
RNA-Seq-Based Transcriptome Analysis of Nitric Oxide Scavenging Response in Neurospora crassa




Abstract
1. Introduction
2. Materials and Methods
2.1. Fungus and Culture Condition
2.2. cPTIO (2-(4-carboxyphenyl)-4,5-dihydro-4,4,5,5-tetramethyl-1H-imidazolyl-1-oxy-3-oxide) Treatment and RNA Preparation for RNA Sequencing
2.3. RNA Sequencing
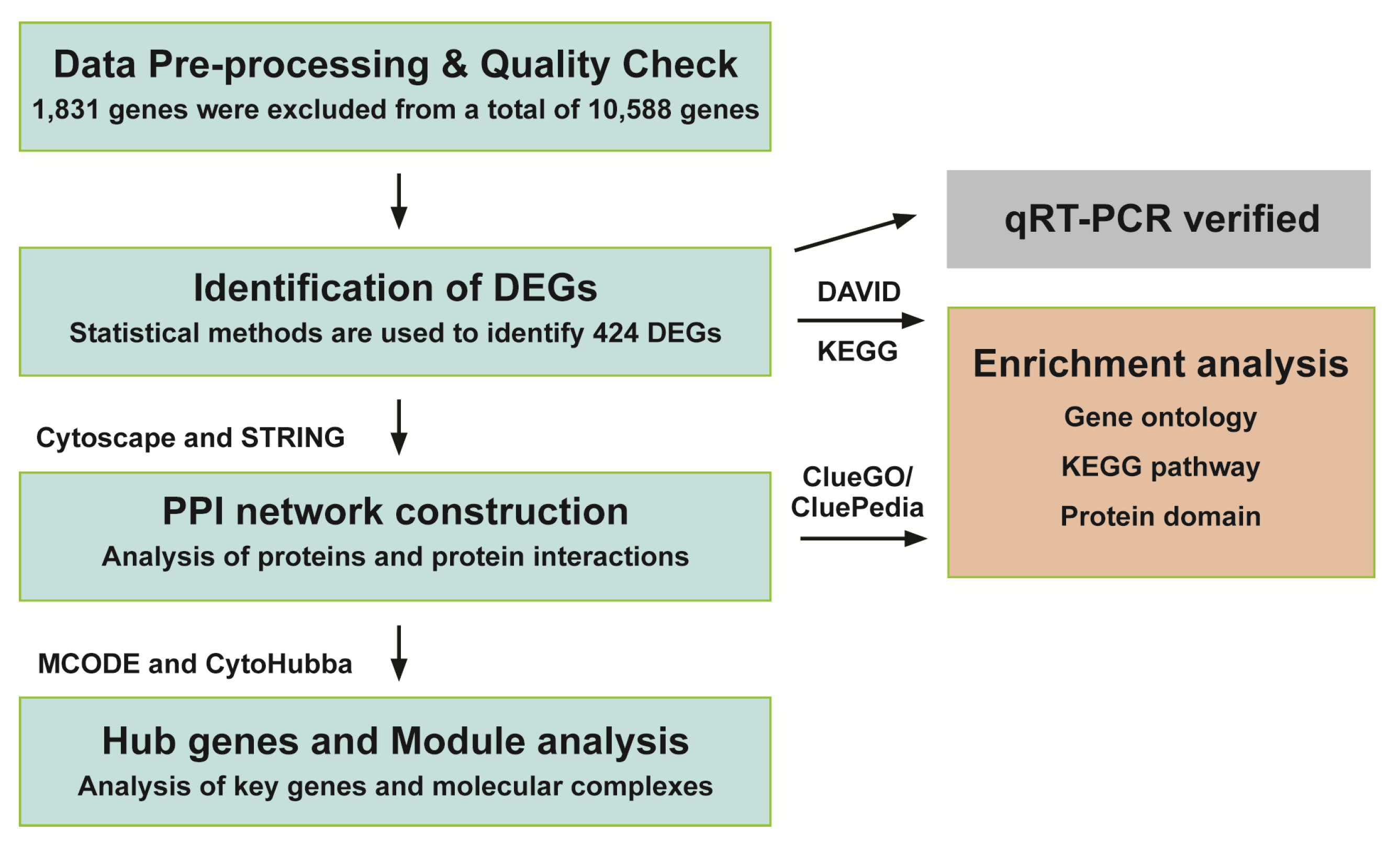
2.4. Data Pre-Processing and Quality Check
2.5. Screening and Functional Enrichment Analysis of DEGs (Differentially Expressed Genes)
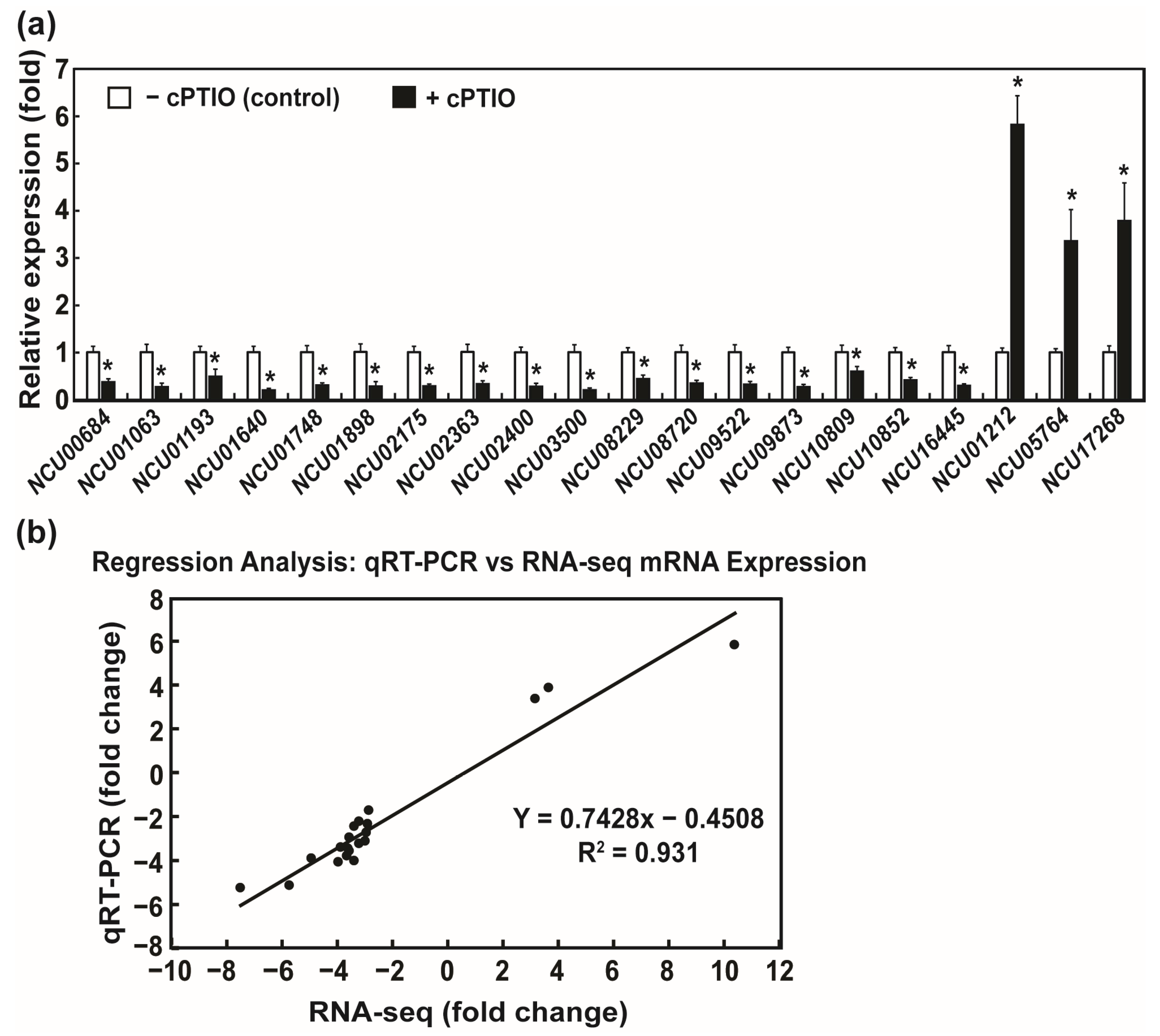
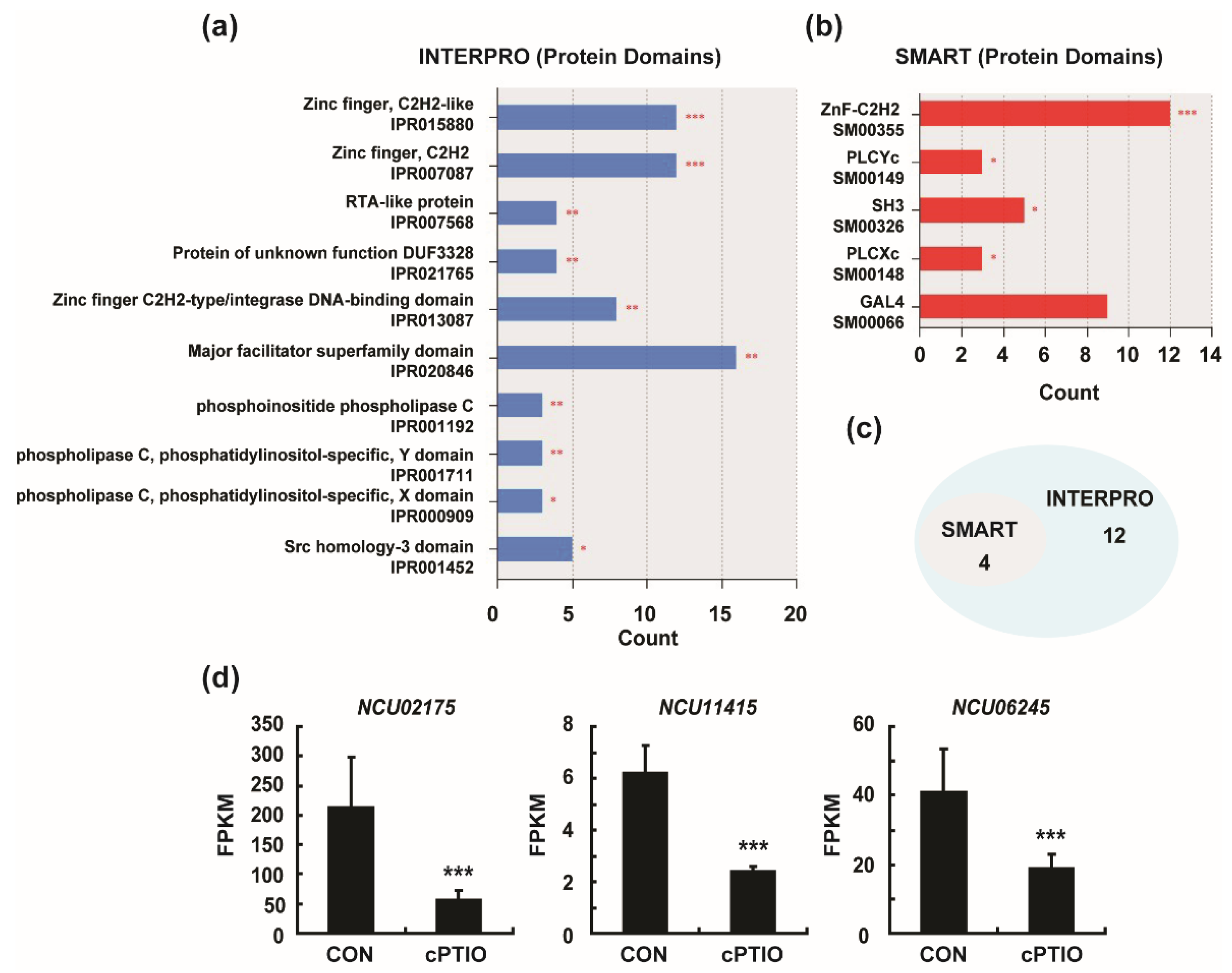
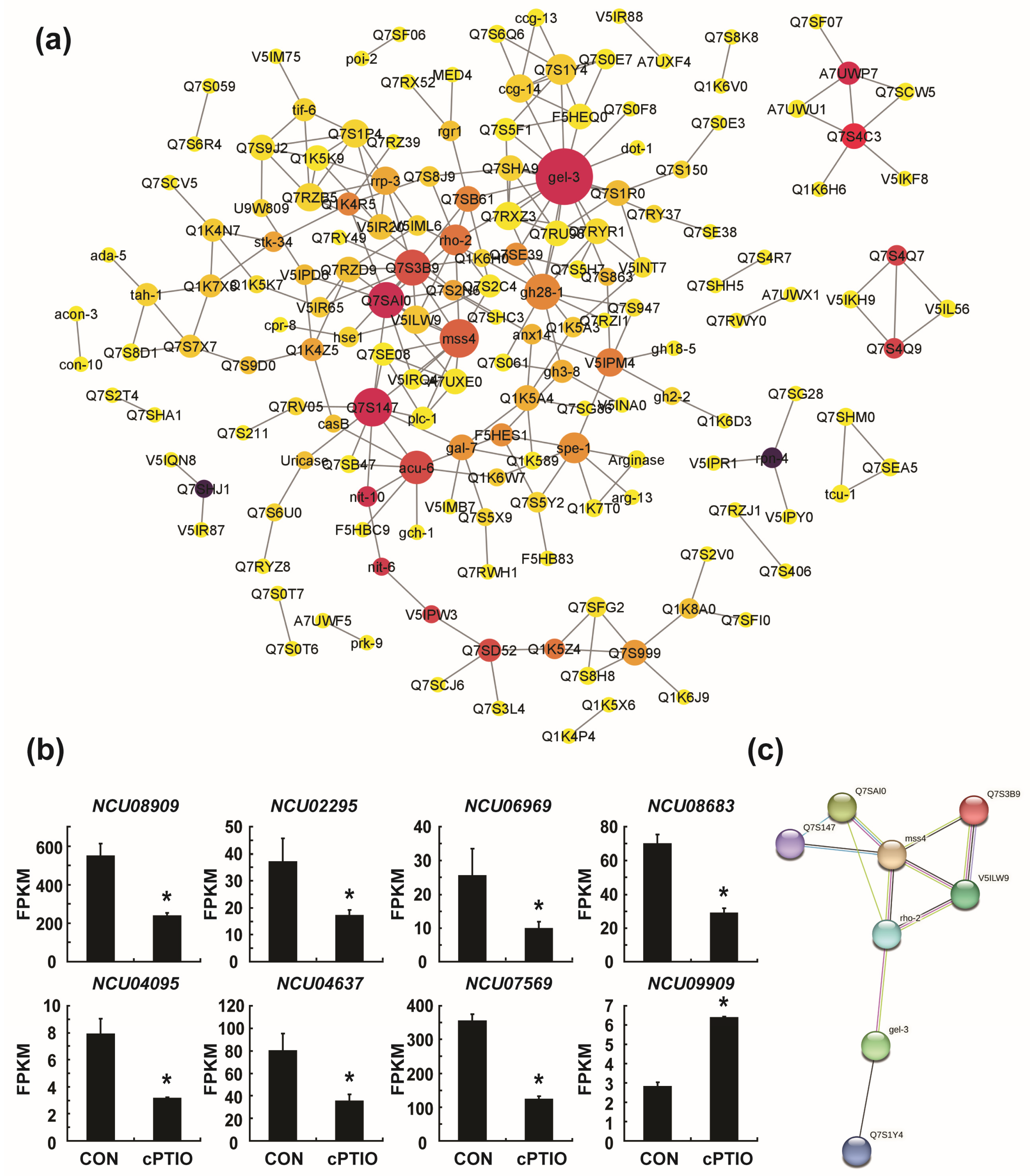
2.6. Validation of DEGs by Real-Time qRT-PCR (Quantitative Reverse Transcription Polymerase Chain Reaction)
2.7. PPI Network Construction and the Identification of Hub Genes
2.8. MCODE (Molecular Complex Detection) Analysis
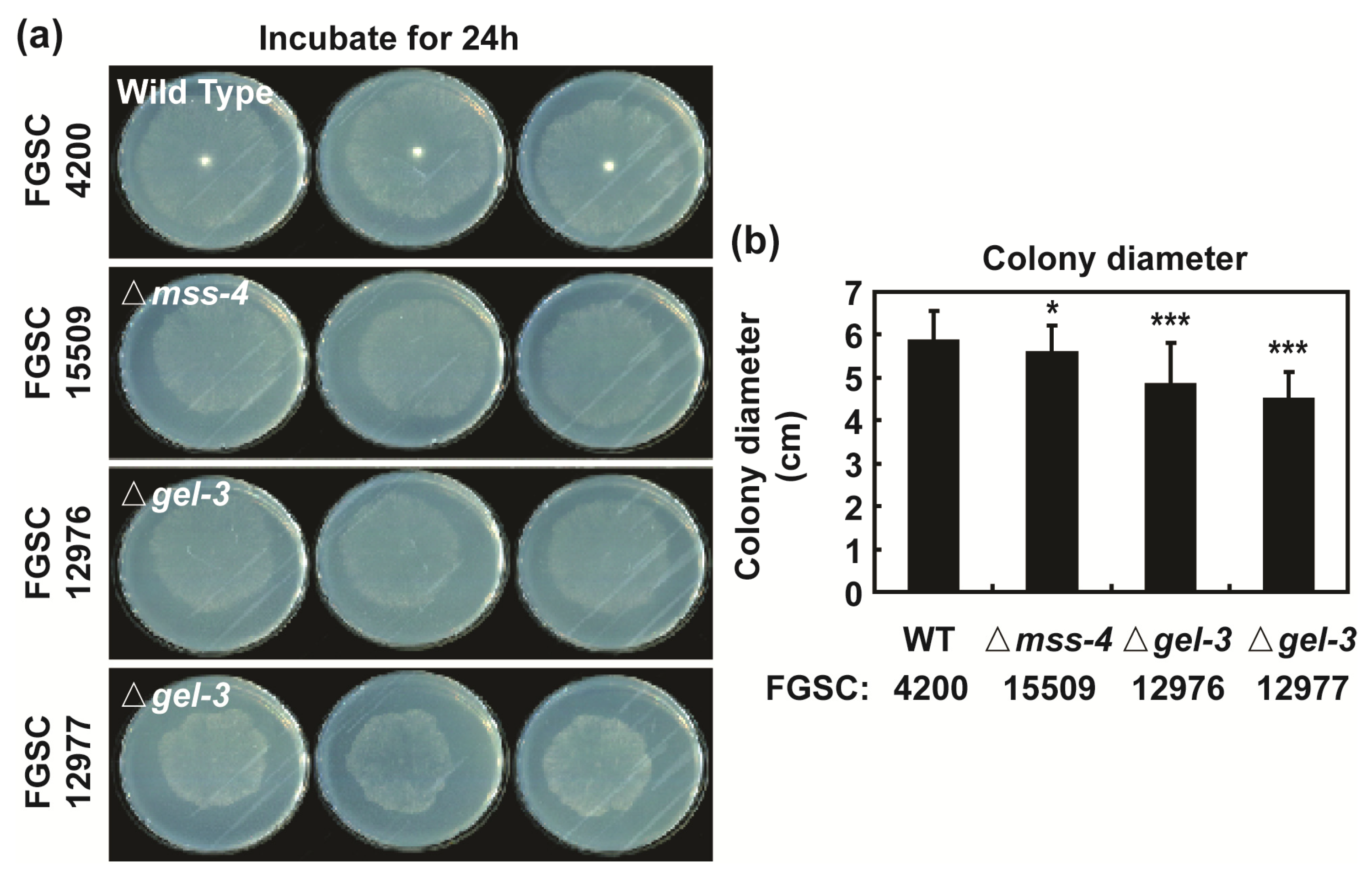
2.9. Phenotypic Analysis of Mutants
2.10. Statistical Analysis
3. Results
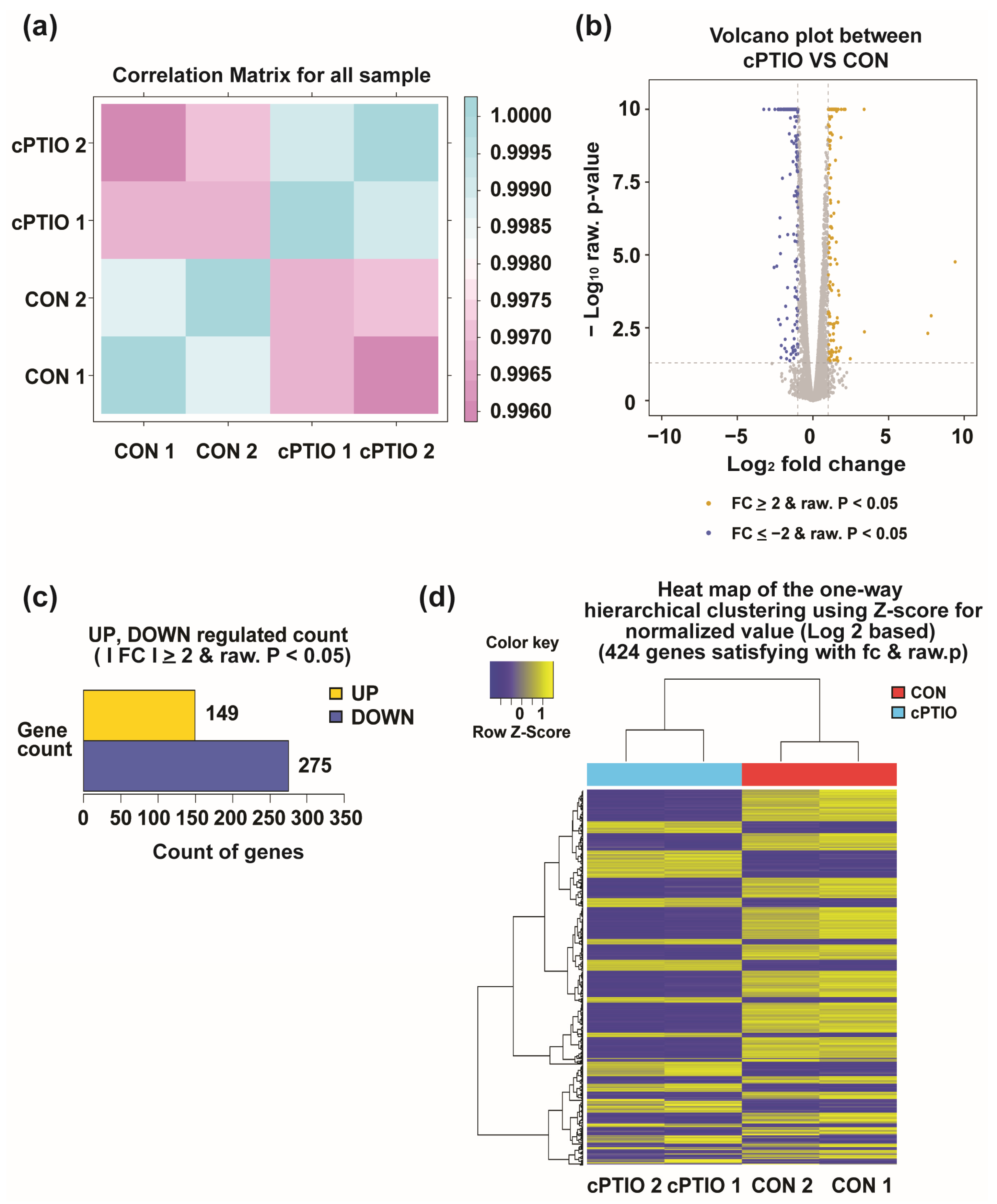
3.1. Identification of DEGs between cPTIO and CON (no cPTIO)
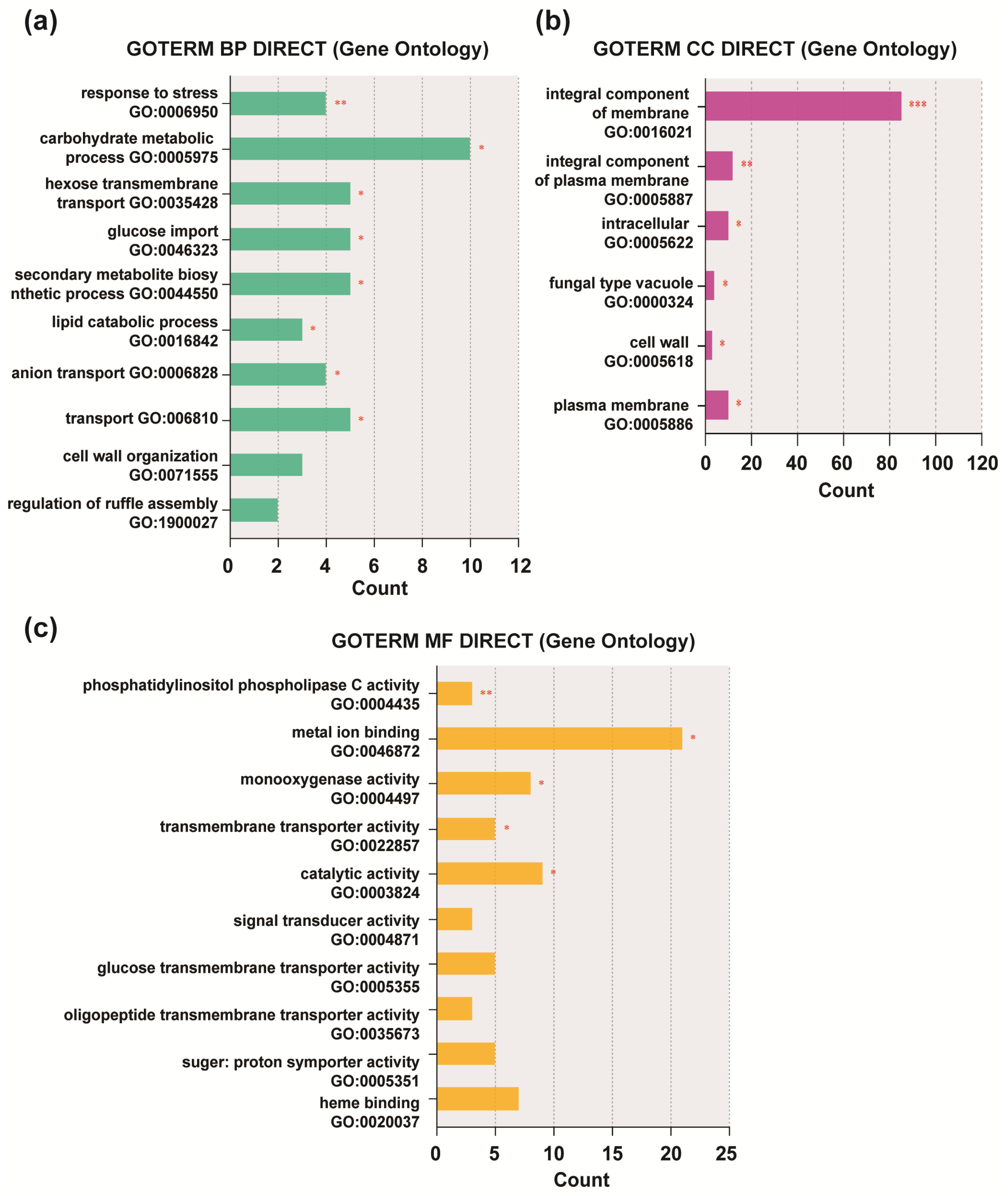
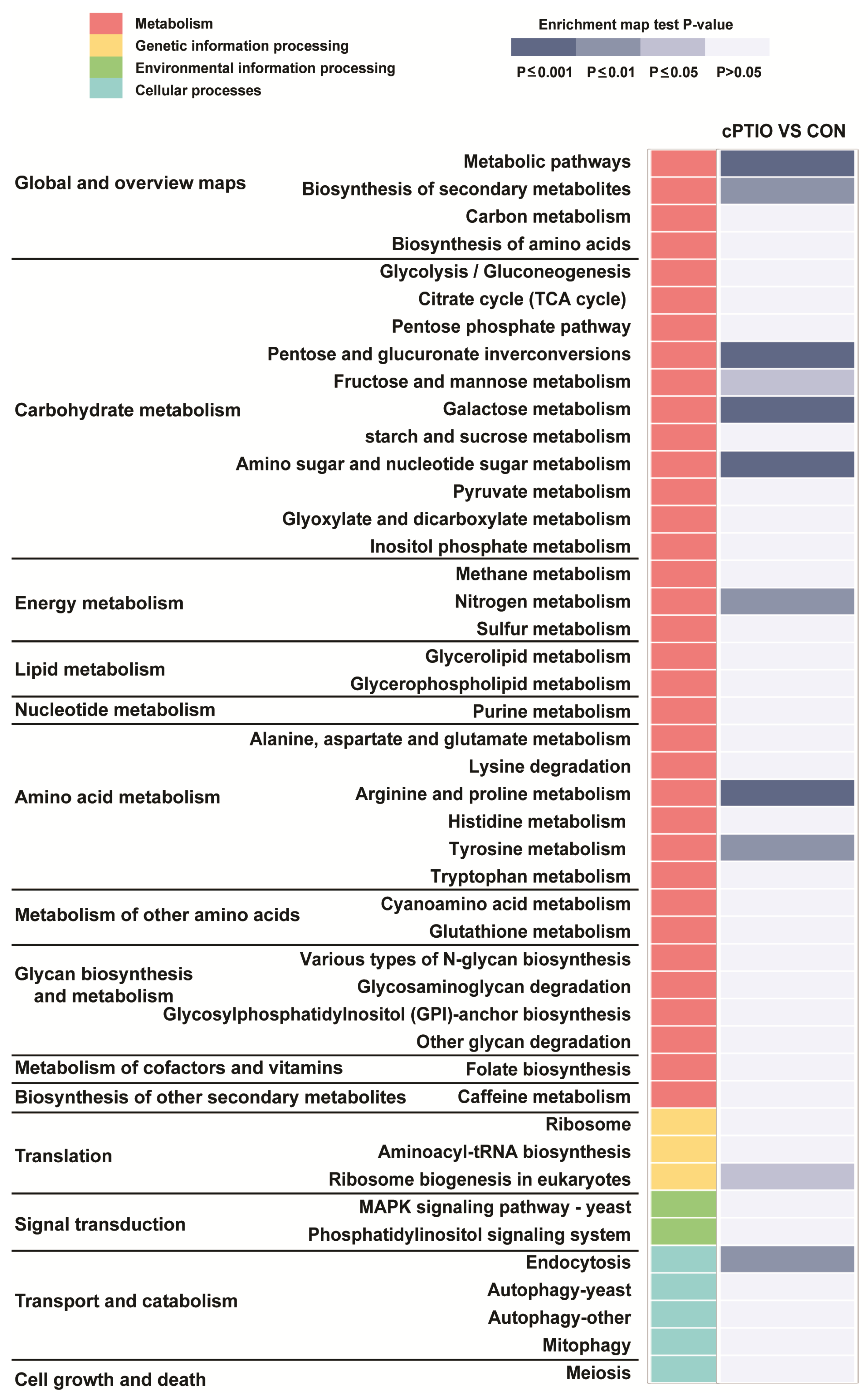
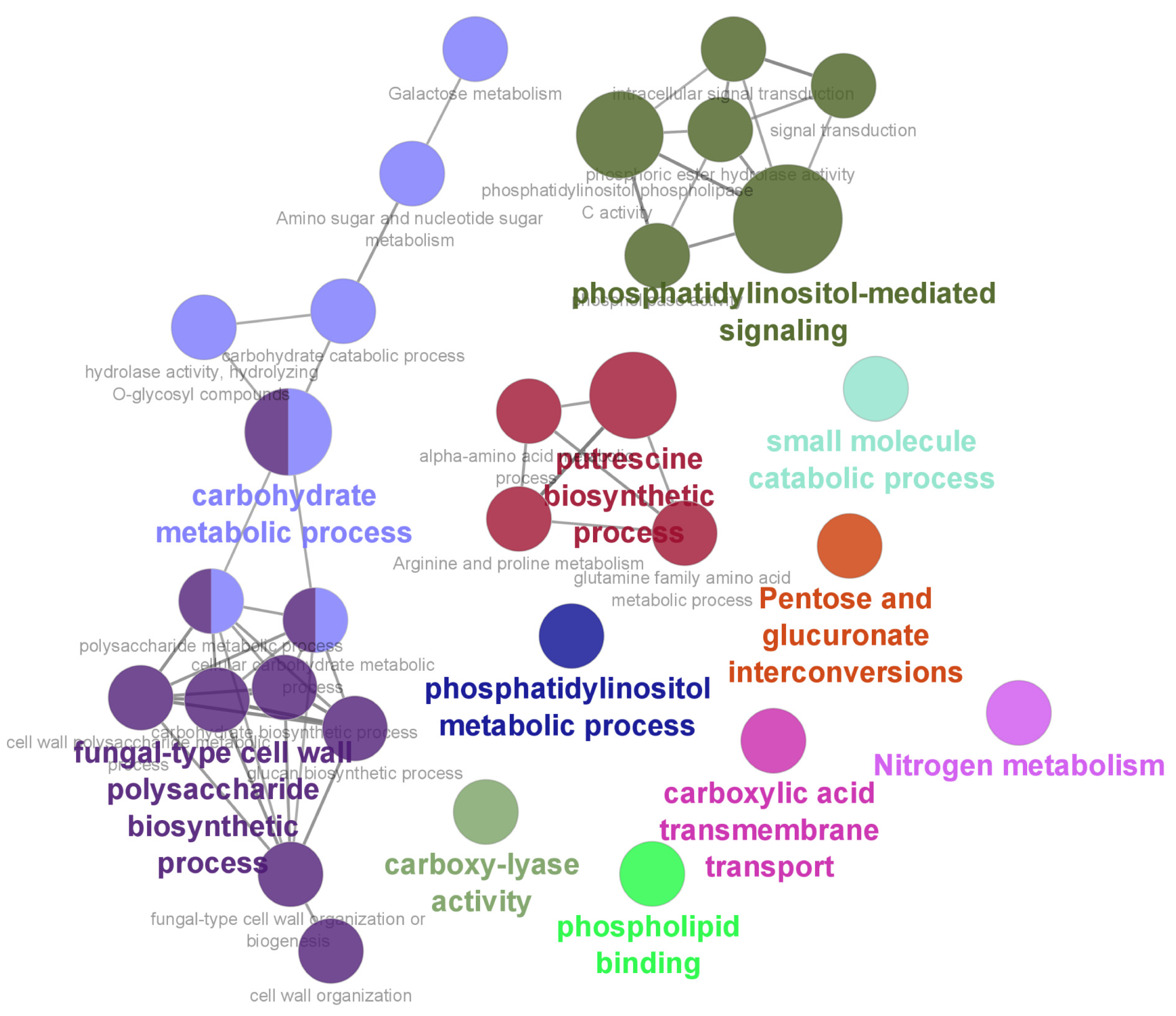
3.2. Functional Enrichment Analysis of DEGs
3.3. Enrichment of Four Protein Domains from DEGs
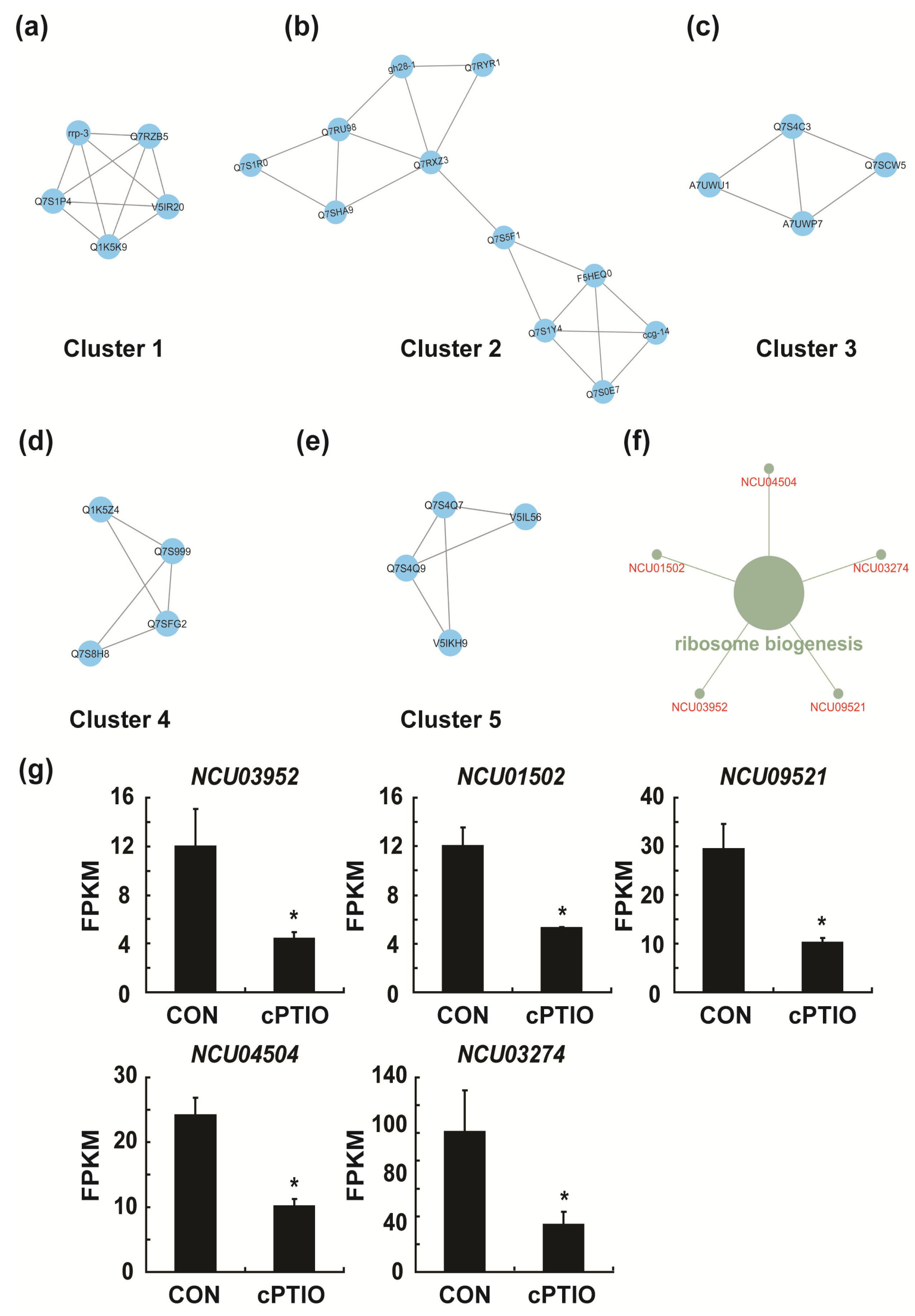
3.4. Identification of Key Genes in the PPI (Protein–Protein Interaction) Network
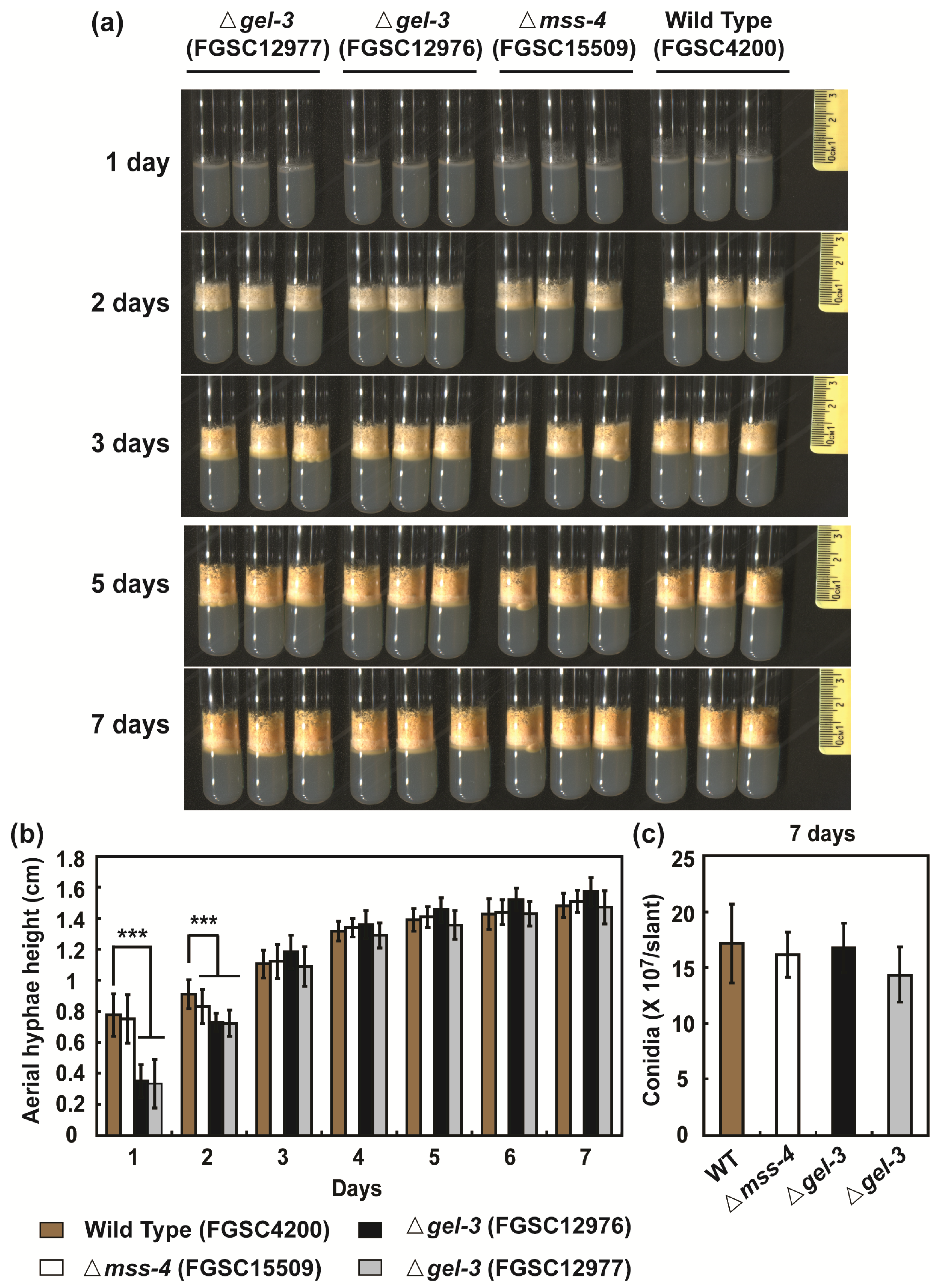
3.5. mss-4 (NCU02295) and gel-3 (NCU08909) Affect Hyphal Extension and Aerial Hyphae Development
3.6. GO and KEGG Analyses of the PPI Network
3.7. NO may Regulate Ribosome Biogenesis through a Molecular Complex
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Dempsey, S.K.; Daneva, Z.; Azam, M.; Li, N.; Li, P.L.; Ritter, J.K. Role of Nitric Oxide in the Cardiovascular and Renal Systems. Int. J. Mol. Sci. 2018, 19, 2605. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, P.; Tripathi, P.; Kashyap, L.; Singh, V. The role of nitric oxide in inflammatory reactions. FEMS Immunol. Med. Microbiol. 2007, 51, 443–452. [Google Scholar] [CrossRef]
