
In Candida glabrata, ERMES Component GEM1 Controls Mitochondrial Morphology, mtROS, and Drug Efflux Pump Expression, Resulting in Azole Susceptibility

 , , , ,
, , , ,  and
and 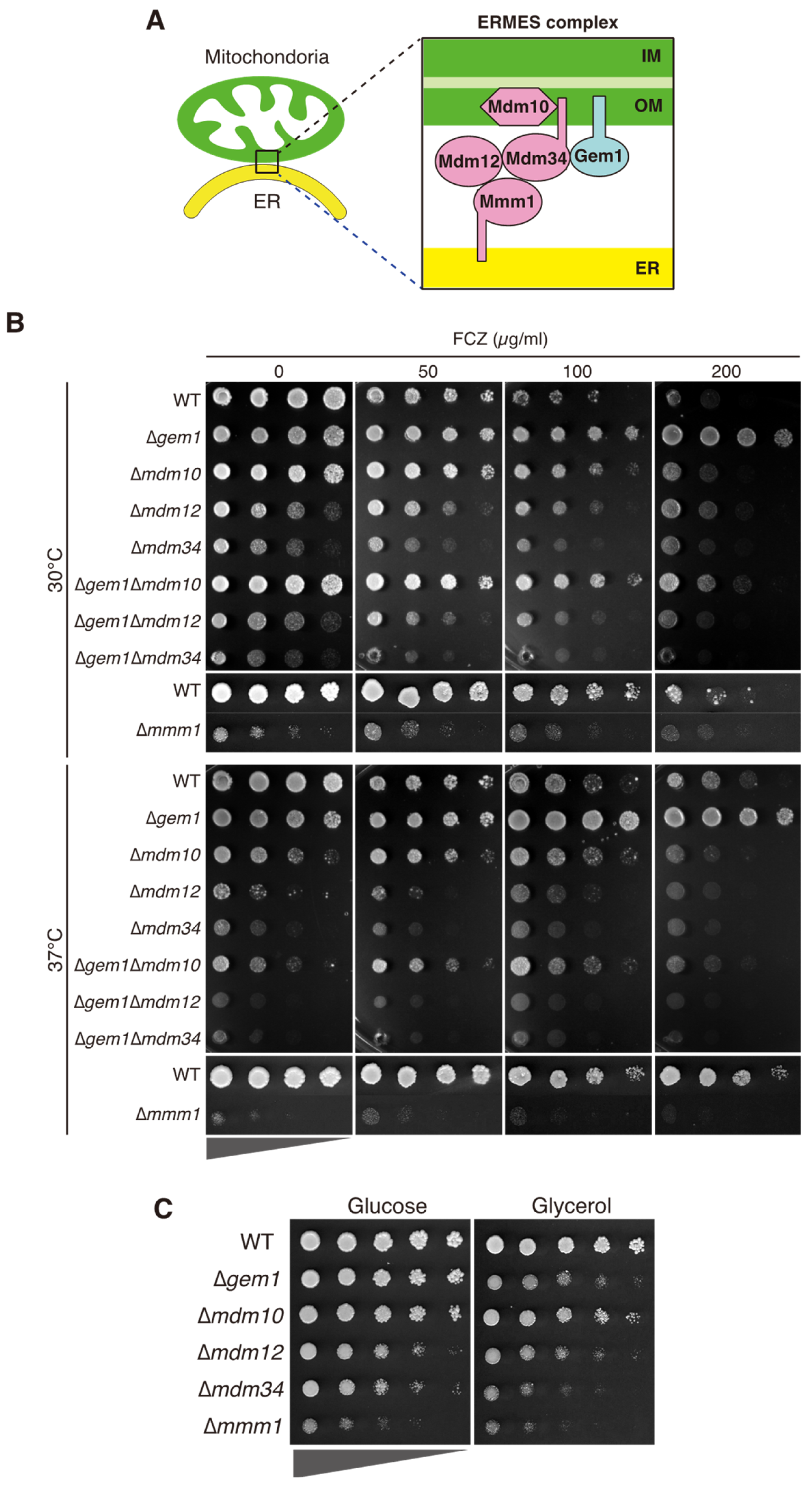{kind=link}
{kind=link}
{kind=link}
{kind=link}
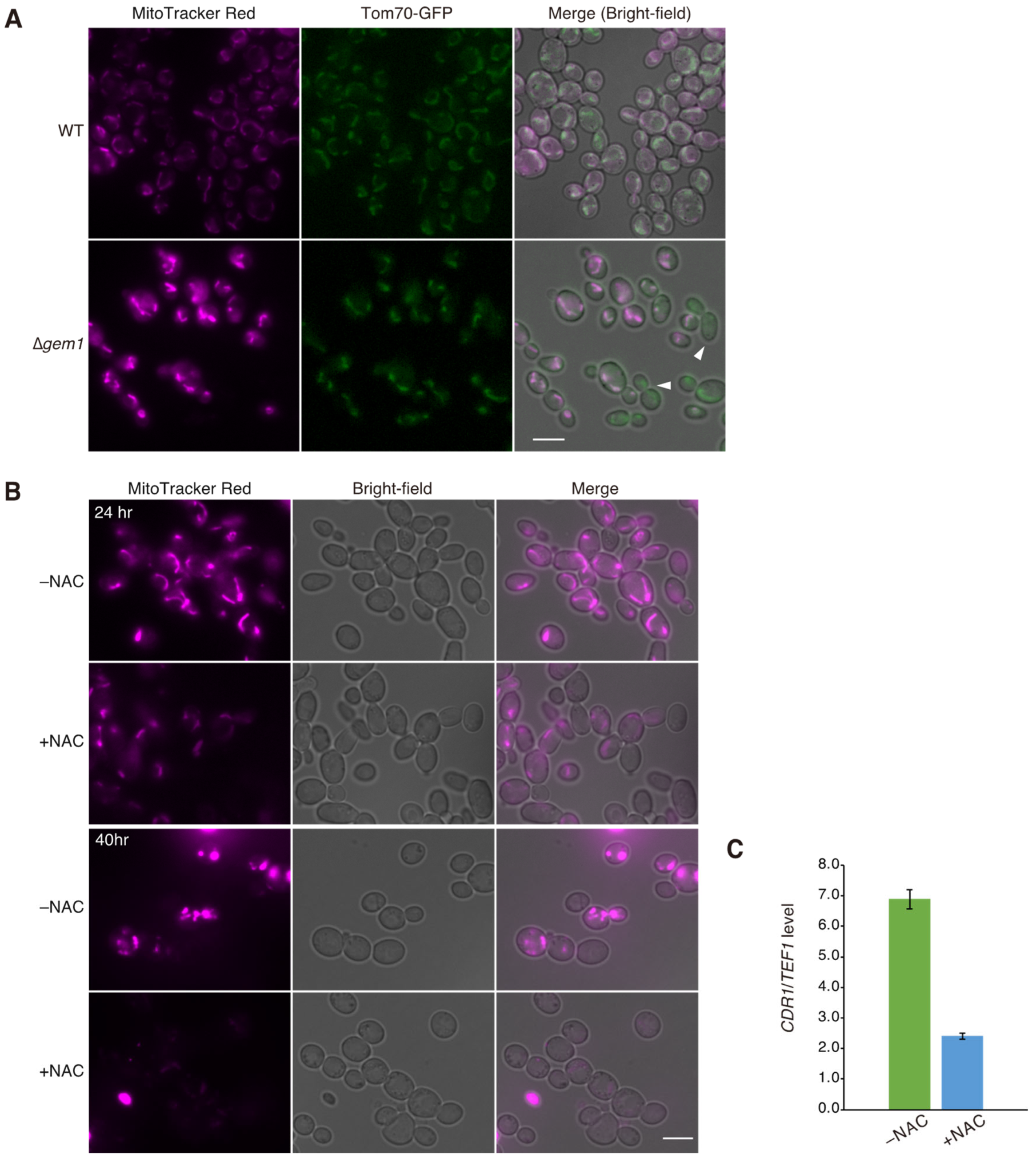{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Strains and Media
2.2. Strain and Plasmid Construction
2.3. Quantitative RT-PCR
2.4. Protein Localization by Fluorescence Microscopy
2.5. Mitochondrial Reactive Oxygen Species Quantification
3. Results
3.1. CgGem1 Is Involved in Azole Susceptibility
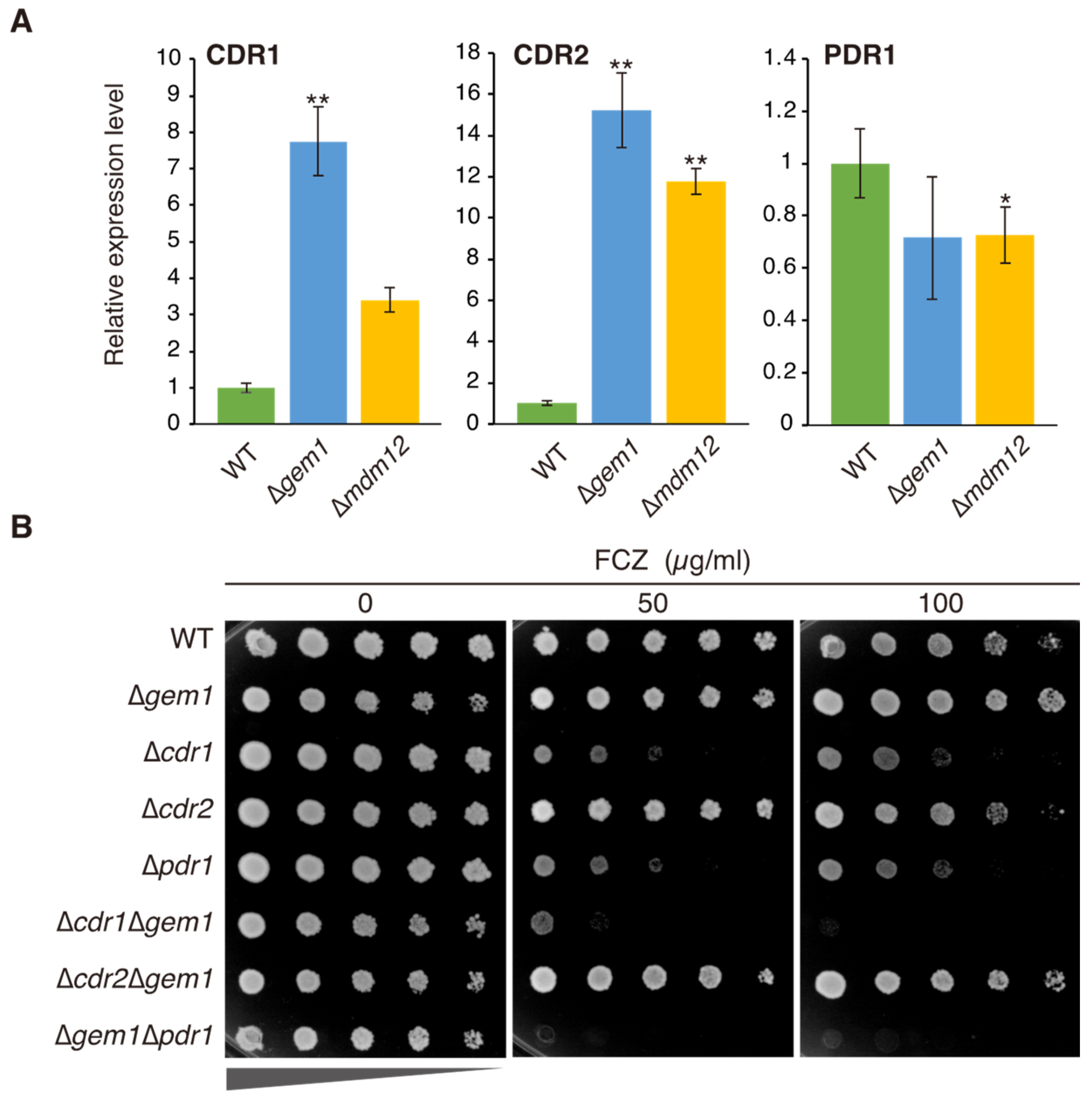
3.2. The Azole Resistance Phenotype of Δgem1 Cells Depends on Pdr1-Mediated CDR1 Upregulation
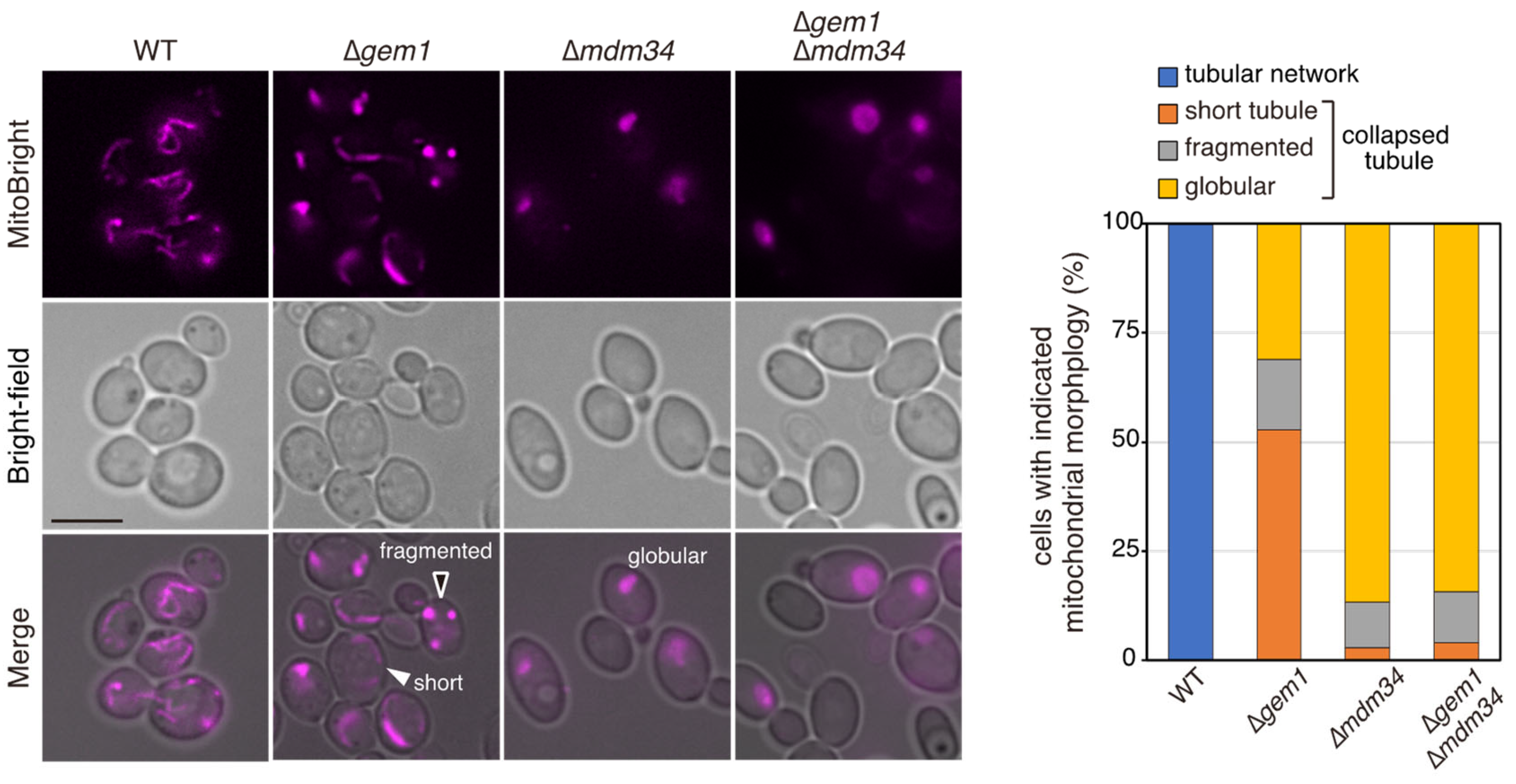
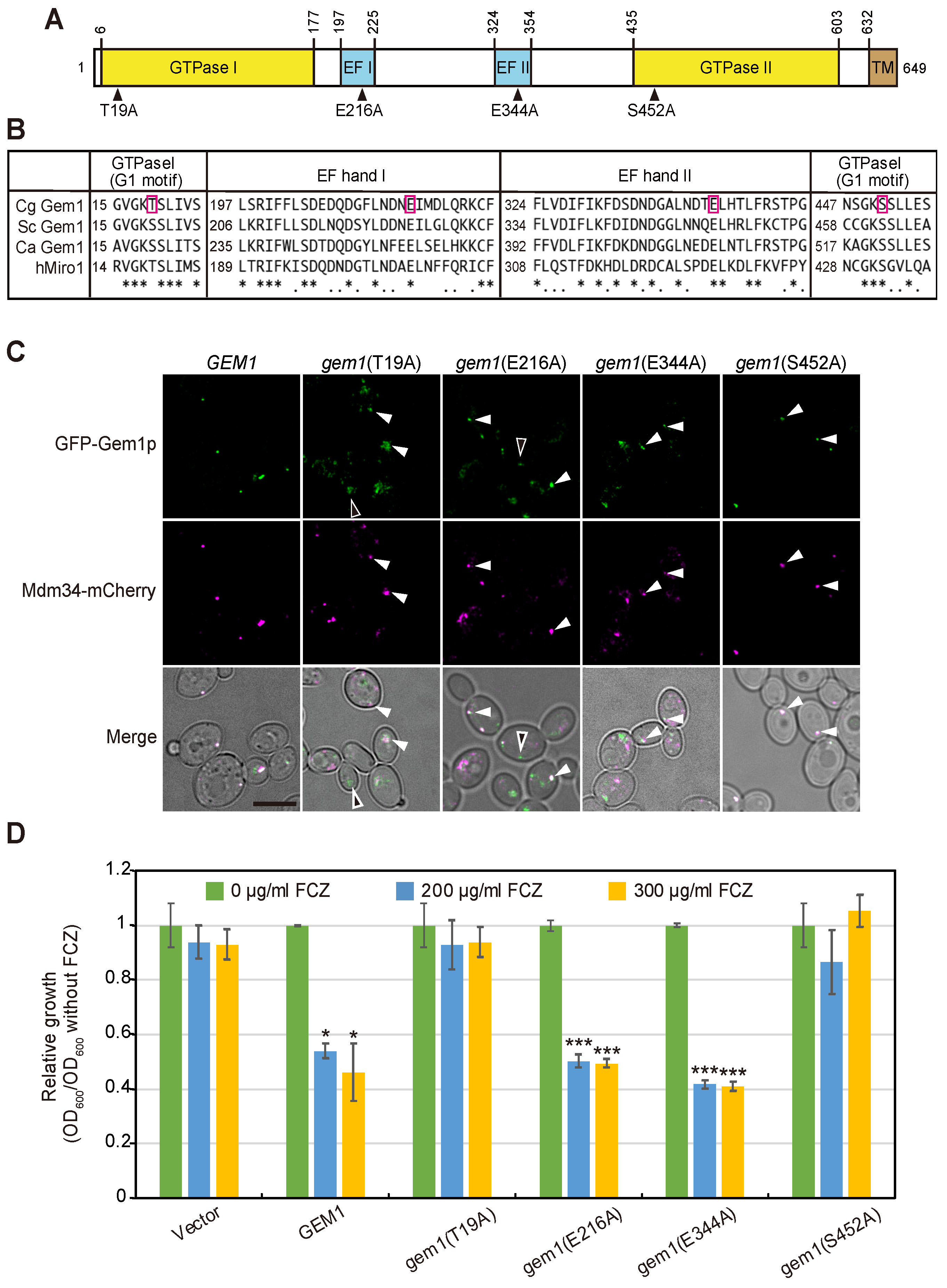
3.3. The GTPase Activity of Gem1 Is Required for Its Interaction with the ERMES Complex and Azole Susceptibility
3.4. Gem1-Dependent Mitochondrial ROS Concentration Affects Azole Susceptibility
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pfaller, M.A.; Diekema, D.J. Epidemiology of Invasive Candidiasis: A Persistent Public Health Problem. Clin. Microbiol. Rev. 2007, 20, 133–163. [Google Scholar] [CrossRef]
- Kullberg, B.J.; Arendrup, M.C. Invasive Candidiasis. N. Engl. J. Med. 2015, 373, 1445–1456. [Google Scholar] [CrossRef]
- Guinea, J. Global Trends in the Distribution of Candida Species Causing Candidemia. Clin. Microbiol. Infect. 2014, 20, 5–10. [Google Scholar] [CrossRef]
- Diekema, D.; Arbefeville, S.; Boyken, L.; Kroeger, J.; Pfaller, M. The Changing Epidemiology of Healthcare-Associated Candidemia over Three Decades. Diagn Microbiol. Infect Dis. 2012, 73, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Tsay, S.V.; Mu, Y.; Williams, S.; Epson, E.; Nadle, J.; Bamberg, W.M.; Barter, D.M.; Johnston, H.L.; Farley, M.M.; Harb, S.; et al. Burden of Candidemia in the United States, 2017. Clin. Infect. Dis. 2020, 71, e449–e453. [Google Scholar] [CrossRef] [PubMed]
