
Comparative Genomic Analysis and Metabolic Potential Profiling of a Novel Culinary-Medicinal Mushroom, Hericium rajendrae (Basidiomycota)


Abstract
1. Introduction
2. Materials and Methods
2.1. Fungal Strain and Strain Culture
2.2. Genome Sequencing, De Novo Assembly, and Annotation
2.2.1. Extraction of Genome DNA
2.2.2. Sequencing and De Novo Assembly
2.2.3. Gene Prediction and Annotation
2.3. Comparative Genomics Analysis
2.4. Transposon Element and the LTR-RT Analysis
2.5. Phylogenomic Analysis and Gene Family Variation Analysis
2.6. CAZy Family, Microsatellite, and Cytochrome P450 Analyses
2.7. Prediction and Cluster Analyses of Gene Clusters Involved in Secondary Metabolites
2.8. Metabolites Profiling of H. rajendrae NPCB A08
2.9. Data Availability
3. Results
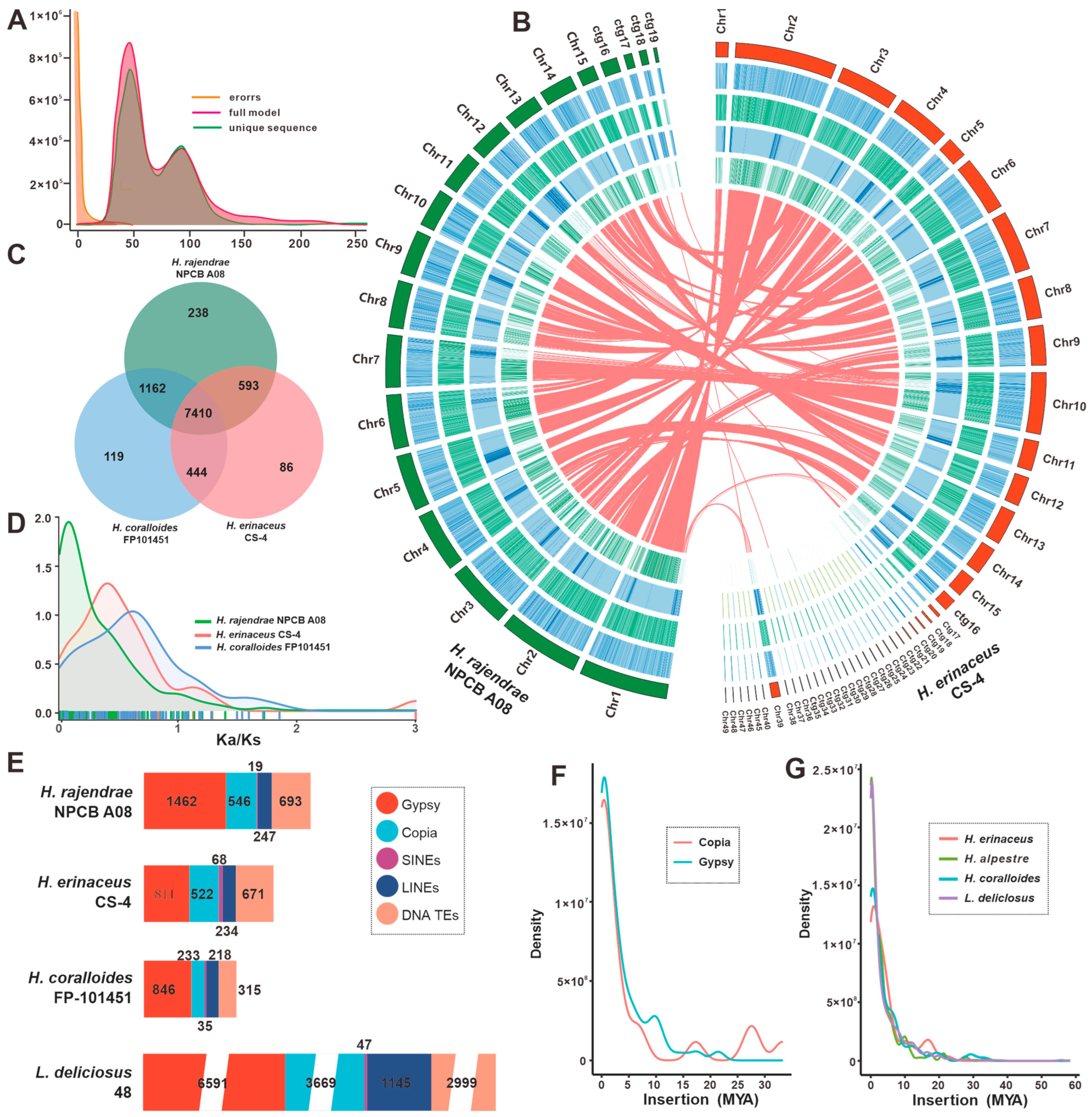
3.1. Genome Sequence Assembly and Annotation of H. rajendrae NPCB A08
3.2. Comparative Genomic Analysis within Hericium Species
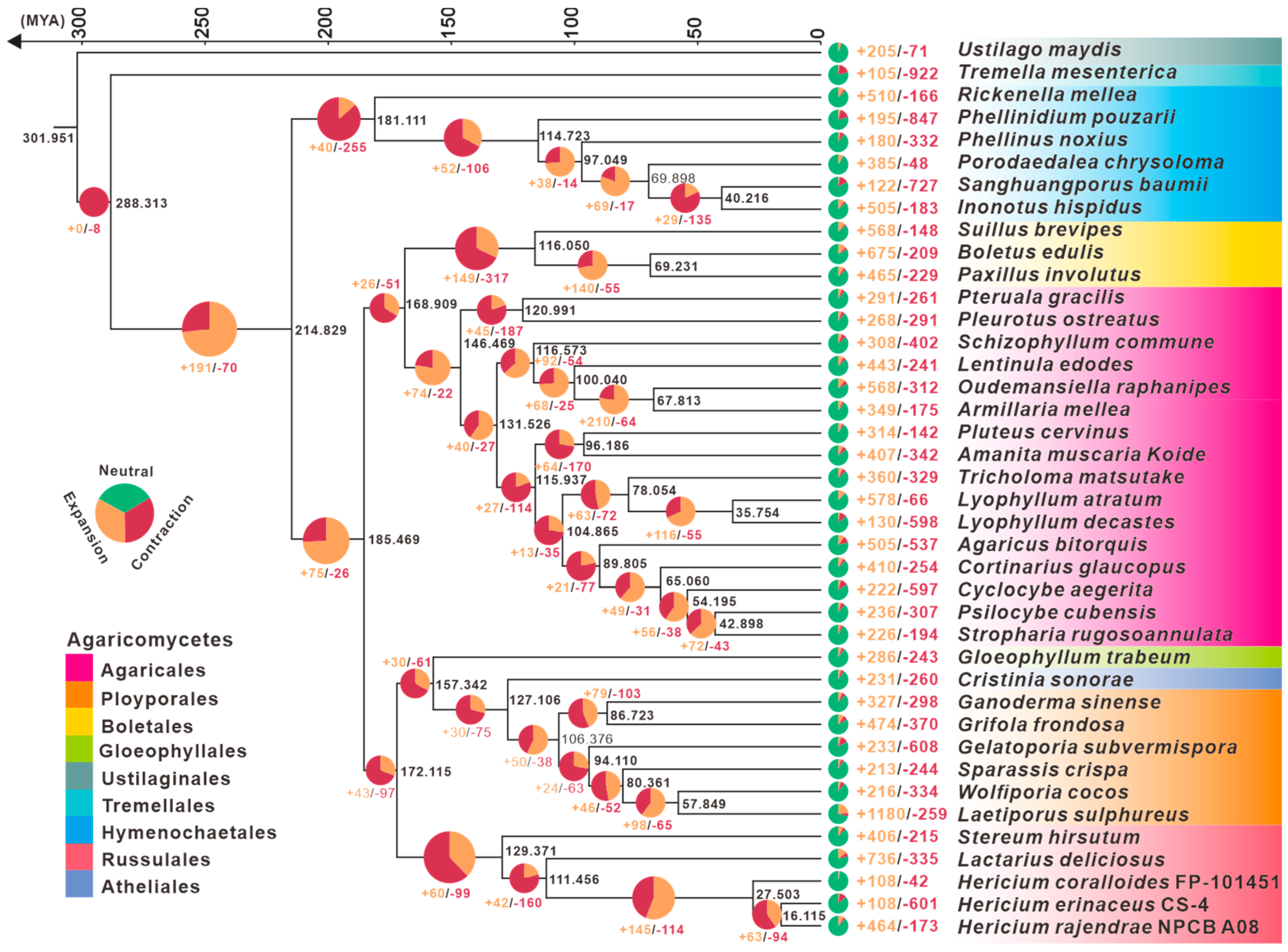