- Picon-Pages, P.; Garcia-Buendia, J.; Munoz, F.J. Functions and dysfunctions of nitric oxide in brain. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1949–1967. [Google Scholar] [CrossRef] [PubMed]
- Planchet, E.; Kaiser, W.M. Nitric oxide production in plants: Facts and fictions. Plant Signal. Behav. 2006, 1, 46–51. [Google Scholar] [CrossRef]
- Crane, B.R.; Sudhamsu, J.; Patel, B.A. Bacterial nitric oxide synthases. Annu. Rev. Biochem. 2010, 79, 445–470. [Google Scholar] [CrossRef]
- Gusarov, I.; Nudler, E. NO-mediated cytoprotection: Instant adaptation to oxidative stress in bacteria. Proc. Natl. Acad. Sci. USA 2005, 102, 13855–13860. [Google Scholar] [CrossRef]
- Gusarov, I.; Shatalin, K.; Starodubtseva, M.; Nudler, E. Endogenous nitric oxide protects bacteria against a wide spectrum of antibiotics. Science 2009, 325, 1380–1384. [Google Scholar] [CrossRef]
- Patel, B.A.; Moreau, M.; Widom, J.; Chen, H.; Yin, L.; Hua, Y.; Crane, B.R. Endogenous nitric oxide regulates the recovery of the radiation-resistant bacterium Deinococcus radiodurans from exposure to UV light. Proc. Natl. Acad. Sci. USA 2009, 106, 18183–18188. [Google Scholar] [CrossRef]
- Buddha, M.R.; Tao, T.; Parry, R.J.; Crane, B.R. Regioselective nitration of tryptophan by a complex between bacterial nitric-oxide synthase and tryptophanyl-tRNA synthetase. J. Biol. Chem. 2004, 279, 49567–49570. [Google Scholar] [CrossRef] [PubMed]
- Cánovas, D.; Marcos, J.F.; Marcos, A.T.; Strauss, J. Nitric oxide in fungi: Is there NO light at the end of the tunnel? Curr. Genet. 2016, 62, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Lim, J.; Xu, J.; Yu, J.H.; Zheng, W. Nitric oxide as a developmental and metabolic signal in filamentous fungi. Mol. Microbiol. 2020, 113, 872–882. [Google Scholar] [CrossRef] [PubMed]
- Ninnemann, H.; Maier, J. Indications for the occurrence of nitric oxide synthases in fungi and plants and the involvement in photoconidiation of Neurospora crassa. Photochem. Photobiol. 1996, 64, 393–398. [Google Scholar] [CrossRef]