- Astvad, K.M.T.; Johansen, H.K.; Røder, B.L.; Rosenvinge, F.S.; Knudsen, J.D.; Lemming, L.; Schønheyder, H.C.; Hare, R.K.; Kristensen, L.; Nielsen, L.; et al. Update from a 12-Year Nationwide Fungemia Surveillance: Increasing Intrinsic and Acquired Resistance Causes Concern. J. Clin. Microbiol. 2018, 56, e01564-17. [Google Scholar] [CrossRef] [PubMed]
- Chapman, B.; Slavin, M.; Marriott, D.; Halliday, C.; Kidd, S.; Arthur, I.; Bak, N.; Heath, C.H.; Kennedy, K.; Morrissey, C.O.; et al. Changing Epidemiology of Candidaemia in Australia. J. Antimicrob. Chemother. 2017, 72, 1103–1108. [Google Scholar] [CrossRef] [PubMed]
- Brunke, S.; Hube, B. Two Unlike Cousins: Candida albicans and C. glabrata Infection Strategies. Cell Microbiol. 2013, 15, 701–708. [Google Scholar] [CrossRef]
- Kelly, S.L.; Lamb, D.C.; Corran, A.J.; Baldwin, B.C.; Kelly, D.E. Mode of Action and Resistance to Azole Antifungals Associated with the Formation of 14α-Methylergosta-8,24(28)-Dien-3β,6α-Diol. Biochem. Biophys. Res. Commun. 1995, 207, 910–915. [Google Scholar] [CrossRef]
- Sanglard, D.; Ischer, F.; Calabrese, D.; Majcherczyk, P.A.; Bille, J. The ATP Binding Cassette Transporter Gene CgCDR1 from Candida glabrata Is Involved in the Resistance of Clinical Isolates to Azole Antifungal Agents. Antimicrob. Agents Chemother. 1999, 43, 2753–2765. [Google Scholar] [CrossRef]
- Sanglard, D.; Ischer, F.; Bille, J. Role of ATP-Binding-Cassette Transporter Genes in High-Frequency Acquisition of Resistance to Azole Antifungals in Candida glabrata. Antimicrob. Agents Chemother. 2001, 45, 1174–1183. [Google Scholar] [CrossRef] [PubMed]