3.3. Phylogenetic and Gene Family Variation Analysis
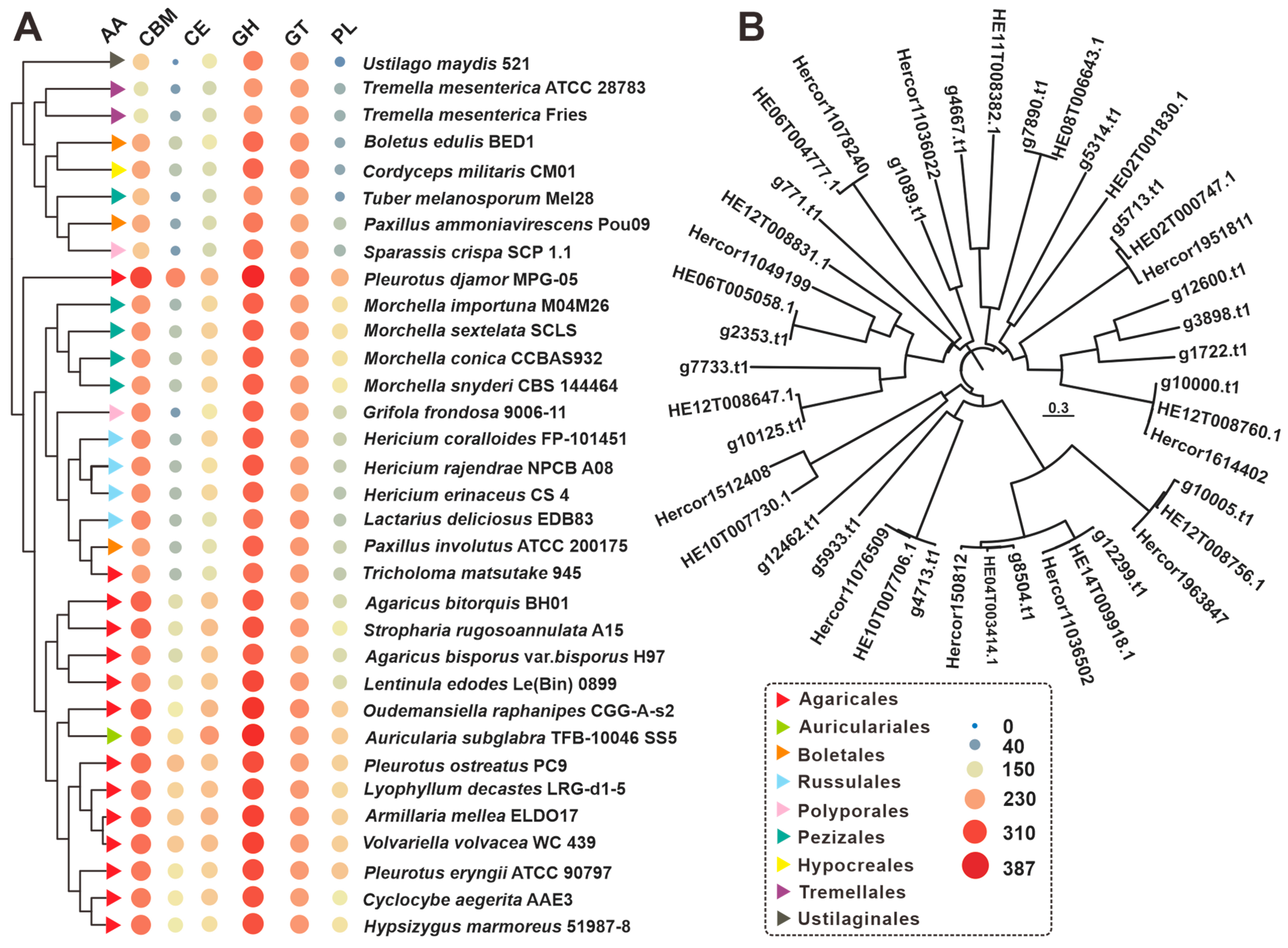
3.4. CAZymes Analysis
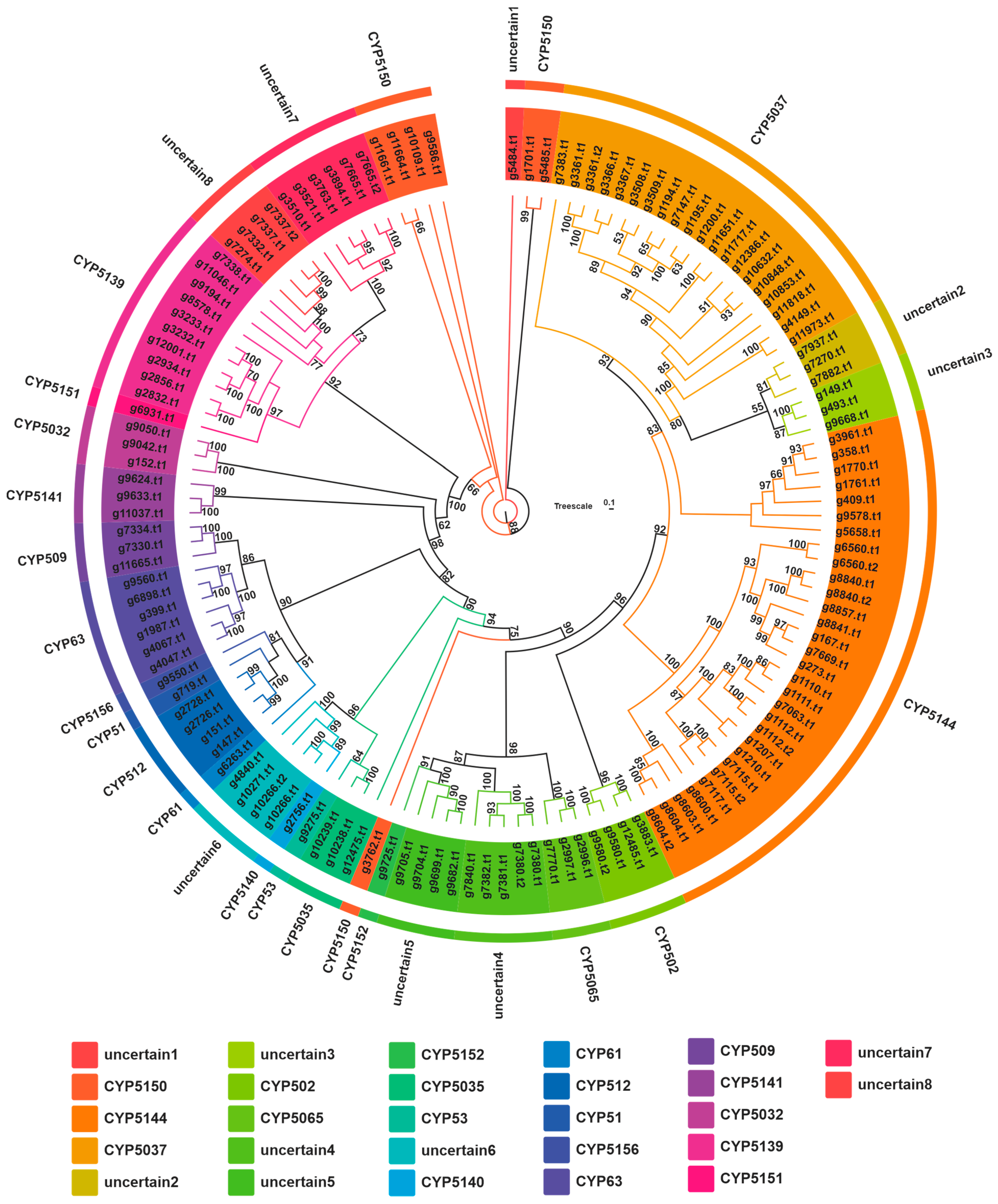
3.5. Cytochrome P450 Family Analysis
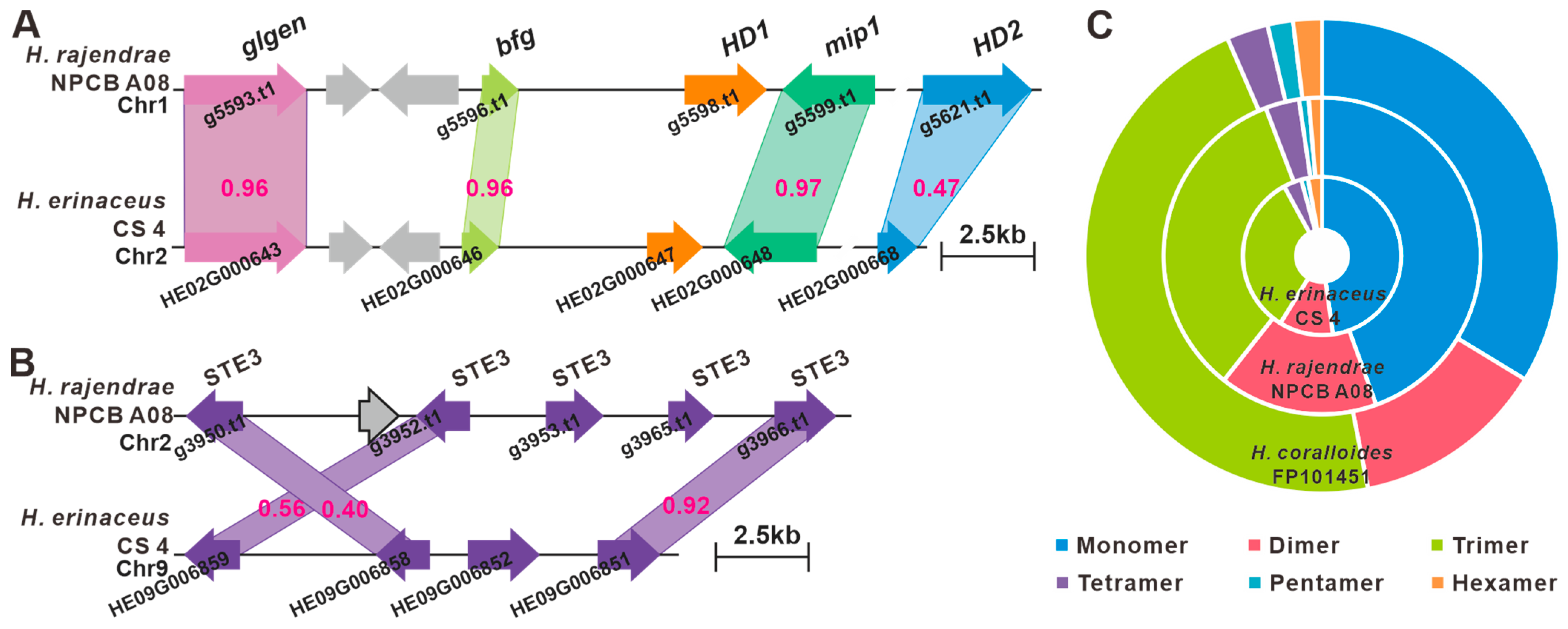
3.6. Identification of the Mating Genes and SSR Marker Development
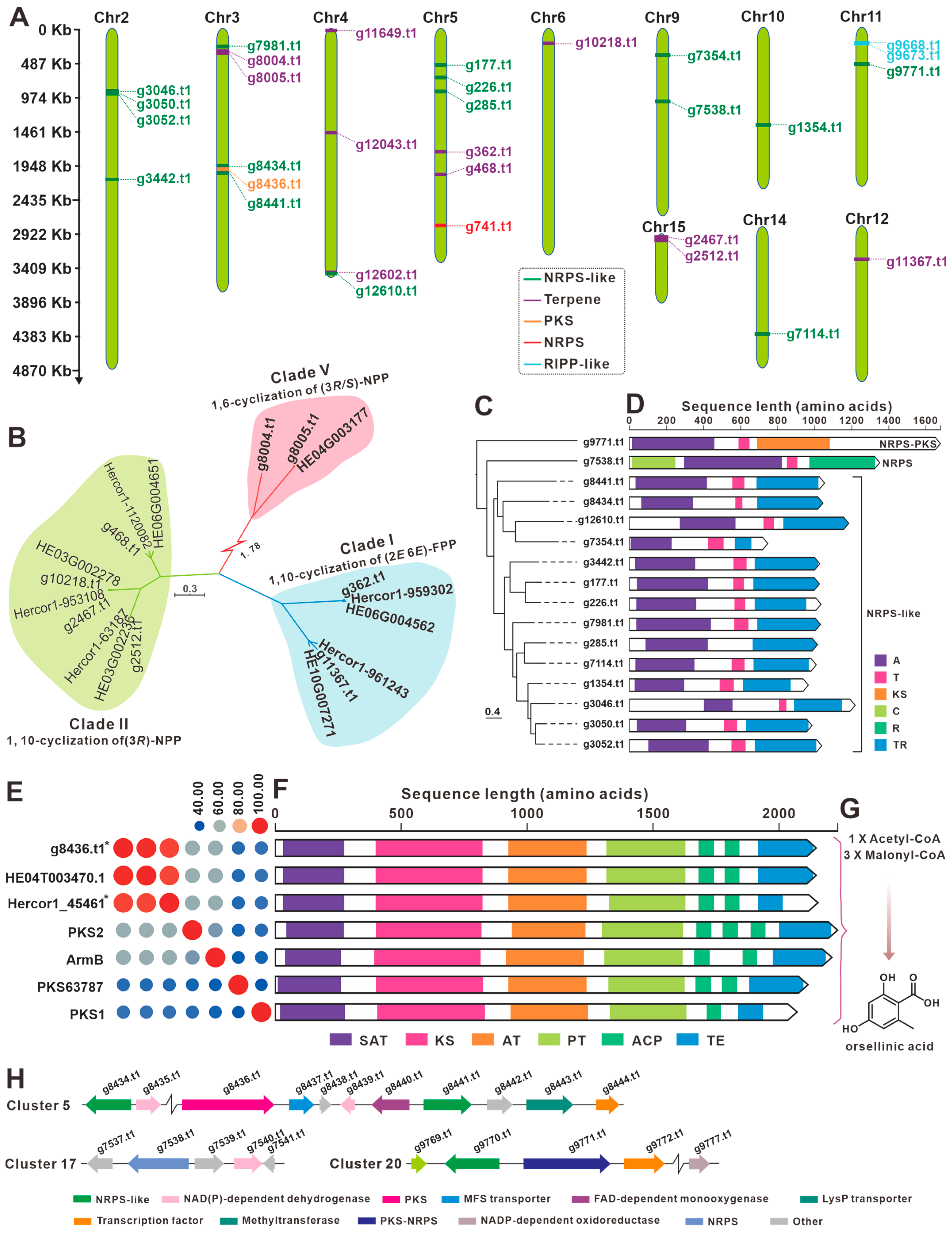
3.7. The BGCs for Secondary Metabolite Analysis
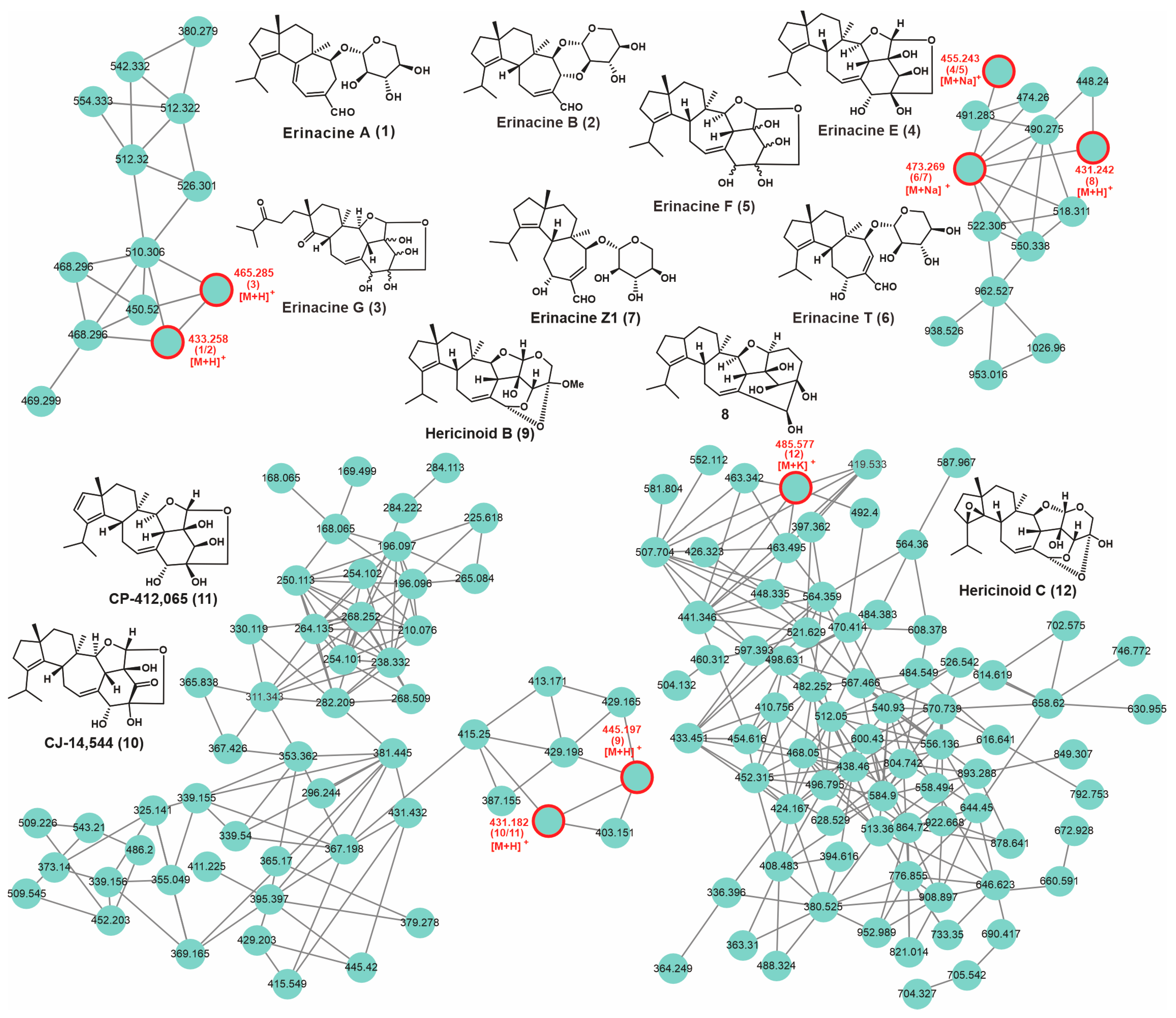
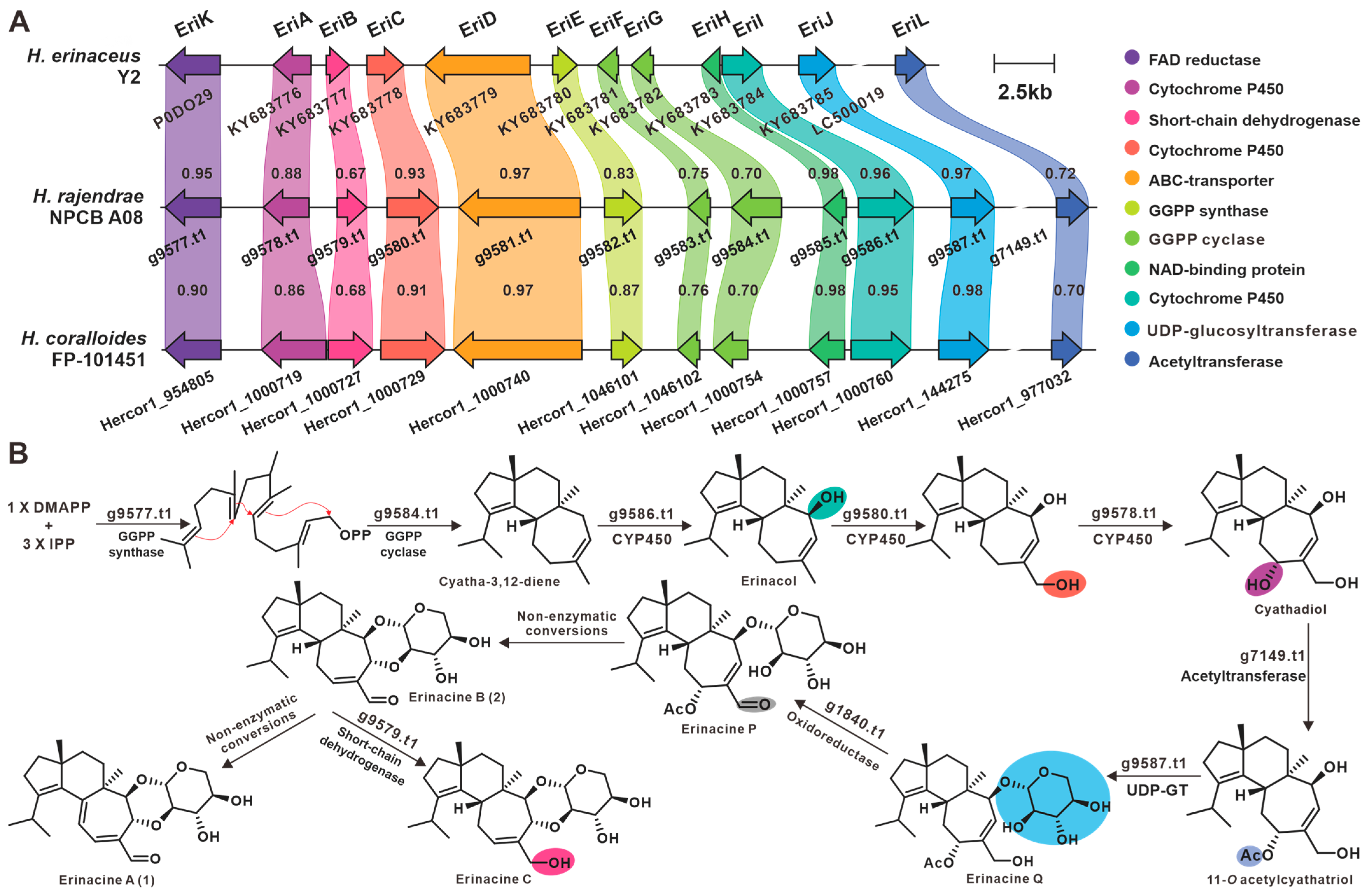
3.8. Secondary Metabolic Profiling and Putative Biosynthesis for Erinacines
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thongbai, B.; Rapior, S.; Hyde, K.D.; Wittstein, K.; Stadler, M. Hericium erinaceus, an amazing medicinal mushroom. Mycol. Prog. 2015, 14, 91. [Google Scholar] [CrossRef]
- Friedman, M. Chemistry, Nutrition, and Health-Promoting Properties of Hericium erinaceus (Lion’s Mane) Mushroom Fruiting Bodies and Mycelia and Their Bioactive Compounds. J. Agric. Food Chem. 2015, 63, 7108–7123. