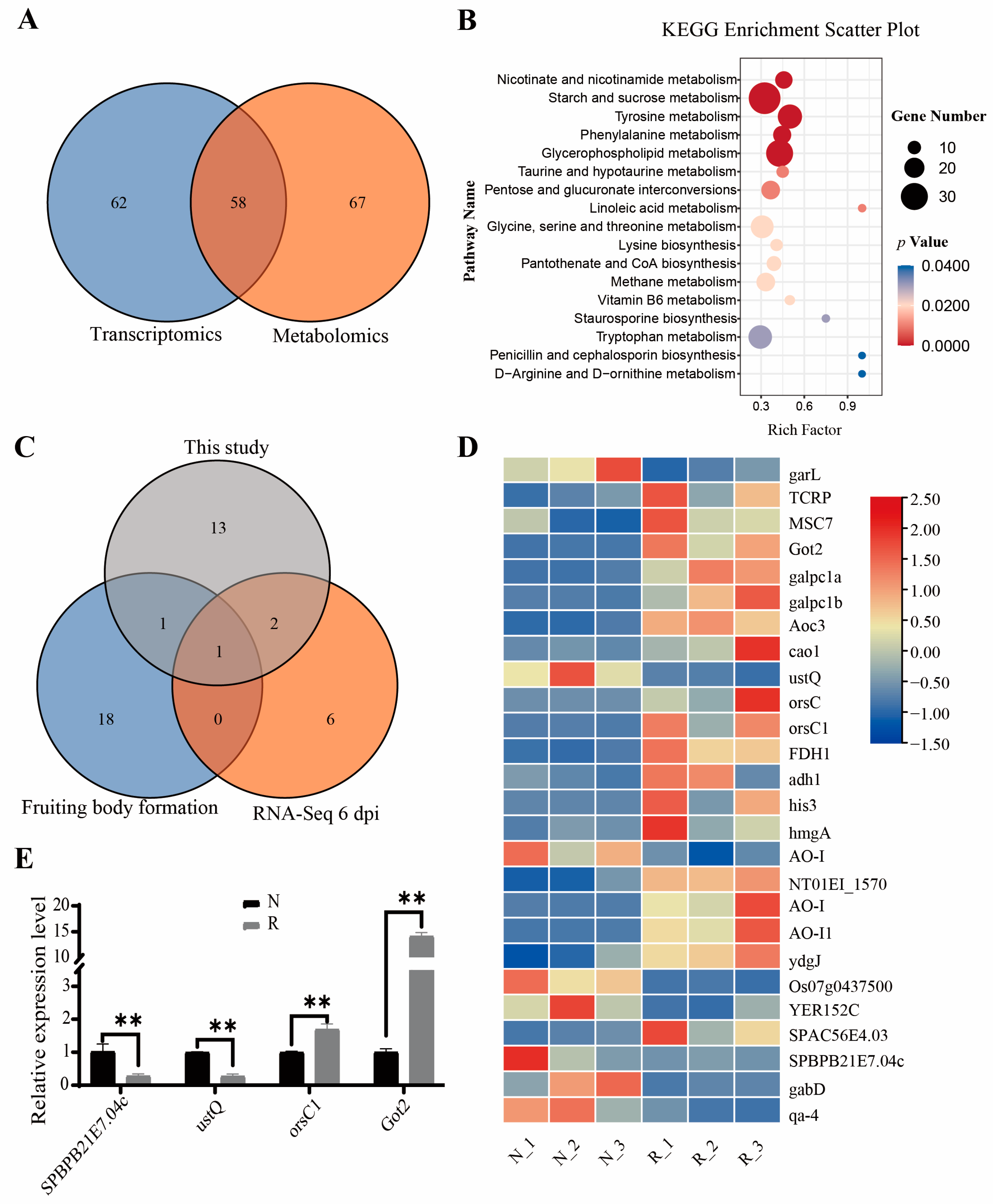
3.4. Identification of Key Pathways and Validation of Gene Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wu, H.; Chen, J.; Li, J.; Liu, Y.; Park, H.J.; Yang, L. Recent advances on bioactive ingredients of Morchella esculenta. Appl. Biochem. Biotechnol. 2021, 193, 4197–4213. [Google Scholar] [CrossRef] [PubMed]
- Sunil, C.; Xu, B. Mycochemical profile and health-promoting effects of morel mushroom Morchella esculenta (l.)—A review. Food Res. Int. 2022, 159, 111571. [Google Scholar] [CrossRef] [PubMed]
- Tietel, Z.; Masaphy, S. True morels (morchella)-nutritional and phytochemical composition, health benefits and flavor: A review. Crit. Rev. Food Sci. Nutr. 2018, 58, 1888–1901. [Google Scholar] [CrossRef]
- Du, X.H.; Zhao, Q.; Yang, Z.L. A review on research advances, issues, and perspectives of morels. Mycology 2015, 6, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Ma, H.; Zhang, Y.; Dong, C. Artificial cultivation of true morels: Current state, issues and perspectives. Crit. Rev. Biotechnol. 2018, 38, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Sambyal, K.; Singh, R.V. A comprehensive review on Morchella importuna: Cultivation aspects, phytochemistry, and other significant applications. Folia Microbiol. (Praha) 2021, 66, 147–157. [Google Scholar] [CrossRef]
- Du, X.H.; Yang, Z.L. Mating systems in true morels (Morchella). Microbiol. Mol. Biol. Rev. 2021, 85, e0022020. [Google Scholar] [CrossRef] [PubMed]
- Masaphy, S. Biotechnology of morel mushrooms: Successful fruiting body formation and development in a soilless system. Biotechnol. Lett. 2010, 32, 1523–1527. [Google Scholar] [CrossRef]
- Liu, W.; Cai, Y.; Zhang, Q.; Shu, F.; Chen, L.; Ma, X.; Bian, Y. Subchromosome-scale nuclear and complete mitochondrial genome characteristics of Morchella crassipes. Int. J. Mol. Sci. 2020, 21, 483. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Lv, M.; Li, L.; Huang, W.; Zhang, Y.; Hao, Z. “Temptation” and “trap” of morchella industry in China. J. Fungal Res. 2021, 19, 232–237. [Google Scholar]
- Shi, X.; Liu, D.; He, X.; Liu, W.; Yu, F. Epidemic identification of fungal diseases in morchella cultivation across China. J. Fungi 2022, 8, 1107. [Google Scholar] [CrossRef] [PubMed]
- Du, X.H.; Zhao, Q.; Xia, E.H.; Gao, L.Z.; Richard, F.; Yang, Z.L. Mixed-reproductive strategies, competitive mating-type distribution and life cycle of fourteen black morel species. Sci. Rep. 2017, 7, 1493. [Google Scholar] [CrossRef] [PubMed]
- Du, X.H.; Wu, D.; Kang, H.; Wang, H.; Xu, N.; Li, T.; Chen, K. Heterothallism and potential hybridization events inferred for twenty-two yellow morel species. IMA Fungus 2020, 11, 4. [Google Scholar] [CrossRef]
