
Targeted NGS-Based Analysis of Pneumocystis jirovecii Reveals Novel Genotypes

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Samples
2.2. DNA Extraction
2.3. Library Preparation
2.4. DNA Sequencing
2.5. Sequencing Analysis
3. Results
3.1. Library Prep Validation
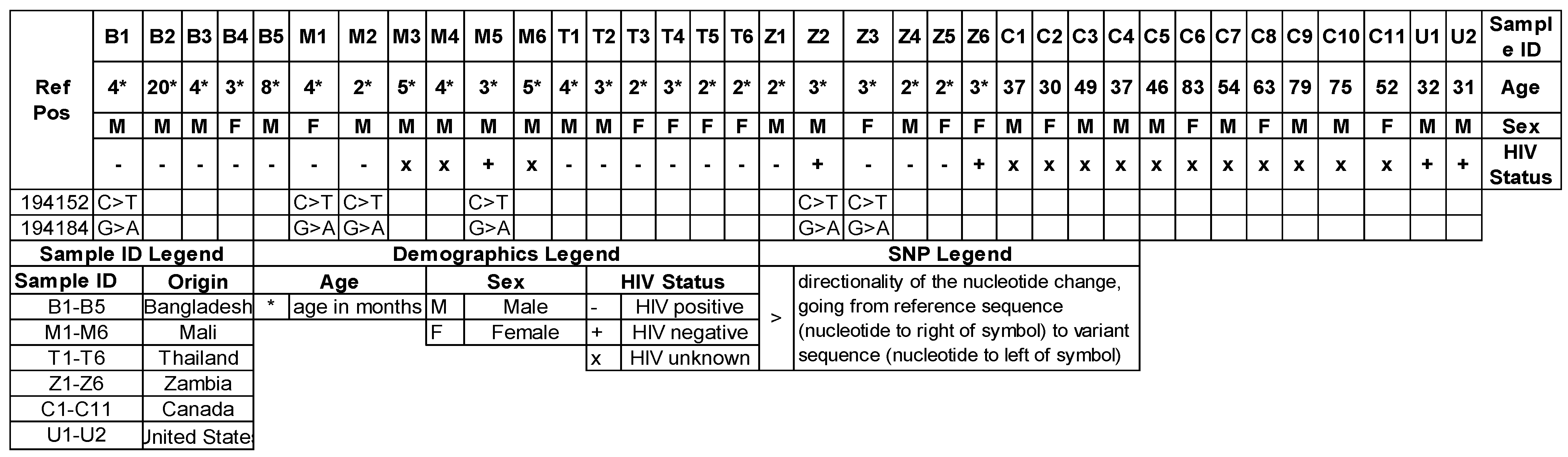
3.2. Meu 10 Genotype
3.3. FourF5 Genotype
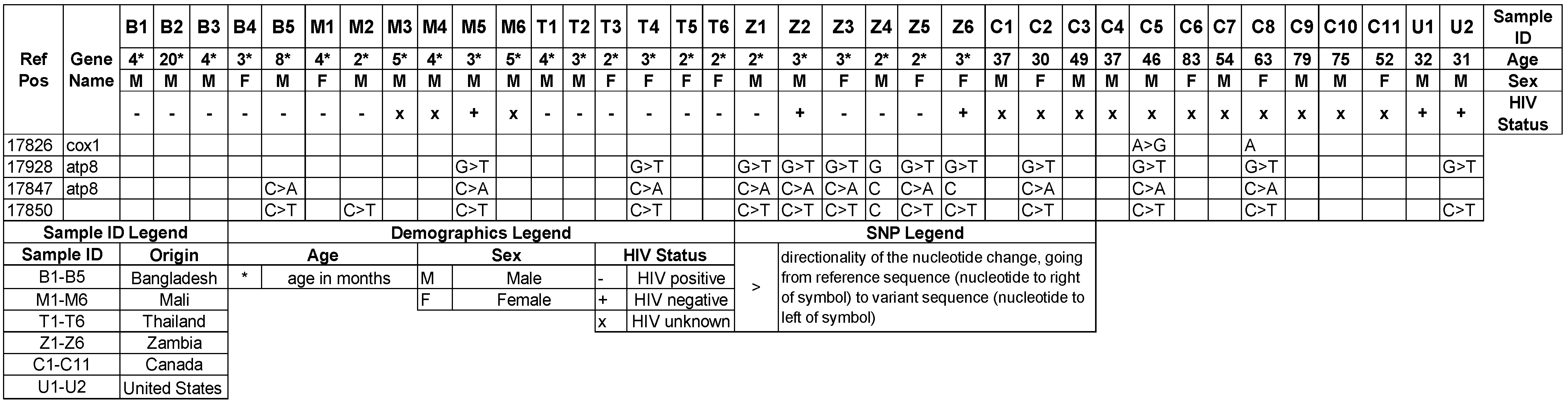
3.4. Mitochondrial Genotypes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pneumonia Etiology Research for Child Health Study Group. Causes of severe pneumonia requiring hospital admission in children without HIV infection from Africa and Asia: The PERCH multi-country case-control study. Lancet 2019, 394, 757–779. [Google Scholar] [CrossRef]
- Elsegeiny, W.; Zheng, M.; Eddens, T.; Gallo, R.L.; Dai, G.; Trevejo-Nunez, G.; Castillo, P.; Kracinovsky, K.; Cleveland, H.; Horne, W.; et al. Murine models of Pneumocystis infection recapitulate human primary immune disorders. JCI Insight 2018, 3, e91894. [Google Scholar] [CrossRef] [PubMed]
- Hoving, J.C.; Kolls, J.K. New advances in understanding the host immune response to Pneumocystis. Curr. Opin. Microbiol. 2017, 40, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Vargas, S.L.; Ponce, C.A.; Gallo, M.; Perez, F.; Astorga, J.F.; Bustamante, R.; Chabe, M.; Durand-Joly, I.; Iturra, P.; Miller, R.F.; et al. Near-universal prevalence of Pneumocystis and associated increase in mucus in the lungs of infants with sudden unexpected death. Clin. Infect. Dis. 2013, 56, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Li, X.L.; Liu, Z.X.; Liu, Z.J.; Li, H.; Wilde, B.; Witzke, O.; Qi, M.; Xu, W.L.; He, Q.; Zhu, J.Q. Pneumocystis pneumonia in liver transplant recipients. Am. J. Transl. Res. 2021, 13, 13981–13992. [Google Scholar] [PubMed]
- Maschmeyer, G.; Helweg-Larsen, J.; Pagano, L.; Robin, C.; Cordonnier, C.; Schellongowski, P.; 6th European Conference on Infections in Leukemia (ECIL-6), a Joint Venture of The European Group for Blood and Marrow Transplantation (EBMT); The European Organization for Research and Treatment of Cancer (EORTC); The International Immunocompromised Host Society (ICHS); The European LeukemiaNet (ELN). ECIL guidelines for treatment of Pneumocystis jirovecii pneumonia in non-HIV-infected haematology patients. J. Antimicrob. Chemother. 2016, 71, 2405–2413. [Google Scholar] [CrossRef] [PubMed]