- Baidya, S.; Cary, J.W.; Grayburn, W.S.; Calvo, A.M. Role of nitric oxide and flavohemoglobin homolog genes in Aspergillus nidulans sexual development and mycotoxin production. Appl. Environ. Microbiol. 2011, 77, 5524–5528. [Google Scholar] [CrossRef]
- Samalova, M.; Johnson, J.; Illes, M.; Kelly, S.; Fricker, M.; Gurr, S. Nitric oxide generated by the rice blast fungus Magnaporthe oryzae drives plant infection. New Phytol. 2013, 197, 207–222. [Google Scholar] [CrossRef] [PubMed]
- Marcos, A.T.; Ramos, M.S.; Marcos, J.F.; Carmona, L.; Strauss, J.; Cánovas, D. Nitric oxide synthesis by nitrate reductase is regulated during development in Aspergillus. Mol. Microbiol. 2016, 99, 15–33. [Google Scholar] [CrossRef]
- Pengkit, A.; Jeon, S.S.; Son, S.J.; Shin, J.H.; Baik, K.Y.; Choi, E.H.; Park, G. Identification and functional analysis of endogenous nitric oxide in a filamentous fungus. Sci. Rep. 2016, 6, 30037. [Google Scholar] [CrossRef][Green Version]
- Yin, S.; Gao, Z.; Wang, C.; Huang, L.; Kang, Z.; Zhang, H. Nitric oxide and reactive oxygen species coordinately regulate the germination of Puccinia striiformis f. sp. tritici urediniospores. Front. Microbiol. 2016, 7, 178. [Google Scholar] [CrossRef]
- Marcos, A.T.; Ramos, M.S.; Schinko, T.; Strauss, J.; Cánovas, D. Nitric oxide homeostasis is required for light-dependent regulation of conidiation in Aspergillus. Fungal Genet. Biol. 2020, 137, 103337. [Google Scholar] [CrossRef]
- Anta-Fernández, F.; Santander-Gordón, D.; Becerra, S.; Santamaría, R.; Díaz-Mínguez, J.M.; Benito, E.P. Nitric Oxide Metabolism Affects Germination in Botrytis cinerea and Is Connected to Nitrate Assimilation. J. Fungi 2022, 8, 699. [Google Scholar] [CrossRef]
- Yu, N.-N.; Ketya, W.; Park, G. Intracellular Nitric Oxide and cAMP Are Involved in Cellulolytic Enzyme Production in Neurospora crassa. Int. J. Mol. Sci. 2023, 24, 4503. [Google Scholar] [CrossRef] [PubMed]
- Egbuta, M.A.; Mwanza, M.; Babalola, O.O. A review of the ubiquity of ascomycetes filamentous fungi in relation to their economic and medical importance. Adv. Microbiol. 2016, 6, 1140–1158. [Google Scholar] [CrossRef]