- Chai, H.; Chen, L.; Chen, W.; Zhao, Q.; Zhang, X.; Su, K.; Zhao, Y. Characterization of mating-type idiomorphs suggests that Morchella importuna, mel-20 and M. sextelata are heterothallic. Mycol. Prog. 2017, 16, 743–752. [Google Scholar] [CrossRef]
- Chai, H.; Chen, W.; Zhang, X.; Su, K.; Zhao, Y. Structural variation and phylogenetic analysis of the mating-type locus in the genus morchella. Mycologia 2019, 111, 551–562. [Google Scholar] [CrossRef]
- Liu, W.; Chen, L.; Cai, Y.; Zhang, Q.; Bian, Y. Opposite polarity monospore genome de novo sequencing and comparative analysis reveal the possible heterothallic life cycle of Morchella importuna. Int. J. Mol. Sci. 2018, 19, 2525. [Google Scholar] [CrossRef]
- Sa, W.; Shang, Q.H.; Yang, T.; Gao, Q.Y.; Liu, M.L.; Liang, J.; Li, Z.H. Characterization of the complete mitochondrial genome of Morchella eohespera. Mitochondrial. DNA B Resour. 2020, 5, 3048–3049. [Google Scholar] [CrossRef]
- Wingfield, B.D.; Bills, G.F.; Dong, Y.; Huang, W.; Nel, W.J.; Swalarsk-Parry, B.S.; Vaghefi, N.; Wilken, P.M.; An, Z.; de Beer, Z.W.; et al. Ima genome-f 9: Draft genome sequence of Annulohypoxylon stygium, Aspergillus mulundensis, Berkeleyomyces basicola (syn. Thielaviopsis basicola), Ceratocystis smalleyi, two Cercospora beticola strains, Coleophoma cylindrospora, Fusarium fracticaudum, Phialophora cf. Hyalina, and Morchella septimelata. IMA Fungus 2018, 9, 199–223. [Google Scholar]
- Liu, W.; Cai, Y.; Zhang, Q.; Chen, L.; Shu, F.; Ma, X.; Bian, Y. The mitochondrial genome of Morchella importuna (272.2 kb) is the largest among fungi and contains numerous introns, mitochondrial non-conserved open reading frames and repetitive sequences. Int. J. Biol. Macromol. 2020, 143, 373–381. [Google Scholar] [CrossRef]
- Li, W.; Zhang, F.; Gao, L.Z. Smrt-based mitochondrial genome of the edible mushroom Morchella conica. Mitochondrial. DNA B Resour. 2020, 5, 3201–3202. [Google Scholar] [CrossRef]
- Mei, H.; Qingshan, W.; Baiyintala; Wuhanqimuge. The whole-genome sequence analysis of Morchella sextelata. Sci. Rep. 2019, 9, 15376. [Google Scholar]
- Longley, R.; Benucci, G.M.N.; Mills, G.; Bonito, G. Fungal and bacterial community dynamics in substrates during the cultivation of morels (morchella rufobrunnea) indoors. FEMS Microbiol. Lett. 2019, 366, fnz215. [Google Scholar] [CrossRef]
- Benucci, G.M.N.; Longley, R.; Zhang, P.; Zhao, Q.; Bonito, G.; Yu, F. Microbial communities associated with the black morel Morchella sextelata cultivated in greenhouses. PeerJ 2019, 7, e7744. [Google Scholar] [CrossRef]
- Lü, B.B.; Wu, G.G.; Sun, Y.; Zhang, L.S.; Wu, X.; Jiang, W.; Li, P.; Huang, Y.N.; Wang, J.B.; Zhao, Y.C.; et al. Comparative transcriptome and endophytic bacterial community analysis of Morchella conica sh. Front. Microbiol. 2021, 12, 682356. [Google Scholar] [CrossRef]
