
Telomere-to-Telomere Genome Sequences across a Single Genus Reveal Highly Variable Chromosome Rearrangement Rates but Absolute Stasis of Chromosome Number


 , , ,
, , ,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Genome Sequencing
2.2. Genome Annotation and Repeat Content Analysis
2.3. Phylogenetic Analysis
2.4. Genome Synteny
2.5. Genome-Scale Multiple Alignment and Ancestral Genome Reconstruction
3. Results
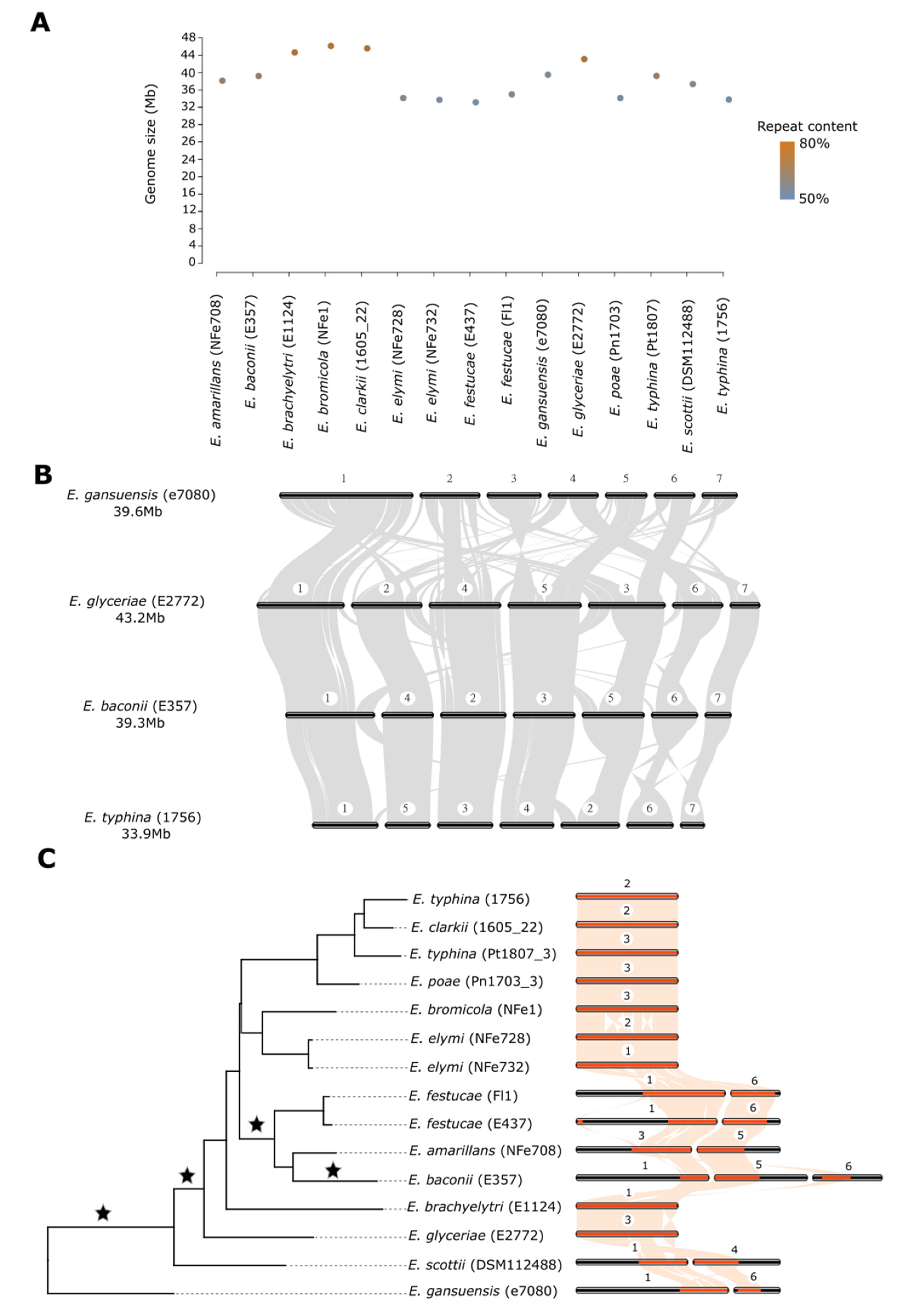
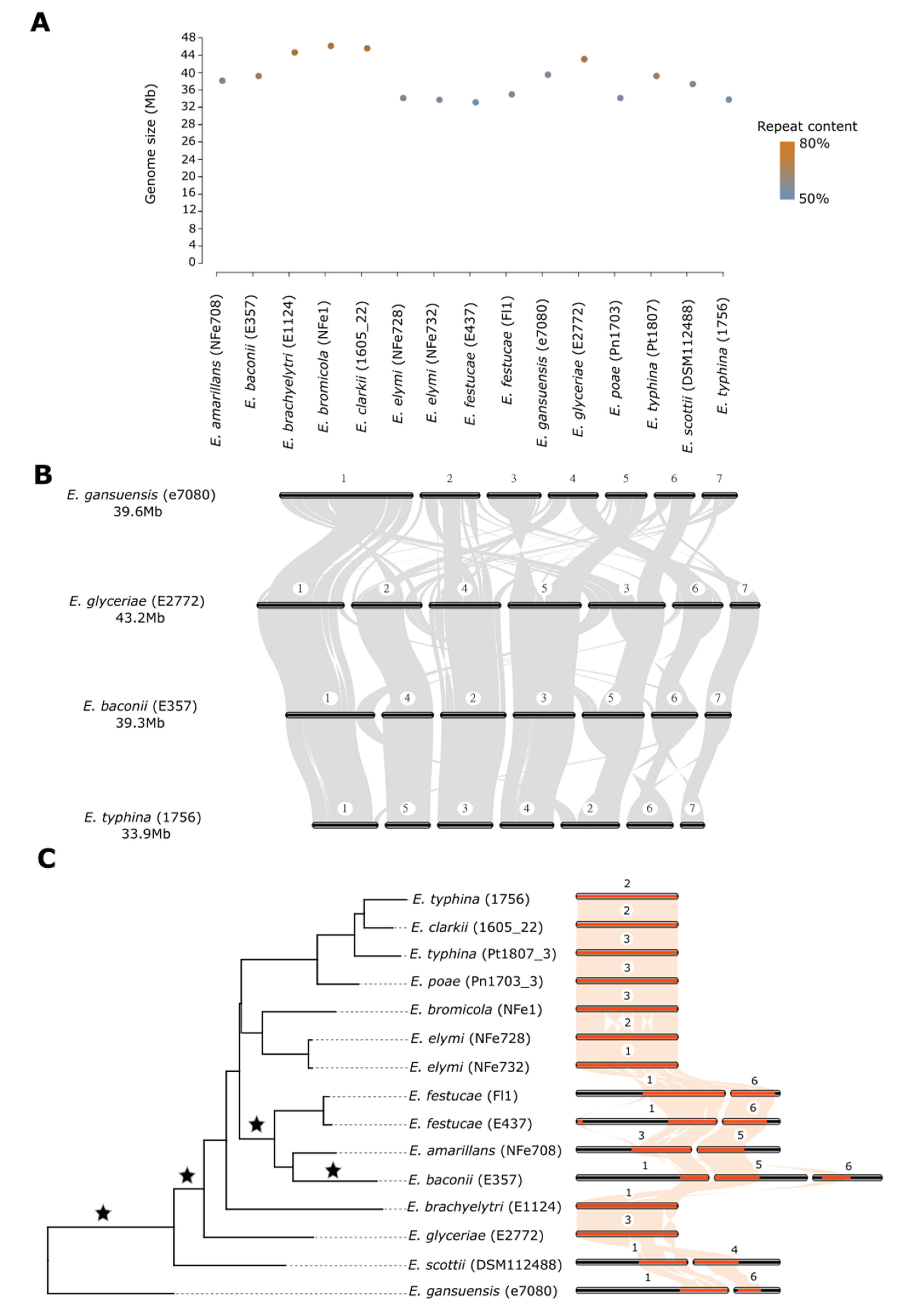
3.1. Telomere-to-Telomere Genome Sequences for 15 Epichloë Species and Strains
3.2. Large Genome Size Variation Is Primarily Due to Repeat Element Dynamics
3.3. Chromosome Structure across the Genus Is Highly Rearranged despite Conservation of Chromosome Number
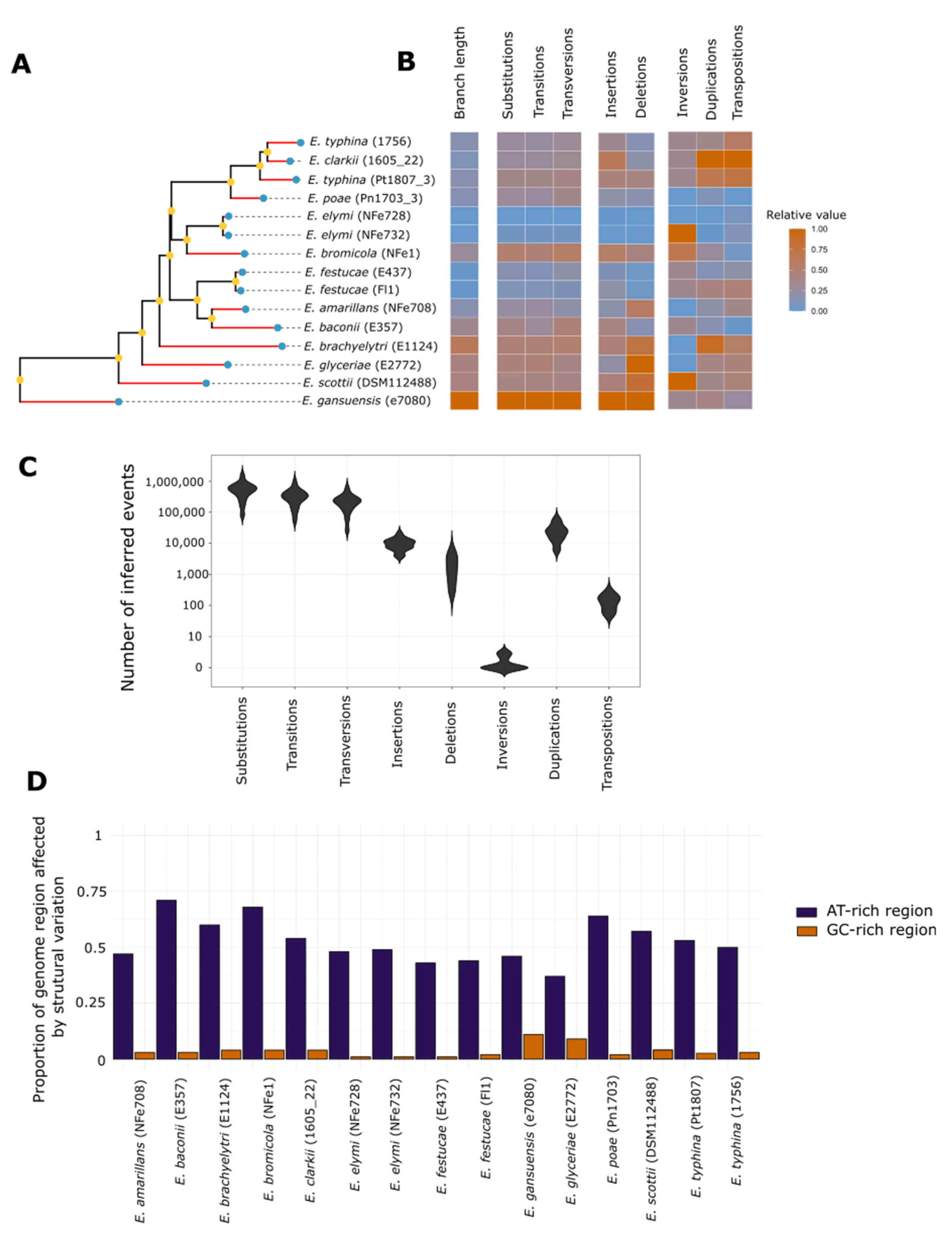
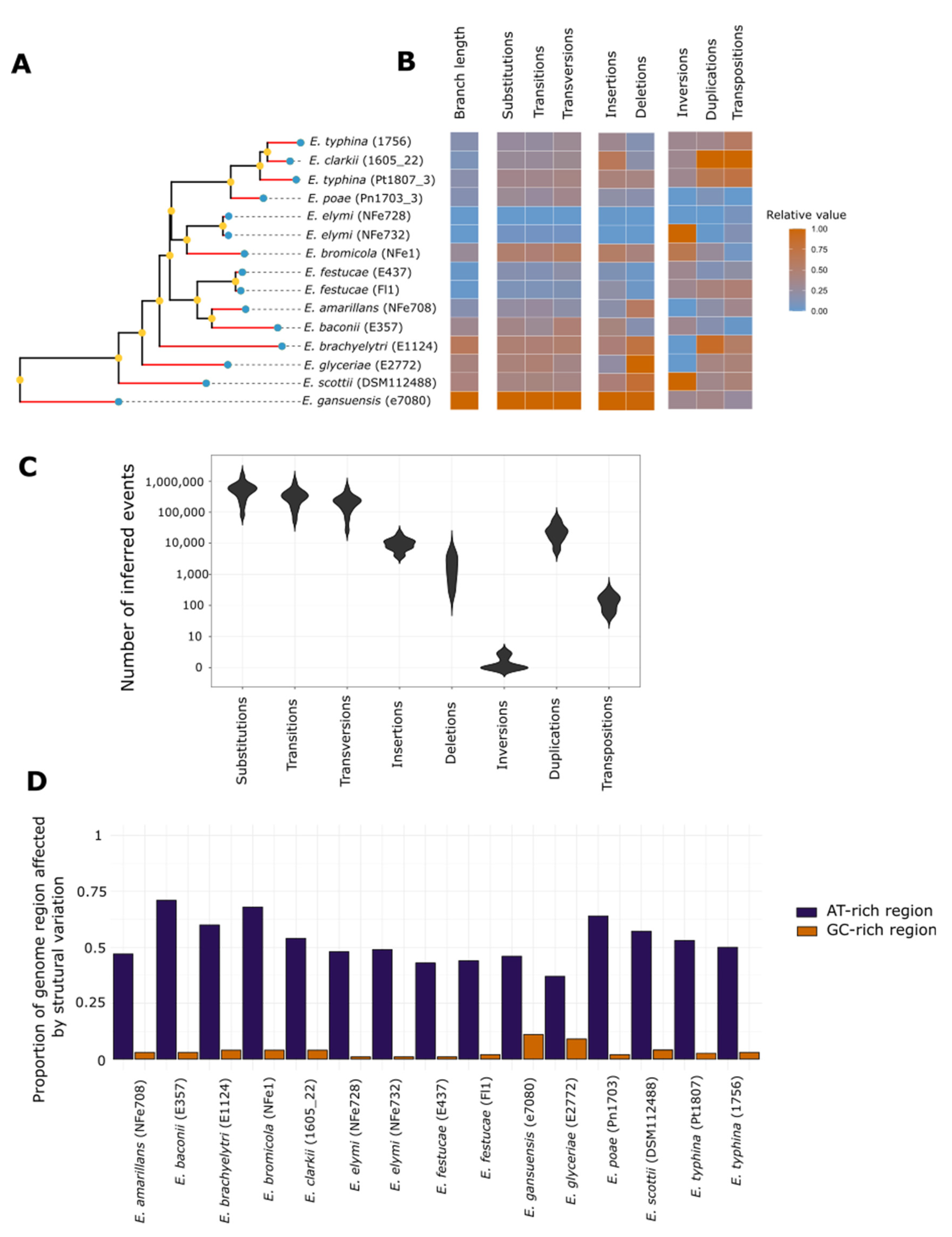