- Füting, P.; Barthel, L.; Cairns, T.C.; Briesen, H.; Schmideder, S. Filamentous fungal applications in biotechnology: A combined bibliometric and patentometric assessment. Fungal Biol. Biotechnol. 2021, 8, 23. [Google Scholar] [CrossRef] [PubMed]
- Adeleke, B.S.; Babalola, O.O. Pharmacological Potential of Fungal Endophytes Associated with Medicinal Plants: A Review. J. Fungi 2021, 7, 147. [Google Scholar] [CrossRef]
- Nutzmann, H.W.; Schroeckh, V.; Brakhage, A.A. Regulatory cross talk and microbial induction of fungal secondary metabolite gene clusters. Methods Enzymol. 2012, 517, 325–341. [Google Scholar] [CrossRef]
- Zhgun, A.A.; Eldarov, M.A. Polyamines Upregulate Cephalosporin C Production and Expression of β-Lactam Biosynthetic Genes in High-Yielding Acremonium chrysogenum Strain. Molecules 2021, 26, 6636. [Google Scholar] [CrossRef] [PubMed]
- Todero Ritter, C.E.; Camassola, M.; Zampieri, D.; Silveira, M.M.; Dillon, A.J. Cellulase and Xylanase Production by Penicillium echinulatum in Submerged Media Containing Cellulose Amended with Sorbitol. Enzym. Res. 2013, 2013, 240219. [Google Scholar] [CrossRef] [PubMed]
- Zaferanloo, B.; Bhattacharjee, S.; Ghorbani, M.M.; Mahon, P.J.; Palombo, E.A. Amylase production by Preussia minima, a fungus of endophytic origin: Optimization of fermentation conditions and analysis of fungal secretome by LC-MS. BMC Microbiol. 2014, 14, 55. [Google Scholar] [CrossRef]
- Chen, C.; Li, Q.; Wang, Q.F.; Lu, D.H.; Zhang, H.; Wang, J.; Fu, R.T. Transcriptional profiling provides new insights into the role of nitric oxide in enhancing Ganoderma oregonense resistance to heat stress. Sci. Rep. 2017, 7, 15694. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Huang, W.L.; Xiong, C.; Zhao, J. Transcriptome analysis reveals the role of nitric oxide in Pleurotus eryngii responses to Cd2+ stress. Chemosphere 2018, 201, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.J.; Li, X.P.; Wang, Y.; Wang, J.W. Nitric oxide donor sodium nitroprusside-induced transcriptional changes and hypocrellin biosynthesis of Shiraia sp. S9. Microb. Cell Factories 2021, 20, 92. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kwon, J.; Kim, D.; Park, M.; Kim, K.; Bae, I.; Kim, H.; Kong, J.; Kim, Y.; Shin, U. Gene expression profiles associated with radio-responsiveness in locally advanced rectal cancer. Biology 2021, 10, 500. [Google Scholar] [CrossRef]