- Orlofsky, E.; Zabari, L.; Bonito, G.; Masaphy, S. Changes in soil bacteria functional ecology associated with morchella rufobrunnea fruiting in a natural habitat. Environ. Microbiol. 2021, 23, 6651–6662. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.; Liu, T.; Yu, Y.; Tang, J.; Jiang, L.; Martin, F.M.; Peng, W. Morel production related to soil microbial diversity and evenness. Microbiol. Spectr. 2021, 9, e0022921. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.; Kohler, A.; Miao, R.; Liu, T.; Zhang, Q.; Zhang, B.; Jiang, L.; Wang, Y.; Xie, L.; Tang, J.; et al. Multi-omic analyses of exogenous nutrient bag decomposition by the black morel morchella importuna reveal sustained carbon acquisition and transferring. Environ. Microbiol. 2019, 21, 3909–3926. [Google Scholar] [CrossRef]
- Liu, Q.; Qu, S.; He, G.; Wei, J.; Dong, C. Mating-type genes play an important role in fruiting body development in Morchella sextelata. J. Fungi 2022, 8, 564. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yang, J.; Wang, H.; Jin, Y.; Liu, J.; Jia, R.; Wang, Z.; Kang, Z. Analysis of primary metabolites of morchella fruit bodies and mycelium based on widely targeted metabolomics. Arch. Microbiol. 2021, 204, 98. [Google Scholar] [CrossRef]
- Hao, H.; Zhang, J.; Wang, H.; Wang, Q.; Chen, M.; Juan, J.; Feng, Z.; Chen, H. Comparative transcriptome analysis reveals potential fruiting body formation mechanisms in Morchella importuna. AMB Express 2019, 9, 103. [Google Scholar] [CrossRef]
- Liu, W.; Cai, Y.; He, P.; Chen, L.; Bian, Y. Comparative transcriptomics reveals potential genes involved in the vegetative growth of Morchella importuna. 3 Biotech 2019, 9, 81. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.F.; Cong, Q.Q.; Wang, Q.W.; Tang, L.N.; Li, X.M.; Yu, Q.W.; Cui, X.; An, X.R.; Yu, C.X.; Kong, F.H.; et al. First report of cladobotryum protrusum causing cobweb disease on cultivated Morchella importuna. Plant Dis. 2020, 104, 977. [Google Scholar] [CrossRef]
- He, P.; Li, C.; Cai, Y.; Zhang, Y.; Bian, Y.; Liu, W. First report of pileus rot disease on cultivated Morchella importuna caused by Diploöspora longispora in China. J. Gen. Plant Pathol. 2018, 84, 65–69. [Google Scholar] [CrossRef]
- Guo, M.P.; Chen, K.; Wang, G.Z.; Bian, Y.B. First report of stipe rot disease on morchella importuna caused by fusarium incarnatum—F. equiseti species complex in China. Plant Dis. 2016, 100, 2530. [Google Scholar] [CrossRef]
- He, X.L.; Peng, W.H.; Miao, R.Y.; Tang, J.; Chen, Y.; Liu, L.X.; Wang, D.; Gan, B.C. White mold on cultivated morels caused by paecilomyces penicillatus. FEMS Microbiol. Lett. 2017, 364, fnx037. [Google Scholar] [CrossRef] [PubMed]