- Vermitsky, J.-P.; Edlind, T.D. Azole Resistance in Candida glabrata: Coordinate Upregulation of Multidrug Transporters and Evidence for a Pdr1-Like Transcription Factor. Antimicrob. Agents Chemother. 2004, 48, 3773–3781. [Google Scholar] [CrossRef]
- Tsai, H.-F.; Krol, A.A.; Sarti, K.E.; Bennett, J.E. Candida glabrata PDR1, a Transcriptional Regulator of a Pleiotropic Drug Resistance Network, Mediates Azole Resistance in Clinical Isolates and Petite Mutants. Antimicrob. Agents Chemother. 2006, 50, 1384–1392. [Google Scholar] [CrossRef]
- Ferrari, S.; Sanguinetti, M.; de Bernardis, F.; Torelli, R.; Posteraro, B.; Vandeputte, P.; Sanglard, D. Loss of Mitochondrial Functions Associated with Azole Resistance in Candida glabrata Results in Enhanced Virulence in Mice. Antimicrob. Agents Chemother. 2011, 55, 1852–1860. [Google Scholar] [CrossRef]
- Bouchara, J.P.; Zouhair, R.; Le Boudouil, S.A.N.D.R.I.N.E.; Renier, G.; Filmon, R.; Chabasse, D.; Hallet, J.N.; Defontaine, A. In-Vivo Selection of an Azole-Resistant Petite Mutant of Candida glabrata. J. Med. Microbiol. 2000, 49, 977–984. [Google Scholar] [CrossRef] [PubMed]
- Posteraro, B.; Tumbarello, M.; La Sorda, M.; Spanu, T.; Trecarichi, E.M.; de Bernardis, F.; Scoppettuolo, G.; Sanguinetti, M.; Fadda, G. Azole Resistance of Candida glabrata in a Case of Recurrent Fungemia. J. Clin. Microbiol. 2006, 44, 3046–3047. [Google Scholar] [CrossRef] [PubMed]
- Kaur, R.; Castanño, I.; Cormack, B.P. Functional Genomic Analysis of Fluconazole Susceptibility in the Pathogenic Yeast Candida glabrata: Roles of Calcium Signaling and Mitochondria. Antimicrob. Agents Chemother. 2004, 48, 1600–1613. [Google Scholar] [CrossRef]
- Neubauer, M.; Zhu, Z.; Penka, M.; Helmschrott, C.; Wagener, N.; Wagener, J. Mitochondrial Dynamics in the Pathogenic Mold Aspergillus fumigatus: Therapeutic and Evolutionary Implications. Mol. Microbiol. 2015, 98, 930–945. [Google Scholar] [CrossRef]