- Yu, N.-N.; Ketya, W.; Choi, E.-H.; Park, G. Plasma Promotes Fungal Cellulase Production by Regulating the Levels of Intracellular NO and Ca2+. Int. J. Mol. Sci. 2022, 23, 6668. [Google Scholar] [CrossRef] [PubMed]
- Raman, K. Construction and analysis of protein-protein interaction networks. Autom. Exp. 2010, 2, 2. [Google Scholar] [CrossRef] [PubMed]
- Talebi, R.; Ahmadi, A.; Afraz, F. Analysis of protein-protein interaction network based on transcriptome profiling of ovine granulosa cells identifies candidate genes in cyclic recruitment of ovarian follicles. J. Anim. Sci. Technol. 2018, 60, 11. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Udhaya Kumar, S.; Thirumal Kumar, D.; Bithia, R.; Sankar, S.; Magesh, R.; Sidenna, M.; George Priya Doss, C.; Zayed, H. Analysis of differentially expressed genes and molecular pathways in familial hypercholesterolemia involved in atherosclerosis: A systematic and bioinformatics approach. Front. Bioeng. Biotechnol. 2020, 11, 734. [Google Scholar] [CrossRef]
- Chin, C.-H.; Chen, S.-H.; Wu, H.-H.; Ho, C.-W.; Ko, M.-T.; Lin, C.-Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8, S11. [Google Scholar] [CrossRef]
- Bader, G.D.; Hogue, C.W. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 2003, 4, 2. [Google Scholar] [CrossRef]
- Chen, J.; Chen, L.; Hua, J.; Song, W. Long-term dynamic compression enhancement TGF-β3-induced chondrogenesis in bovine stem cells: A gene expression analysis. BMC Genom. Data 2021, 22, 13. [Google Scholar] [CrossRef]
- Spirin, V.; Mirny, L.A. Protein complexes and functional modules in molecular networks. Proc. Natl. Acad. Sci. USA 2003, 100, 12123–12128. [Google Scholar] [CrossRef]