- Lv, B.; Sun, Y.; Chen, Y.; Yu, H.; Mo, Q. First report of Lecanicillium aphanocladii causing rot of Morchella sextelata in China. Plant Dis. 2022, 106, 3202. [Google Scholar] [CrossRef] [PubMed]
- Sjaarda, C.P.; Abubaker, K.S.; Castle, A.J. Induction of lcc2 expression and activity by Agaricus bisporus provides defence against trichoderma aggressivum toxic extracts. Microb. Biotechnol. 2015, 8, 918–929. [Google Scholar] [CrossRef]
- Fu, Y.; Wang, X.; Li, D.; Liu, Y.; Song, B.; Zhang, C.; Wang, Q.; Chen, M.; Zhang, Z.; Li, Y. Identification of resistance to wet bubble disease and genetic diversity in wild and cultivated strains of Agaricus bisporus. Int. J. Mol. Sci. 2016, 17, 1568. [Google Scholar] [CrossRef]
- Foulongne-Oriol, M.; Rodier, A.; Savoie, J.M. Relationship between yield components and partial resistance to lecanicillium fungicola in the button mushroom, Agaricus bisporus, assessed by quantitative trait locus mapping. Appl. Environ. Microbiol. 2012, 78, 2435–2442. [Google Scholar] [CrossRef]
- Yu, Y.; Tan, H.; Liu, T.; Liu, L.; Tang, J.; Peng, W. Dual rna-seq analysis of the interaction between edible fungus Morchella sextelata and its pathogenic fungus Paecilomyces penicillatus uncovers the candidate defense and pathogenic factors. Front. Microbiol. 2021, 12, 760444. [Google Scholar] [CrossRef]
- Yu, Y.; Liu, T.; Liu, L.; Chen, Y.; Tang, J.; Peng, W.; Tan, H. Application of the mushroom volatile 1-octen-3-ol to suppress a morel disease caused by Paecilomyces penicillatus. Appl. Microbiol. Biotechnol. 2022, 106, 4787–4799. [Google Scholar] [CrossRef]
- Liu, W.; Cai, Y.; He, P.; Ma, X.; Bian, Y. Occurrence regularity and control measures of diseases and insect pests in morel cultivation. Acta Edulis Fungi 2019, 26, 123–125+128–134. [Google Scholar]
- Liu, T.; Zhou, J.; Wang, D.; He, X.; Tang, J.; Chen, Y.; Wang, J.; Peng, W. A new stipe rot disease of the cultivated Morchella sextelata. Mycosystema 2021, 40, 15. [Google Scholar]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. Embnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from rna-seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using diamond. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by rna-seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. Edger: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for rna-seq: Accounting for selection bias. Genome. Biol. 2010, 11, R14. [Google Scholar] [CrossRef]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the kegg orthology (ko) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. Kegg for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. Kegg: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein. Sci. 2019, 28, 1947–1951. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Fang, J.; Qi, X.; Lin, M.; Zhong, Y.; Sun, L.; Cui, W. Combined analysis of the fruit metabolome and transcriptome reveals candidate genes involved in flavonoid biosynthesis in Actinidia arguta. Int. J. Mol. Sci. 2018, 19, 1471. [Google Scholar] [CrossRef]