- Elbaz, Y.; Schuldiner, M. Staying in Touch: The Molecular Era of Organelle Contact Sites. Trends Biochem. Sci. 2011, 36, 616–623. [Google Scholar] [CrossRef]
- Helle, S.C.J.; Kanfer, G.; Kolar, K.; Lang, A.; Michel, A.H.; Kornmann, B. Organization and Function of Membrane Contact Sites. Biochim. Et Biophys. Acta BBA-Mol. Cell Res. 2013, 1833, 2526–2541. [Google Scholar] [CrossRef] [PubMed]
- Kornmann, B.; Currie, E.; Collins, S.R.; Schuldiner, M.; Nunnari, J.; Weissman, J.S.; Walter, P. An ER-Mitochondria Tethering Complex Revealed by a Synthetic Biology Screen. Science 2009, 325, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Kornmann, B.; Osman, C.; Walter, P. The Conserved GTPase Gem1 Regulates Endoplasmic Reticulum–Mitochondria Connections. Proc. Natl. Acad. Sci. USA 2011, 108, 14151–14156. [Google Scholar] [CrossRef] [PubMed]
- Stroud, D.A.; Oeljeklaus, S.; Wiese, S.; Bohnert, M.; Lewandrowski, U.; Sickmann, A.; Guiard, B.; van der Laan, M.; Warscheid, B.; Wiedemann, N. Composition and Topology of the Endoplasmic Reticulum–Mitochondria Encounter Structure. J. Mol. Biol. 2011, 413, 743–750. [Google Scholar] [CrossRef]
- Kopec, K.O.; Alva, V.; Lupas, A.N. Homology of SMP Domains to the TULIP Superfamily of Lipid-Binding Proteins Provides a Structural Basis for Lipid Exchange between ER and Mitochondria. Bioinformatics 2010, 26, 1927–1931. [Google Scholar] [CrossRef] [PubMed]
- Kawano, S.; Tamura, Y.; Kojima, R.; Bala, S.; Asai, E.; Michel, A.H.; Kornmann, B.; Riezman, I.; Riezman, H.; Sakae, Y.; et al. Structure–Function Insights into Direct Lipid Transfer between Membranes by Mmm1–Mdm12 of ERMES. J. Cell Biol. 2018, 217, 959–974. [Google Scholar] [CrossRef] [PubMed]
- Kojima, R.; Endo, T.; Tamura, Y. A Phospholipid Transfer Function of ER-Mitochondria Encounter Structure Revealed in Vitro. Sci. Rep. 2016, 6, 30777. [Google Scholar] [CrossRef] [PubMed]