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Li, J.; Feng, X.; Zhang, Y.; Hu, X.; Hui, H.; Xue, X.; Qi, J. Unprecedented Neoverrucosane and Cyathane Diterpenoids with Anti-Neuroinflammatory Activity from Cultures of the Culinary-Medicinal Mushroom Hericium erinaceus. Molecules 2023, 28, 6380. [Google Scholar] [CrossRef]
- Bailly, C.; Gao, J.M. Erinacine A and related cyathane diterpenoids: Molecular diversity and mechanisms underlying their neuroprotection and anticancer activities. Pharmacol. Res. 2020, 159, 104953. [Google Scholar] [CrossRef] [PubMed]
- Rupcic, Z.; Rascher, M.; Kanaki, S.; Köster, R.W.; Stadler, M.; Wittstein, K. Two New Cyathane Diterpenoids from Mycelial Cultures of the Medicinal Mushroom Hericium erinaceus and the Rare Species, Hericium flagellum. Int. J. Mol. Sci. 2018, 19, 740. [Google Scholar] [CrossRef]
- Li, L.-N.; Wang, L.; Guo, X.-L. Chemical constituents from the culture of the fungus Hericium alpestre. J. Asian Nat. Prod. Res. 2019, 21, 735–741. [Google Scholar] [CrossRef]