- Spies, D.; Ciaudo, C. Dynamics in transcriptomics: Advancements in RNA-seq time course and downstream analysis. Comput. Struct. Biotechnol. J. 2015, 13, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Gene Ontology Consortium. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004, 32, D258–D261. [Google Scholar] [CrossRef]
- Itoh, M.; Nacher, J.C.; Kuma, K.-i.; Goto, S.; Kanehisa, M. Evolutionary history and functional implications of protein domains and their combinations in eukaryotes. Genome Biol. 2007, 8, R121. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, H.; Zhong, H.; Xue, Z. Protein domain identification methods and online resources. Comput. Struct. Biotechnol. J. 2021, 19, 1145–1153. [Google Scholar] [CrossRef]
- Leon, O.; Roth, M. Zinc fingers: DNA binding and protein-protein interactions. Biol. Res. 2000, 33, 21–30. [Google Scholar] [CrossRef]
- Morton, C.; Campbell, I. SH3 domains: Molecular ‘velcro’. Curr. Biol. 1994, 4, 615–617. [Google Scholar] [CrossRef] [PubMed]
- Singer, W.D.; Brown, H.A.; Sternweis, P.C. Regulation of eukaryotic phosphatidylinositol-specific phospholipase C and phospholipase D. Annu. Rev. Biochem. 1997, 66, 475–509. [Google Scholar] [CrossRef]
- Seo, C.H.; Kim, J.-R.; Kim, M.-S.; Cho, K.-H. Hub genes with positive feedbacks function as master switches in developmental gene regulatory networks. Bioinformatics 2009, 25, 1898–1904. [Google Scholar] [CrossRef]
- He, X.; Zhang, J. Why do hubs tend to be essential in protein networks? PLoS Genet. 2006, 2, e88. [Google Scholar] [CrossRef]
- Park, G.; Pan, S.; Borkovich, K.A. Mitogen-activated protein kinase cascade required for regulation of development and secondary metabolism in Neurospora crassa. Eukaryot. Cell 2008, 7, 2113–2122. [Google Scholar] [CrossRef]
- Bindea, G.; Galon, J.; Mlecnik, B. CluePedia Cytoscape plugin: Pathway insights using integrated experimental and in silico data. Bioinformatics 2013, 29, 661–663. [Google Scholar] [CrossRef]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.-H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [PubMed]