- Eddens, T.; Elsegeiny, W.; Ricks, D.; Goodwin, M.; Horne, W.T.; Zheng, M.; Kolls, J.K. Transcriptomic and Proteomic Approaches to Finding Novel Diagnostic and Immunogenic Candidates in Pneumocystis. mSphere 2019, 4, e00488-19. [Google Scholar] [CrossRef] [PubMed]
- Cushion, M.T.; Ashbaugh, A.; Hendrix, K.; Linke, M.J.; Tisdale, N.; Sayson, S.G.; Porollo, A. Gene Expression of Pneumocystis murina after Treatment with Anidulafungin Results in Strong Signals for Sexual Reproduction, Cell Wall Integrity, and Cell Cycle Arrest, Indicating a Requirement for Ascus Formation for Proliferation. Antimicrob. Agents Chemother. 2018, 62, e02513-17. [Google Scholar] [CrossRef] [PubMed]
- Cushion, M.T.; Linke, M.J.; Ashbaugh, A.; Sesterhenn, T.; Collins, M.S.; Lynch, K.; Brubaker, R.; Walzer, P.D. Echinocandin treatment of pneumocystis pneumonia in rodent models depletes cysts leaving trophic burdens that cannot transmit the infection. PLoS ONE 2010, 5, e8524. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.F., Jr.; Limper, A.H. Current insights into the biology and pathogenesis of Pneumocystis pneumonia. Nat. Rev. Microbiol. 2007, 5, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Bendl, J.; Stourac, J.; Salanda, O.; Pavelka, A.; Wieben, E.D.; Zendulka, J.; Brezovsky, J.; Damborsky, J. PredictSNP: Robust and accurate consensus classifier for prediction of disease-related mutations. PLoS Comput. Biol. 2014, 10, e1003440. [Google Scholar] [CrossRef] [PubMed]
- Tougan, T.; Chiba, Y.; Kakihara, Y.; Hirata, A.; Nojima, H. Meu10 is required for spore wall maturation in Schizosaccharomyces pombe. Genes Cells 2002, 7, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Cisse, O.H.; Pagni, M.; Hauser, P.M. De novo assembly of the Pneumocystis jirovecii genome from a single bronchoalveolar lavage fluid specimen from a patient. mBio 2012, 4, e00428-12. [Google Scholar] [CrossRef] [PubMed]
- Cushion, M.T.; Keely, S.P. Assembly and annotation of Pneumocystis jirovecii from the human lung microbiome. mBio 2013, 4, e00224. [Google Scholar] [CrossRef] [PubMed]