- Li, L.-N.; Wang, L.; Cheng, Y.-N.; Cao, Z.-Q.; Zhang, X.-K.; Guo, X.-L. Discovery and Characterization of 4-Hydroxy-2-pyridone Derivative Sambutoxin as a Potent and Promising Anticancer Drug Candidate: Activity and Molecular Mechanism. Mol. Pharm. 2018, 15, 4898–4911. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Xu, J.; Liu, C.; Zhu, Y.; Nelson, D.R.; Zhou, S.; Li, C.; Wang, L.; Guo, X.; Sun, Y.; et al. Genome sequence of the model medicinal mushroom Ganoderma lucidum. Nat. Commun. 2012, 3, 913. [Google Scholar] [CrossRef]
- Huo, J.; Zhong, S.; Du, X.; Cao, Y.; Wang, W.; Sun, Y.; Tian, Y.; Zhu, J.; Chen, J.; Xuan, L.; et al. Whole-genome sequence of Phellinus gilvus (mulberry Sanghuang) reveals its unique medicinal values. J. Adv. Res. 2020, 24, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Gong, W.; Wang, Y.; Xie, C.; Zhou, Y.; Zhu, Z.; Peng, Y. Whole genome sequence of an edible and medicinal mushroom, Hericium erinaceus (Basidiomycota, Fungi). Genomics 2020, 112, 2393–2399. [Google Scholar] [CrossRef]