- Berger, K.H.; Sogo, L.F.; Yaffe, M.P. Mdm12p, a Component Required for Mitochondrial Inheritance That Is Conserved between Budding and Fission Yeast. J. Cell Biol. 1997, 136, 545–553. [Google Scholar] [CrossRef]
- Burgess, S.M.; Delannoy, M.; Jensen, R.E. MMM1 Encodes a Mitochondrial Outer Membrane Protein Essential for Establishing and Maintaining the Structure of Yeast Mitochondria. J. Cell Biol. 1994, 126, 1375–1391. [Google Scholar] [CrossRef]
- Sogo, L.F.; Yaffe, M.P. Regulation of Mitochondrial Morphology and Inheritance by Mdm10p, a Protein of the Mitochondrial Outer Membrane. J. Cell Biol. 1994, 126, 1361–1373. [Google Scholar] [CrossRef] [PubMed]
- Youngman, M.J.; Hobbs, A.E.A.; Burgess, S.M.; Srinivasan, M.; Jensen, R.E. Mmm2p, a Mitochondrial Outer Membrane Protein Required for Yeast Mitochondrial Shape and Maintenance of mtDNA Nucleoids. J. Cell Biol. 2004, 164, 677–688. [Google Scholar] [CrossRef]
- Boldogh, I.R.; Nowakowski, D.W.; Yang, H.-C.; Chung, H.; Karmon, S.; Royes, P.; Pon, L.A. A Protein Complex Containing Mdm10p, Mdm12p, and Mmm1p Links Mitochondrial Membranes and DNA to the Cytoskeleton-Based Segregation Machinery. Mol. Biol. Cell 2003, 14, 4618–4627. [Google Scholar] [CrossRef] [PubMed]
- Böckler, S.; Westermann, B. Mitochondrial ER Contacts Are Crucial for Mitophagy in Yeast. Dev. Cell 2014, 28, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Murley, A.; Lackner, L.L.; Osman, C.; West, M.; Voeltz, G.K.; Walter, P.; Nunnari, J. ER-Associated Mitochondrial Division Links the Distribution of Mitochondria and Mitochondrial DNA in Yeast. eLife 2013, 2, e00422. [Google Scholar] [CrossRef] [PubMed]
- Koch, B.; Tucey, T.M.; Lo, T.L.; Novakovic, S.; Boag, P.; Traven, A. The Mitochondrial GTPase Gem1 Contributes to the Cell Wall Stress Response and Invasive Growth of Candida albicans. Front. Microbiol. 2017, 8, 2555. [Google Scholar] [CrossRef] [PubMed]