- Yu, C.; Luo, X.; Zhan, X.; Hao, J.; Zhang, L.; YB, L.S.; Shen, C.; Dong, M. Comparative metabolomics reveals the metabolic variations between two endangered taxus species (T. fuana and T. yunnanensis) in the himalayas. BMC Plant Biol. 2018, 18, 197. [Google Scholar] [CrossRef]
- Yang, C.; Ma, L.; Ying, Z.H.; Jiang, X.L.; Lin, Y.Q. Sequence analysis and expression of a blue-light photoreceptor gene, slwc-1 from the cauliflower mushroom Sparassis latifolia. Curr. Microbiol. 2017, 74, 469–475. [Google Scholar] [CrossRef]
- Zhang, Q.; Liu, W.; Cai, Y.; Lan, A.F.; Bian, Y. Validation of internal control genes for quantitative real-time pcr gene expression analysis in morchella. Molecules 2018, 23, 2331. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Kang, R.; Kroemer, G.; Tang, D. Ferroptosis in infection, inflammation, and immunity. J. Exp. Med. 2021, 218, 518. [Google Scholar] [CrossRef]
- Xu, Y.; Tang, J.; Wang, Y.; He, X.; Tan, H.; Yu, Y.; Chen, Y.; Peng, W. Large-scale commercial cultivation of morels: Current state and perspectives. Appl. Microbiol. Biotechnol. 2022, 106, 4401–4412. [Google Scholar] [CrossRef]
- Fu, Y.; Xu, X.; Wu, H.; Li, L.; Wang, J.; Sun, Y.; Gu, L.; Yu, Q. First report of Clonostachys rosea causing rot of Morchella sextelata in anhui province, China. Plant Dis. 2022. Available online: https://doi.org/10.1094/PDIS-08-22-1794-PDN (accessed on 12 March 2023). [CrossRef] [PubMed]
- Tanaka, Y.; Sasaki, N.; Ohmiya, A. Biosynthesis of plant pigments: Anthocyanins, betalains and carotenoids. Plant J. 2008, 54, 733–749. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Huang, Q.; Lin, C.; Lin, L.; Zhou, Q.; Lin, F.; He, E. Transcriptome comparison reveals candidate genes responsible for the betalain-/anthocyanidin-production in bougainvilleas. Funct Plant Biol. 2016, 43, 278–286. [Google Scholar] [CrossRef]
- Li, Y.; Fang, J.; Qi, X.; Lin, M.; Zhong, Y.; Sun, L. A key structural gene, aaldox, is involved in anthocyanin biosynthesis in all red-fleshed kiwifruit (Actinidia arguta) based on transcriptome analysis. Gene 2018, 648, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Osorio-Guarín, J.A.; Gopaulchan, D.; Quanckenbush, C.; Lennon, A.M.; Umaharan, P.; Cornejo, O.E. Comparative transcriptomic analysis reveals key components controlling spathe color in Anthurium andraeanum (hort.). PLoS ONE 2021, 16, e0261364. [Google Scholar] [CrossRef]
- Doehlemann, G.; Ökmen, B.; Zhu, W.; Sharon, A. Plant pathogenic fungi. Microbiol. Spectr. 2017, 5, 14. [Google Scholar] [CrossRef]
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2018, 14, 207–215. [Google Scholar] [CrossRef]