3.4. Chromosome Rearrangements Are Not Correlated with Nucleotide Scale Changes
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rhie, A.; McCarthy, S.A.; Fedrigo, O.; Damas, J.; Formenti, G.; Koren, S.; Uliano-Silva, M.; Chow, W.; Fungtammasan, A.; Kim, J.; et al. Towards complete and error-free genome assemblies of all vertebrate species. Nature 2021, 592, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Seixas, F.A.; Edelman, N.B.; Mallet, J. Synteny-based genome assembly for 16 species of Heliconius butterflies, and an assessment of structural variation across the genus. Genome Biol. Evol. 2021, 13, evab069. [Google Scholar] [CrossRef] [PubMed]
- Gordon, J.L.; Byrne, K.P.; Wolfe, K.H. Mechanisms of chromosome number evolution in yeast. PLoS Genet. 2011, 7, e1002190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arneodo, A.; Vaillant, C.; Audit, B.; Argoul, F.; D’aubenton-Carafa, Y.; Thermes, C. Multi-scale coding of genomic information: From DNA sequence to genome structure and function. Phys. Rep. 2011, 498, 45–188. [Google Scholar] [CrossRef]
- Heitman, J.; Priest, S.J.; Yadav, V. Advances in understanding the evolution of fungal genome architecture. F1000Research 2020, 9, 776. [Google Scholar] [CrossRef]
- Ho, S.S.; Urban, A.E.; Mills, R.E. Structural variation in the sequencing era. Nat. Rev. Genet. 2019, 21, 171–189. [Google Scholar] [CrossRef]
- Engel, S.R.; Dietrich, F.S.; Fisk, D.G.; Binkley, G.; Balakrishnan, R.; Costanzo, M.C.; Dwight, S.S.; Hitz, B.C.; Karra, K.; Nash, R.S.; et al. The reference genome sequence of Saccharomyces cerevisiae: Then and now. G3 Genes Genomes Genet. 2014, 4, 389–398. [Google Scholar] [CrossRef] [Green Version]