- Tucey, T.M.; Verma-Gaur, J.; Nguyen, J.; Hewitt, V.L.; Lo, T.L.; Shingu-Vazquez, M.; Robertson, A.A.B.; Hill, J.R.; Pettolino, F.A.; Beddoe, T.; et al. The Endoplasmic Reticulum-Mitochondrion Tether ERMES Orchestrates Fungal Immune Evasion, Illuminating Inflammasome Responses to Hyphal Signals. Msphere 2016, 1, e00074-16. [Google Scholar] [CrossRef]
- Geißel, B.; Penka, M.; Neubauer, M.; Wagener, J. The ER-Mitochondria Encounter Structure Contributes to Hyphal Growth, Mitochondrial Morphology and Virulence of the Pathogenic Mold Aspergillus fumigatus. Int. J. Med. Microbiol. 2017, 307, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Yang, G.; Li, C.; Yang, F.; Chang, X. Requirement of a Putative Mitochondrial GTPase, GemA, for Azole Susceptibility, Virulence, and Cell Wall Integrity in Aspergillus fumigatus. Front. Microbiol. 2022, 13, 67. [Google Scholar] [CrossRef]
- Ueno, K.; Matsumoto, Y.; Uno, J.; Sasamoto, K.; Sekimizu, K.; Kinjo, Y.; Chibana, H. Intestinal Resident Yeast Candida glabrata Requires Cyb2p-Mediated Lactate Assimilation to Adapt in Mouse Intestine. PLoS ONE 2011, 9, e86980. [Google Scholar] [CrossRef] [PubMed]
- Vu, B.G.; Moye-Rowley, W.S. Construction and Use of a Recyclable Marker to Examine the Role of Major Facilitator Superfamily Protein Members in Candida glabrata Drug Resistance Phenotypes. Msphere 2018, 3, e00099-18. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, M.; Takahashi-Nakaguchi, A.; Tejima, K.; Sasamoto, K.; Yamaguchi, M.; Aoyama, T.; Nagi, M.; Tanabe, K.; Miyazaki, Y.; Nakayama, H.; et al. Erg25 Controls Host-Cholesterol Uptake Mediated by Aus1p-Associated Sterol-Rich Membrane Domains in Candida glabrata. Front. Cell Dev. Biol. 2022, 10, 820675. [Google Scholar] [CrossRef]