- Veneault-Fourrey, C.; Barooah, M.; Egan, M.; Wakley, G.; Talbot, N.J. Autophagic fungal cell death is necessary for infection by the rice blast fungus. Science 2006, 312, 580–583. [Google Scholar] [CrossRef]
- Zhu, X.M.; Li, L.; Wu, M.; Liang, S.; Shi, H.B.; Liu, X.H.; Lin, F.C. Current opinions on autophagy in pathogenicity of fungi. Virulence 2019, 10, 481–489. [Google Scholar] [CrossRef]
- Ren, Z.; Tang, B.; Xing, J.; Liu, C.; Cai, X.; Hendy, A.; Kamran, M.; Liu, H.; Zheng, L.; Huang, J.; et al. Mta1-mediated rna m(6) a modification regulates autophagy and is required for infection of the rice blast fungus. New Phytol. 2022, 235, 247–262. [Google Scholar] [CrossRef]
- Tang, L.; Chu, T.; Shang, J.; Yang, R.; Song, C.; Bao, D.; Tan, Q.; Jian, H. Oxidative stress and autophagy are important processes in post ripeness and brown film formation in mycelium of lentinula edodes. Front. Microbiol. 2022, 13, 811673. [Google Scholar] [CrossRef] [PubMed]
- Hopke, A.; Brown, A.J.P.; Hall, R.A.; Wheeler, R.T. Dynamic fungal cell wall architecture in stress adaptation and immune evasion. Trends Microbiol. 2018, 26, 284–295. [Google Scholar] [CrossRef]
- Taborda, C.P.; da Silva, M.B.; Nosanchuk, J.D.; Travassos, L.R. Melanin as a virulence factor of Paracoccidioides brasiliensis and other dimorphic pathogenic fungi: A minireview. Mycopathologia 2008, 165, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Tan, H.; Wang, B.; Peng, W.; Sun, Q.; Yu, Y. Integrated multi-omic analyses on yellow Flammulina filiformis cultivar reveal postharvest oxidative damage responses. Postharvest Biol. Technol. 2023, 195, 112111. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, C.; Jiang, X.; Ma, L.; Xiao, D.; Liu, X.; Ying, Z.; Li, Y.; Lin, Y. Transcriptomic and Metabolomic Profiles Provide Insights into the Red-Stipe Symptom of Morel Fruiting Bodies. J. Fungi 2023, 9, 373. https://doi.org/10.3390/jof9030373
Yang C, Jiang X, Ma L, Xiao D, Liu X, Ying Z, Li Y, Lin Y. Transcriptomic and Metabolomic Profiles Provide Insights into the Red-Stipe Symptom of Morel Fruiting Bodies. Journal of Fungi. 2023; 9(3):373. https://doi.org/10.3390/jof9030373
Chicago/Turabian StyleYang, Chi, Xiaoling Jiang, Lu Ma, Donglai Xiao, Xiaoyu Liu, Zhenghe Ying, Yaru Li, and Yanquan Lin. 2023. "Transcriptomic and Metabolomic Profiles Provide Insights into the Red-Stipe Symptom of Morel Fruiting Bodies" Journal of Fungi 9, no. 3: 373. https://doi.org/10.3390/jof9030373
APA StyleYang, C., Jiang, X., Ma, L., Xiao, D., Liu, X., Ying, Z., Li, Y., & Lin, Y. (2023). Transcriptomic and Metabolomic Profiles Provide Insights into the Red-Stipe Symptom of Morel Fruiting Bodies. Journal of Fungi, 9(3), 373. https://doi.org/10.3390/jof9030373