- Dwane, S.; Kiely, P.A. Tools used to study how protein complexes are assembled in signaling cascades. Bioeng. Bugs 2011, 2, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Patyshakuliyeva, A.; Jurak, E.; Kohler, A.; Baker, A.; Battaglia, E.; De Bruijn, W.; Burton, K.S.; Challen, M.P.; Coutinho, P.M.; Eastwood, D.C. Carbohydrate utilization and metabolism is highly differentiated in Agaricus bisporus. BMC Genom. 2013, 14, 663. [Google Scholar] [CrossRef] [PubMed]
- Mähs, A.; Ischebeck, T.; Heilig, Y.; Stenzel, I.; Hempel, F.; Seiler, S.; Heilmann, I. The essential phosphoinositide kinase MSS-4 is required for polar hyphal morphogenesis, localizing to sites of growth and cell fusion in Neurospora crassa. PLoS ONE 2012, 7, e51454. [Google Scholar] [CrossRef]
- Kamei, M.; Yamashita, K.; Takahashi, M.; Fukumori, F.; Ichiishi, A.; Fujimura, M. Deletion and expression analysis of beta-(1, 3)-glucanosyltransferase genes in Neurospora crassa. Fungal Genet. Biol. 2013, 52, 65–72. [Google Scholar] [CrossRef]
- Woolford, J.L., Jr.; Baserga, S.J. Ribosome biogenesis in the yeast Saccharomyces cerevisiae. Genetics 2013, 195, 643–681. [Google Scholar] [CrossRef] [PubMed]
- Lew, R.R.; Giblon, R.E.; Lorenti, M.S. The phenotype of a phospholipase C (plc-1) mutant in a filamentous fungus, Neurospora crassa. Fungal Genet. Biol. 2015, 82, 158–167. [Google Scholar] [CrossRef]
- Dong, J.; Zhang, M.; Lu, L.; Sun, L.; Xu, M. Nitric oxide fumigation stimulates flavonoid and phenolic accumulation and enhances antioxidant activity of mushroom. Food Chem. 2012, 135, 1220–1225. [Google Scholar] [CrossRef]
- Khalil, Z.G.; Kalansuriya, P.; Capon, R.J. Lipopolysaccharide (LPS) stimulation of fungal secondary metabolism. Mycology 2014, 5, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Liao, X.-Z.; Wang, J.-X.; Ning, Y.-N.; Li, C.-X.; Liao, L.-S.; Liu, Q.; Jiang, Q.; Gu, L.-S.; Fu, L.-H. Transcription factor Atf1 regulates expression of cellulase and xylanase genes during solid-state fermentation of ascomycetes. Appl. Environ. Microbiol. 2019, 85, e01226-19. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Domain | InterPro Count p-Value | SMART Count p-Value | Gene ID |
|---|---|---|---|