- Zordan, R.E.; Ren, Y.; Pan, S.; Rotondo, G.; las Peñas, A.D.; Iluore, J.; Cormack, B.P. Expression Plasmids for Use in Candida glabrata. G3 Genes Genomes Genet. 2013, 3, 1675–1686. [Google Scholar] [CrossRef]
- Koshiba, T.; Holman, H.A.; Kubara, K.; Yasukawa, K.; Kawabata, S.I.; Okamoto, K.; Macfarlane, J.; Shaw, J.M. Structure-Function Analysis of the Yeast Mitochondrial Rho GTPase, Gem1p: Implications for Mitochondrial Inheritance. J. Biol. Chem. 2011, 286, 354–362. [Google Scholar] [CrossRef]
- Benhamou, R.I.; Bibi, M.; Steinbuch, K.B.; Engel, H.; Levin, M.; Roichman, Y.; Berman, J.; Fridman, M. Real-Time Imaging of the Azole Class of Antifungal Drugs in Live Candida Cells. ACS Chem. Biol. 2017, 12, 1769–1777. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER Tubules Mark Sites of Mitochondrial Division. Science 2011, 334, 358–362. [Google Scholar] [CrossRef]
- Hanekamp, T.; Thorsness, M.K.; Rebbapragada, I.; Fisher, E.M.; Seebart, C.; Darland, M.R.; Coxbill, J.A.; Updike, D.L.; Thorsness, P.E. Maintenance of Mitochondrial Morphology Is Linked to Maintenance of the Mitochondrial Genome in Saccharomyces cerevisiae. Genetics 2002, 162, 1147–1156. [Google Scholar] [CrossRef] [PubMed]
- Vu, B.G.; Thomas, G.H.; Moye-Rowley, W.S. Evidence That Ergosterol Biosynthesis Modulates Activity of the Pdr1 Transcription Factor in Candida glabrata. MBio 2019, 10, e00934-19. [Google Scholar] [CrossRef]
- Betinova, V.; Toth Hervay, N.; Elias, D.; Horvathova, A.; Gbelska, Y. The UPC2 Gene in Kluyveromyces lactis Stress Adaptation. Folia Microbiol. 2022, 67, 641–647. [Google Scholar] [CrossRef]
- Ban-Ishihara, R.; Ishihara, T.; Sasaki, N.; Mihara, K.; Ishihara, N. Dynamics of Nucleoid Structure Regulated by Mitochondrial Fission Contributes to Cristae Reformation and Release of Cytochrome c. Proc. Natl. Acad. Sci. USA 2013, 110, 11863–11868. [Google Scholar] [CrossRef]