- Dong, W.-G.; Wang, Z.-X.; Feng, X.-L.; Zhang, R.-Q.; Shen, D.-Y.; Du, S.; Gao, J.-M.; Qi, J. Chromosome-Level Genome Sequences, Comparative Genomic Analyses, and Secondary-Metabolite Biosynthesis Evaluation of the Medicinal Edible Mushroom Laetiporus sulphureus. Microbiol. Spectr. 2022, 10, e0243922. [Google Scholar] [CrossRef]
- Zhang, R.-Q.; Feng, X.-L.; Wang, Z.-X.; Xie, T.-C.; Duan, Y.; Liu, C.; Gao, J.-M.; Qi, J. Genomic and Metabolomic Analyses of the Medicinal Fungus Inonotus hispidus for Its Metabolite & Biosynthesis and Medicinal Application. J. Fungi 2022, 8, 1245. [Google Scholar]
- Yu, H.; Zhang, M.; Sun, Y.; Li, Q.; Liu, J.; Song, C.; Shang, X.; Tan, Q.; Zhang, L.; Yu, H. Whole-genome sequence of a high-temperature edible mushroom Pleurotus giganteus (zhudugu). Front. Microbiol. 2022, 13, 941889. [Google Scholar] [CrossRef]
- Ling, Z.L.; Cao, B.; Hu, S.N.; Geng, J.N.; Liu, F.; Liu, D.M.; Zhao, R.L. Insights into the genomic evolution and the alkali tolerance mechanisms of Agaricus sinodeliciosus by comparative genomic and transcriptomic analyses. Microb. Genom. 2023, 9, mgen000928. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Feng, X.-l.; Wang, Z.-x.; Qi, J. The First Whole Genome Sequencing of Agaricus bitorquis and Its Metabolite Profiling. J. Fungi 2023, 9, 485. [Google Scholar] [CrossRef]
- Chen, J.; Zeng, X.; Yang, Y.L.; Xing, Y.M.; Zhang, Q.; Li, J.M.; Ma, K.; Liu, H.W.; Guo, S.X. Genomic and transcriptomic analyses reveal differential regulation of diverse terpenoid and polyketides secondary metabolites in Hericium erinaceus. Sci. Rep. 2017, 7, 10151. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Xu, L.; Li, J.; Chen, J.; Yang, M. Genome Sequencing of Hericium coralloides by a Combination of PacBio RS II and Next-Generation Sequencing Platforms. Int. J. Genom. 2022, 2022, 4017654. [Google Scholar] [CrossRef]
- JGI. Hericium Coralloides FP-101451 v1.0. Available online: https://genome.jgi.doe.gov/portal/Hercor1/Hercor1.download.html (accessed on 10 July 2023).