- Alanio, A.; Gits-Muselli, M.; Guigue, N.; Desnos-Ollivier, M.; Calderon, E.J.; Di Cave, D.; Dupont, D.; Hamprecht, A.; Hauser, P.M.; Helweg-Larsen, J.; et al. Diversity of Pneumocystis jirovecii Across Europe: A Multicentre Observational Study. EBioMedicine 2017, 22, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Charpentier, E.; Garnaud, C.; Wintenberger, C.; Bailly, S.; Murat, J.B.; Rendu, J.; Pavese, P.; Drouet, T.; Augier, C.; Malvezzi, P.; et al. Added Value of Next-Generation Sequencing for Multilocus Sequence Typing Analysis of a Pneumocystis jirovecii Pneumonia Outbreak1. Emerg. Infect Dis. 2017, 23, 1237–1245. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Sequence Name | Target: Gene Name/Murina Ortholog | Target Length | Sequence 5′ to 3′ | Primer Notes |
|---|---|---|---|---|
| RHH.W4300B6A5BDF041 | N/A | N/A | N/A | Custom rhAmpSeq™ FWD Panel |
| RH.CF78E0180F584D9Z0Z.F | T551_03199/Arp9 | 134 | /rhSeq-f/TCA TAG CCC ACA TGA ATA ACA ATT rCCA GA/GT4/ | Forward |
| RH.A2C31F6013C24B7Z0Z.F | T551_03199/Arp9 | 148–152 | /rhSeq-f/GTT CAA TGC TGA CAC TGT TTA TCrG CCA T/GT4/ | Forward |
| RH.1B63D59F4F974A1Z0Z.F | T551_03038/meiotically up-regulated gene 117 protein | 138–142 | /rhSeq-f/GGT TTC TGA GAA AGA TTT AGC CAT ATrG TAA C/GT1/ | Forward |
| RH.FD38EF6B8AEA421Z0Z.F | T551_00303/GTP binding | 148–150 | /rhSeq-f/GAT CCA CTG TCC AGA CAA TTrG CAT C/GT1/ | Forward |
| RH.103374AC8028435Z0Z.F | T551_02533/Meu10 | 148 | /rhSeq-f/ATA GGA GTG GTT TCC GTC TTT rCTT TC/GT3/ | Forward |
| RH.E0CE477C4B19464Z0Z.F | T551_00896/Four F5 protein | 150–153 | /rhSeq-f/GAG AAA ATG ATA GAC TTA GGG CTrC AAA A/GT4/ | Forward |
| RH.316DF1391E794B9Z0Z.F | T551_01756/Acrl | 148–149 | /rhSeq-f/CAG AAG ATG AAT TGC CAC CAA rACA TT/GT4/ | Forward |
| RH.E45C51A8E1AB462Z0Z.F | T551_01755/Serine protease | 110–112 | /rhSeq-f/CAT CTG CAG GTA AAG ACA AGA AArC CAG A/GT1/ | Forward |
| RH.AD8B9A3AFA344A3Z0Z.F | T551_01830/heat shock protein 70 | 142–144 | /rhSeq-f/CCC GAA TTT CCG TCC AAT rCAG AC/GT2/ | Forward |
| RH.47196AD4997D471Z0Z.F | T551_02650/SUMO-targeted ubiquitin-protein ligase E3 Slx8 | 136–138 | /rhSeq-f/TGA CAC ACA AAG GCC ATT TAT TrCT GTC/GT1/ | Forward |