- Kitada, K.; Yamaguchi, E.; Arisawa, M. Cloning of the Candida glabrata TRP1 and HIS3 Genes, and Construction of Their Disruptant Strains by Sequential Integrative Transformation. Gene 1995, 165, 203–206. [Google Scholar] [CrossRef]
- Ueno, K.; Uno, J.; Nakayama, H.; Sasamoto, K.; Mikami, Y.; Chibana, H. Development of a Highly Efficient Gene Targeting System Induced by Transient Repression of YKU80 Expression in Candida glabrata. Eukaryotic Cell 2007, 6, 1239–1247. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Okamoto, M.; Nakano, K.; Takahashi-Nakaguchi, A.; Sasamoto, K.; Yamaguchi, M.; Teixeira, M.C.; Chibana, H. In Candida glabrata, ERMES Component GEM1 Controls Mitochondrial Morphology, mtROS, and Drug Efflux Pump Expression, Resulting in Azole Susceptibility. J. Fungi 2023, 9, 240. https://doi.org/10.3390/jof9020240
Okamoto M, Nakano K, Takahashi-Nakaguchi A, Sasamoto K, Yamaguchi M, Teixeira MC, Chibana H. In Candida glabrata, ERMES Component GEM1 Controls Mitochondrial Morphology, mtROS, and Drug Efflux Pump Expression, Resulting in Azole Susceptibility. Journal of Fungi. 2023; 9(2):240. https://doi.org/10.3390/jof9020240
Chicago/Turabian StyleOkamoto, Michiyo, Keiko Nakano, Azusa Takahashi-Nakaguchi, Kaname Sasamoto, Masashi Yamaguchi, Miguel Cacho Teixeira, and Hiroji Chibana. 2023. "In Candida glabrata, ERMES Component GEM1 Controls Mitochondrial Morphology, mtROS, and Drug Efflux Pump Expression, Resulting in Azole Susceptibility" Journal of Fungi 9, no. 2: 240. https://doi.org/10.3390/jof9020240
APA StyleOkamoto, M., Nakano, K., Takahashi-Nakaguchi, A., Sasamoto, K., Yamaguchi, M., Teixeira, M. C., & Chibana, H. (2023). In Candida glabrata, ERMES Component GEM1 Controls Mitochondrial Morphology, mtROS, and Drug Efflux Pump Expression, Resulting in Azole Susceptibility. Journal of Fungi, 9(2), 240. https://doi.org/10.3390/jof9020240