- Gilchrist, C.L.M.; Chooi, Y.-H. Synthaser: A CD-Search enabled Python toolkit for analysing domain architecture of fungal secondary metabolite megasynth(et)ases. Fun. Biol. Biotech. 2021, 8, 13. [Google Scholar] [CrossRef]
- Gilchrist, C.L.M.; Chooi, Y.-H. Clinker & clustermap.js: Automatic generation of gene cluster comparison figures. Bioinformatics 2021, 37, 2473–2475. [Google Scholar] [PubMed]
- Sista Kameshwar, A.K.; Qin, W. Comparative study of genome-wide plant biomass-degrading CAZymes in white rot, brown rot and soft rot fungi. Mycology 2018, 9, 93–105. [Google Scholar] [CrossRef]
- Rytioja, J.; Hildén, K.; Yuzon, J.; Hatakka, A.; Vries, R.P.d.; Mäkelä, M.R. Plant-Polysaccharide-Degrading Enzymes from Basidiomycetes. Microbiol. Mol. Biol. Rev. 2014, 78, 614–649. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.J.; Casselton, L.A. Mating in mushrooms: Increasing the chances but prolonging the affair. Trends Genet. 2001, 17, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Bao, D.-P. Research progress on the mating-typing locus structures of basidiomycete mushrooms. Mycosystema 2019, 38, 2061–2077. [Google Scholar]
- Han, H.; Yu, C.; Qi, J.; Wang, P.; Zhao, P.; Gong, W.; Xie, C.; Xia, X.; Liu, C. High-efficient production of mushroom polyketide compounds in a platform host Aspergillus oryzae. Microb. Cell Fact. 2023, 22, 60. [Google Scholar] [CrossRef] [PubMed]
- Kawagishi, H.; Shimada, A.; Shirai, R.; Okamoto, K.; Ojima, F.; Sakamoto, H.; Ishiguro, Y.; Furukawa, S. Erinacines A, B and C, strong stimulators of nerve growth factor (NGF)-synthesis, from the mycelia of Hericium erinaceum. Tetrahedron Lett. 1994, 35, 1569–1572. [Google Scholar] [CrossRef]
- Kawagishi, H.; Shimada, A.; Hosokawa, S.; Mori, H.; Sakamoto, H.; Ishiguro, Y.; Sakemi, S.; Bordner, J.; Kojima, N.; Furukawa, S. Erinacines E, F, and G, stimulators of nerve growth factor (NGF)-synthesis, from the mycelia of Hericium erinaceum. Tetrahedron Lett. 1996, 37, 7399–7402. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, L.; Bao, L.; Yang, Y.; Ma, K.; Liu, H. Three new cyathane diterpenes with neurotrophic activity from the liquid cultures of Hericium erinaceus. J. Antibiot. 2018, 71, 818–821. [Google Scholar] [CrossRef]
- Kawagishi, H.; Y Mori, Y.; Sakamoto, H.; Ishiguro, Y. Cyathane Derivative and Inducer for Nerve Growth Factor Production Containing the Same as Active Ingredient. JP Patent JPH09241921A, 1996. [Google Scholar]
- Chen, L.; Yao, J.-N.; Chen, H.-P.; Zhao, Z.-Z.; Li, Z.-H.; Feng, T.; Liu, J.-K. Hericinoids A–C, cyathane diterpenoids from culture of mushroom Hericium erinaceus. Phytochem. Lett. 2018, 27, 94–100. [Google Scholar] [CrossRef]
- Saito, T.; Aoki, F.; Hirai, H.; Inagaki, T.; Matsunaga, Y.; Sakakibara, T.; Sakemi, S.; Suzuki, Y.; Watanabe, S.; Suga, O.; et al. Erinacine E as a kappa opioid receptor agonist and its new analogs from a basidiomycete, Hericium ramosum. J. Antibiot. 1998, 51, 983–990. [Google Scholar] [CrossRef][Green Version]
- Liu, C.; Minami, A.; Ozaki, T.; Wu, J.; Kawagishi, H.; Maruyama, J.I.; Oikawa, H. Efficient Reconstitution of Basidiomycota Diterpene Erinacine Gene Cluster in Ascomycota Host Aspergillus oryzae Based on Genomic DNA Sequences. J. Am. Chem. Soc. 2019, 141, 15519–15523. [Google Scholar] [CrossRef]
- Yang, Y.-l.; Zhang, S.; Ma, K.; Xu, Y.; Tao, Q.; Chen, Y.; Chen, J.; Guo, S.; Ren, J.; Wang, W.; et al. Discovery and Characterization of a New Family of Diterpene Cyclases in Bacteria and Fungi. Angew. Chem. Int. Edit. 2017, 56, 4749–4752. [Google Scholar] [CrossRef]
- An, H.; Jo, I.H.; Oh, Y.L.; Jang, K.Y.; Kong, W.S.; Sung, J.K.; So, Y.S.; Chung, J.W. Molecular Characterization of 170 New gDNA-SSR Markers for Genetic Diversity in Button Mushroom (Agaricus bisporus). Mycobiology 2019, 47, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.; Lee, H.Y.; Shim, D.; Kim, M.; Ka, K.H.; Ryoo, R.; Ko, H.G.; Koo, C.D.; Chung, J.W.; Ryu, H. Development and Molecular Characterization of Novel Polymorphic Genomic DNA SSR Markers in Lentinula edodes. Mycobiology 2017, 45, 105–109. [Google Scholar] [CrossRef]
- Liu, X.B.; Li, J.; Yang, Z.L. Genetic diversity and structure of core collection of winter mushroom (Flammulina velutipes) developed by genomic SSR markers. Hereditas 2018, 155, 3. [Google Scholar] [CrossRef]
- Fu, Y.; Dai, Y.; Yang, C.; Wei, P.; Song, B.; Yang, Y.; Sun, L.; Zhang, Z.W.; Li, Y. Comparative Transcriptome Analysis Identified Candidate Genes Related to Bailinggu Mushroom Formation and Genetic Markers for Genetic Analyses and Breeding. Sci. Rep. 2017, 7, 9266. [Google Scholar] [CrossRef] [PubMed]
- Angulo-Sanchez, L.T.; López-Peña, D.; Torres-Moreno, H.; Gutiérrez, A.; Gaitán-Hernández, R.; Esqueda, M. Biosynthesis, Gene Expression, and Pharmacological Properties of Triterpenoids of Ganoderma Species (Agaricomycetes): A Review. Int. J. Med. Mushrooms 2022, 24, 1–17. [Google Scholar] [CrossRef]