| RH.AEC03A6FD9A64E0Z0Z.F | T551_03158/DNAJ | 135–136 | /rhSeq-f/CCG AAT TGG AAC TCA CTT TAC TrCG AAG/GT3/ | Forward |
| RH.E8A3CF8496644ACZ0Z.F | T551_02309/GSC-1 | 129–134 | /rhSeq-f/TCT GGA GTA AAT CGA ACT TGA TTT rGCT TC/GT4/ | Forward |
| RH.8DF9507B0841432Z0Z.F | mito NAD6 | 149–153 | /rhSeq-f/CAA TTC TGG TTG TAT CTT CAA GAA ATC rCTG TA/GT2/ | Forward |
| RH.3E370038B0CB4B3Z0Z.F | mito NAD1 | 151–156 | /rhSeq-f/GCT TTA TTA GGT TCC TTG TGA AGT ArCT GCT/GT3/ | Forward |
| RH.DDFCD6AD33494FBZ0Z.F | cytochrome c oxidase subunit 1/ atp8 | 155–160 | /rhSeq-f/TGA TTT CAC CAC CAG CTT TCrC ATG C/GT1/ | Forward |
| RH.FF72BC52B1E14BCZ0Z.F | atp6 | 155–160 | /rhSeq-f/CTT CAC ATC TGG TCT TTA CTG TCrG CTT T/GT3/ | Forward |
| RH.E67274DA8AD04D9Z0Z.F | cytochrome c oxidase subunit 3 | 151–156 | /rhSeq-f/CTA GCT TTT ACT CTT TTA CAG GGT rGTG GA/GT4/ | Forward |
| RH.EF9CF32F289649AZ0Z.F | atp9 | 149–152 | /rhSeq-f/GGT TCA GGG TTA GCT ACA ATT rGGA TT/GT4/ | Forward |
| RH.99229D6013E14CBZ0Z.F | NADH dehydrogenase subunit 4L/nad5 | 151–152 | /rhSeq-f/TCC ATG GCT TTA GAT GAT TTA GAA GrGA CAG/GT1/ | Forward |
| RH.795964E1E42344EZ0Z.F | Nad4 | 125–128 | /rhSeq-f/TCA CTC CTT TGG TCT ATA CTG TrCT GTG/GT1/ | Forward |
| RHH.W4300B6A5BDF041 | N/A | N/A | N/A | Custom rhAmpSeq™ REV Panel |
| RH.CF78E0180F584D9Z0Z.R | T551_03199/Arp9 | 134 | /rhSeq-r/CGT GAT AAA GAA CTT GCA ACA TGrC ACA T/GT1/ | Reverse |
| RH.A2C31F6013C24B7Z0Z.R | T551_03199/Arp9 | 148–152 | /rhSeq-r/GAG AGT ATT TGG CTC AAA AAG AAG rCGG AG/GT4/ | Reverse |
| RH.1B63D59F4F974A1Z0Z.R | T551_03038/meiotically up-regulated gene 117 protein | 138–142 | /rhSeq-r/TCT ACT CTA TTA ATG TAT TCT GAC CCA rCAA GG/GT4/ | Reverse |
| RH.FD38EF6B8AEA421Z0Z.R | T551_00303/GTP binding | 148–150 | /rhSeq-r/GGT TAT GAC AAT TTC GCC GArC CGT T/GT2/ | Reverse |
| RH.103374AC8028435Z0Z.R | T551_02533/Meu10 | 148 | /rhSeq-r/TGA CCA AGT CGC TGT TAT TGT rATA TG/GT2/ | Reverse |
| RH.E0CE477C4B19464Z0Z.R | T551_00896/Four F5 protein | 150–153 | /rhSeq-r/GAT GTT TGC TTG GCT AAG GTA rCAG AT/GT1/ | Reverse |
| RH.316DF1391E794B9Z0Z.R | T551_01756/Acrl | 148–149 | /rhSeq-r/TCC CAT CGT AAA AGG CGT AAT rCTT GA/GT1/ | Reverse |
| RH.E45C51A8E1AB462Z0Z.R | T551_01755/Serine protease | 110–112 | /rhSeq-r/TGA ACA AAT GAA TGC GAC AAT AGT rCCC AA/GT2/ | Reverse |