| Zinc finger C2H2 a | 8 0.0026 | 12 0.0001 | NCU03878, NCU05022, NCU09333, NCU03552, NCU04848, NCU00694, NCU06186, NCU01629 |
| PLCYc b | 3 0.0098 | 3 0.0103 | NCU02175, NCU11415, NCU06245 |
| SH3 c | 5 0.0160 | 5 0.0129 | NCU03888, NCU01883, NCU04095, NCU04841, NCU04637 |
| PLCXc d | 3 0.0158 | 3 0.0168 | NCU02175, NCU11415, NCU06245 |
| Category a | MCC b | MNC c | Degree d | EPC e | Closeness | Radiality |
|---|---|---|---|---|---|---|
| Gene top 10 | NCU08909 a | NCU08909 | NCU08909 | NCU02295 | NCU08909 | NCU06969 |
| NCU09521 | NCU02295 | NCU09909 | NCU08683 | NCU06969 | NCU02295 | |
| NCU03952 | NCU07569 | NCU02295 | NCU06969 | NCU02295 | NCU07569 | |
| NCU01502 | NCU08683 | NCU04637 | NCU08909 | NCU09909 | NCU08683 | |
| NCU02295 | NCU04095 | NCU02369 | NCU04095 | NCU08683 | NCU06252 | |
| NCU04504 | NCU00451 | NCU06969 | NCU07569 | NCU04637 | NCU04637 | |
| NCU03274 | NCU09521 | NCU09873 | NCU02369 | NCU06252 | NCU08909 | |
| NCU07569 | NCU03952 | NCU08683 | NCU00451 | NCU02369 | NCU04095 | |
| NCU00451 | NCU07787 | NCU07569 | NCU04637 | NCU02137 | NCU02137 | |
| NCU03950 | NCU06969 | NCU01271 | NCU06252 | NCU04095 | NCU09873 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, N.-N.; Veerana, M.; Ketya, W.; Sun, H.-N.; Park, G. RNA-Seq-Based Transcriptome Analysis of Nitric Oxide Scavenging Response in Neurospora crassa. J. Fungi 2023, 9, 985. https://doi.org/10.3390/jof9100985
Yu N-N, Veerana M, Ketya W, Sun H-N, Park G. RNA-Seq-Based Transcriptome Analysis of Nitric Oxide Scavenging Response in Neurospora crassa. Journal of Fungi. 2023; 9(10):985. https://doi.org/10.3390/jof9100985
Chicago/Turabian StyleYu, Nan-Nan, Mayura Veerana, Wirinthip Ketya, Hu-Nan Sun, and Gyungsoon Park. 2023. "RNA-Seq-Based Transcriptome Analysis of Nitric Oxide Scavenging Response in Neurospora crassa" Journal of Fungi 9, no. 10: 985. https://doi.org/10.3390/jof9100985
APA StyleYu, N.-N., Veerana, M., Ketya, W., Sun, H.-N., & Park, G. (2023). RNA-Seq-Based Transcriptome Analysis of Nitric Oxide Scavenging Response in Neurospora crassa. Journal of Fungi, 9(10), 985. https://doi.org/10.3390/jof9100985