- Zhen-xin, W.; Xi-long, F.; Chengwei, L.; Gao, J.-m.; Jianzhao, Q. Diverse Metabolites and Pharmacological Effects from the Basidiomycetes Inonotus hispidus. Antibiotics 2022, 11, 1097. [Google Scholar]
- Lee, I.K.; Yun, B.S. Styrylpyrone-class compounds from medicinal fungi Phellinus and Inonotus spp., and their medicinal importance. J. Antibiot. 2011, 64, 349–359. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | H. rajendrae NPCB A08 | H. erinaceus CS-4 | H. coralloides FP101451 |
|---|---|---|---|
| Sequencing technology | Illumina NovaSeq 6000 Nanopore PromethION | PacBio RSII, Illumina Hiseq X-Ten | PacBio |
| Sequencing depth | 118× | 750× | 122× |
| No. of contig | 19 | 52 | 125 |
| No. of chromosome | 15 | 15 | NA |
| Total length (bp) | 46,767,965 | 41,880,340 | 55,905,675 |
| Largest length (bp) | 5,307,752 | 6,077,030 | 1,366,710 |
| Contig N50 (bp) | 3,238,877 | 3,208,415 | 711,881 |
| BUSCO (%) | 91.6 | 96.4 | 97.5 |
| Heterozygosity (%) | 3.61 | 0.0226 | 0.847 |
| GC content (%) | 52.57 | 52.30 | 53.64 |
| No. of protein-coding genes | 13,418 | 10,620 | 12,369 |
| GenBank accession No. | PRJNA1018320 | GCA_006506795.2 | JGI_Hercor1 |
| References | This study | [10] | [18] |
| Cluster No. | Location | Start (bp) | End (bp) | Core Gene ID | Core Gene Type |
|---|---|---|---|---|---|
| 1 | Chr2 | 860,561 | 963,214 | g3046.t1 | NRPS-like |
| g3050.t1 | NRPS-like | ||||
| g3052.t1 | NRPS-like | ||||
| 2 | Chr2 | 2,123,606 | 2,187,764 | g3442.t1 | NRPS-like |
| 3 | Chr3 | 228,685 | 291,856 | g7981.t1 | NRPS-like |
| 4 | Chr3 | 318,507 | 352,028 | g8004.t1 | terpene |
| g8005.t1 | terpene | ||||
| 5 | Chr3 | 1,992,954 | 2,074,528 | g8434.t1 | NRPS-like |
| g8436.t1 | T1PKS | ||||
| g8441.t1 | NRPS-like | ||||
| 6 | Chr4 | 26,215 | 52,908 | g11649.t1 | terpene |
| 7 | Chr4 | 1,486,121 | 1,513,187 | g12043.t1 | terpene |
| 8 | Chr4 | 3,476,834 | 3,542,535 | g12602.t1 | terpene |
| g12610.t1 | NRPS-like | ||||
| 9 | Chr5 | 493,923 | 554,595 | g177.t1 | NRPS-like |
| 10 | Chr5 | 674,234 | 733,369 | g226.t1 | NRPS-like |
| 11 | Chr5 | 868,813 | 933,044 | g285.t1 | NRPS-like |
| 12 | Chr5 | 1,747,264 | 1,775,280 | g362.t1 | terpene |
| 13 | Chr5 | 2,071,028 | 2,102,333 | g468.t1 | terpene |
| 14 | Chr5 | 2,804,202 | 2,824,688 | g741.t1 | NRPS |
| 15 | Chr6 | 198,427 | 229,557 | g10218.t1 | terpene |
| 16 | Chr9 | 354,078 | 414,897 | g7354.t1 | NRPS-like |
| 17 | Chr9 | 1,012,150 | 1,076,675 | g7538.t1 | NRPS-like |
| 18 | Chr10 | 1,369,643 | 1,433,801 | g1354.t1 | NRPS-like |
| 19 | Chr11 | 197,542 | 257,730 | g9668.t1 | fungal-RiPP |
| g9673.t1 | fungal-RiPP | ||||
| 20 | Chr11 | 492,774 | 559,259 | g9771.t1 | NRPS-like |
| 21 | Chr12 | 460,518 | 492,887 | g11367.t1 | terpene |
| 22 | Chr14 | 1,503,013 | 1,567,208 | g7114.t1 | NRPS-like |
| 23 | Chr15 | 2 | 11,721 | g2467.t1 | terpene |
| 24 | Chr15 | 115,836 | 146,976 | g2512.t1 | terpene |
| No. | Putative Metabolite | Molecular Formula | Adduct | m/z | Reference |
|---|---|---|---|---|---|
| 1 | Erinacine A | C25H36O6 | [M + H]+ | 433.258 | [26] |
| 2 | Erinacine B | C25H36O6 | [M + H]+ | 433.258 | [26] |
| 3 | Erinacine G | C25H36O8 | [M + H]+ | 465.285 | [27] |
| 4 | Erinacine E | C25H36O6 | [M + Na]+ | 455.243 | [27] |
| 5 | Erinacine F | C25H36O6 | [M + Na]+ | 455.243 | [27] |
| 6 | Erinacines T | C25H38O7 | [M + Na]+ | 473.269 | [28] |
| 7 | Erinacine Z1 | C25H38O7 | [M + Na]+ | 473.269 | [5] |
| 8 | 8 | C26H38O5 | [M + H]+ | 431.242 | [29] |
| 9 | Hericinoid B | C26H36O6 | [M + H]+ | 445.197 | [30] |
| 10 | CJ-14,544 | C25H34O6 | [M + H]+ | 431.182 | [31] |
| 11 | CP-412,065 | C25H34O6 | [M + H]+ | 431.182 | [30] |
| 12 | Hericinoid C | C25H34O7 | [M + K]+ | 485.577 | [30] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, J.; Cheng, M.; Zhu, J.-f.; Zhang, Y.; Cui, K.; Wang, X.; Qi, J. Comparative Genomic Analysis and Metabolic Potential Profiling of a Novel Culinary-Medicinal Mushroom, Hericium rajendrae (Basidiomycota). J. Fungi 2023, 9, 1018. https://doi.org/10.3390/jof9101018
Wei J, Cheng M, Zhu J-f, Zhang Y, Cui K, Wang X, Qi J. Comparative Genomic Analysis and Metabolic Potential Profiling of a Novel Culinary-Medicinal Mushroom, Hericium rajendrae (Basidiomycota). Journal of Fungi. 2023; 9(10):1018. https://doi.org/10.3390/jof9101018
Chicago/Turabian StyleWei, Jing, Min Cheng, Jian-fang Zhu, Yilin Zhang, Kun Cui, Xuejun Wang, and Jianzhao Qi. 2023. "Comparative Genomic Analysis and Metabolic Potential Profiling of a Novel Culinary-Medicinal Mushroom, Hericium rajendrae (Basidiomycota)" Journal of Fungi 9, no. 10: 1018. https://doi.org/10.3390/jof9101018
APA StyleWei, J., Cheng, M., Zhu, J.-f., Zhang, Y., Cui, K., Wang, X., & Qi, J. (2023). Comparative Genomic Analysis and Metabolic Potential Profiling of a Novel Culinary-Medicinal Mushroom, Hericium rajendrae (Basidiomycota). Journal of Fungi, 9(10), 1018. https://doi.org/10.3390/jof9101018