| RH.AD8B9A3AFA344A3Z0Z.R | T551_01830/heat shock protein 70 | 142–144 | /rhSeq-r/TCA AGG GAA TCG AAC CAC ArCC ATC/GT4/ | Reverse |
| RH.47196AD4997D471Z0Z.R | T551_02650/SUMO-targeted ubiquitin-protein ligase E3 Slx8 | 136–138 | /rhSeq-r/TTT GTT TGG TGC AAG CAT TAT TTrC AAG G/GT2/ | Reverse |
| RH.AEC03A6FD9A64E0Z0Z.R | T551_03158/DNAJ | 135–136 | /rhSeq-r/CGT GAC CAG GAT ACG ATA TCT rCAT GG/GT3/ | Reverse |
| RH.E8A3CF8496644ACZ0Z.R | T551_02309/GSC-1 | 129–134 | /rhSeq-r/CCT TTA GAA TCT GCA ATA TAT CGT TrGG AAG/GT4/ | Reverse |
| RH.8DF9507B0841432Z0Z.R | mito NAD6 | 149–153 | /rhSeq-r/GCT ATA GCT CCA ACA TAT ACA GTA ATrA TAT G/GT4/ | Reverse |
| RH.3E370038B0CB4B3Z0Z.R | mito NAD1 | 151–156 | /rhSeq-r/GGC AAA ACA AAC CAA ATA GCT TTT TrGT TCT/GT1/ | Reverse |
| RH.DDFCD6AD33494FBZ0Z.R | cytochrome c oxidase subunit 1/ atp8 | 155–160 | /rhSeq-r/TGT CTA GGC AGA ACC CAT TTA rGCG AA/GT2/ | Reverse |
| RH.FF72BC52B1E14BCZ0Z.R | atp6 | 155–160 | /rhSeq-r/ACA AAG AAG GAG AAG AAT TTC AGA rCCA TG/GT1/ | Reverse |
| RH.E67274DA8AD04D9Z0Z.R | cytochrome c oxidase subunit 3 | 151–156 | /rhSeq-r/AGT CCT ACA GAT AAA AAG ATA GCA rCCC AC/GT1/ | Reverse |
| RH.EF9CF32F289649AZ0Z.R | atp9 | 149–152 | /rhSeq-r/GCT TCT GCT AGA GCA AAT CCrC AAG A/GT1/ | Reverse |
| RH.99229D6013E14CBZ0Z.R | NADH dehydrogenase subunit 4L/nad5 | 151–152 | /rhSeq-r/ACA CCG ATT TTA TGT CCT AAC AAA rCCT AC/GT4/ | Reverse |
| RH.795964E1E42344EZ0Z.R | Nad4 | 125–128 | /rhSeq-r/GCC AAA GCC ATA TGT CCA ATA rGAA GA/GT4/ | Reverse |
| i5_IDT-1 | N/A | N/A | AAT GAT ACG GCG ACC ACC GAG ATC TAC ACA TAT GCG CAC ACT CTT TCC CTA CAC GAC | rhAmpSeq™ Index 1 (i5) Primer |
| i5_IDT-2 | N/A | N/A | AAT GAT ACG GCG ACC ACC GAG ATC TAC ACT GGT ACA GAC ACT CTT TCC CTA CAC GAC | rhAmpSeq™ Index 2 (i5) Primer |
| i7_IDT-1 | N/A | N/A | CAA GCA GAA GAC GGC ATA CGA GAT ACG ATC AGG TGA CTG GAG TTC AGA CGT GT | rhAmpSeq™ Index 1 (i7) Primer |
| i7_IDT-2 | N/A | N/A | CAA GCA GAA GAC GGC ATA CGA GAT TCG AGA GTG TGA CTG GAG TTC AGA CGT GT | rhAmpSeq™ Index 2 (i7) Primer |
| i7_IDT-3 | N/A | N/A | CAA GCA GAA GAC GGC ATA CGA GAT CTA GCT CAG TGA CTG GAG TTC AGA CGT GT | rhAmpSeq™ Index 3 (i7) Primer |
| i7_IDT-4 | N/A | N/A | CAA GCA GAA GAC GGC ATA CGA GAT ATC GTC TCG TGA CTG GAG TTC AGA CGT GT | rhAmpSeq™ Index 4 (i7) Primer |
| i7_IDT-5 | N/A | N/A | CAA GCA GAA GAC GGC ATA CGA GAT TCG ACA AGG TGA CTG GAG TTC AGA CGT GT | rhAmpSeq™ Index 5 (i7) Primer |
| i7_IDT-6 | N/A | N/A | CAA GCA GAA GAC GGC ATA CGA GAT CCT TGG AAG TGA CTG GAG TTC AGA CGT GT | rhAmpSeq™ Index 6 (i7) Primer |
| i7_IDT-7 | N/A | N/A | CAA GCA GAA GAC GGC ATA CGA GAT ATC ATG CGG TGA CTG GAG TTC AGA CGT GT | rhAmpSeq™ Index 7 (i7) Primer |
| i7_IDT-8 | N/A | N/A | CAA GCA GAA GAC GGC ATA CGA GAT TGT TCC GTG TGA CTG GAG TTC AGA CGT GT | rhAmpSeq™ Index 8 (i7) Primer |
| i7_IDT-9 | N/A | N/A | CAA GCA GAA GAC GGC ATA CGA GAT ATT AGC CGG TGA CTG GAG TTC AGA CGT GT | rhAmpSeq™ Index 9 (i7) Primer |
| i7_IDT-10 | N/A | N/A | CAA GCA GAA GAC GGC ATA CGA GAT CGA TCG ATG TGA CTG GAG TTC AGA CGT GT | rhAmpSeq™ Index 10 (i7) Primer |
| i7_IDT-11 | N/A | N/A | CAA GCA GAA GAC GGC ATA CGA GAT GAT CTT GCG TGA CTG GAG TTC AGA CGT GT | rhAmpSeq™ Index 11 (i7) Primer |
| i7_IDT-12 | N/A | N/A | CAA GCA GAA GAC GGC ATA CGA GAT AGG ATA GCG TGA CTG GAG TTC AGA CGT GT | rhAmpSeq™ Index 12 (i7) Primer |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pungan, D.; Eddens, T.; Song, K.; Lakey, M.A.; Crovetto, N.S.; Arora, S.K.; Husain, S.; Kolls, J.K. Targeted NGS-Based Analysis of Pneumocystis jirovecii Reveals Novel Genotypes. J. Fungi 2022, 8, 863. https://doi.org/10.3390/jof8080863
Pungan D, Eddens T, Song K, Lakey MA, Crovetto NS, Arora SK, Husain S, Kolls JK. Targeted NGS-Based Analysis of Pneumocystis jirovecii Reveals Novel Genotypes. Journal of Fungi. 2022; 8(8):863. https://doi.org/10.3390/jof8080863
Chicago/Turabian StylePungan, Dora, Taylor Eddens, Kejing Song, Meredith A. Lakey, Nicolle S. Crovetto, Simran K. Arora, Shahid Husain, and Jay K. Kolls. 2022. "Targeted NGS-Based Analysis of Pneumocystis jirovecii Reveals Novel Genotypes" Journal of Fungi 8, no. 8: 863. https://doi.org/10.3390/jof8080863
APA StylePungan, D., Eddens, T., Song, K., Lakey, M. A., Crovetto, N. S., Arora, S. K., Husain, S., & Kolls, J. K. (2022). Targeted NGS-Based Analysis of Pneumocystis jirovecii Reveals Novel Genotypes. Journal of Fungi, 8(8), 863. https://doi.org/10.3390/jof8080863