- Gorkovskiy, A.; Verstrepen, K.J. The role of structural variation in adaptation and evolution of yeast and other fungi. Genes 2021, 12, 699. [Google Scholar] [CrossRef]
- Shi-Kunne, X.; Faino, L.; van den Berg, G.C.M.; Thomma, B.P.H.J.; Seidl, M.F. Evolution within the fungal genus Verticillium is characterized by chromosomal rearrangement and gene loss. Environ. Microbiol. 2018, 20, 1362–1373. [Google Scholar] [CrossRef] [Green Version]
- Tadych, M.; Bergen, M.S.; White, J.F. Epichloë spp. associated with grasses: New insights on life cycles, dissemination and evolution. Mycologia 2014, 106, 181–201. [Google Scholar] [CrossRef]
- Pan, J.; Bhardwaj, M.; Nagabhyru, P.; Grossman, R.B.; Schardl, C.L.; Wilson, R.A. Enzymes from fungal and plant origin required for chemical diversification of insecticidal loline alkaloids in grass-Epichloë symbiota. PLoS ONE 2014, 9, e115590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leuchtmann, A.; Bacon, C.W.; Schardl, C.L.; White, J.F.; Tadych, M. Nomenclatural realignment of Neotyphodium species with genus Epichloë. Mycologia 2017, 106, 202–215. [Google Scholar] [CrossRef] [PubMed]
- Thünen, T.; Becker, Y.; Cox, M.P.; Ashrafi, S. Epichloë scottii sp. nov., a new endophyte isolated from Melica uniflora is the missing ancestor of Epichloë disjuncta. IMA Fungus 2022, 13, 2. [Google Scholar] [CrossRef] [PubMed]
- Shymanovich, T.; Charlton, N.D.; Musso, A.M.; Scheerer, J.; Cech, N.B.; Faeth, S.H.; Young, C.A. Interspecific and intraspecific hybrid Epichloë species symbiotic with the North American native grass Poa alsodes. Mycologia 2017, 109, 459–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, M.A.; Tapper, B.A.; Simpson, W.R.; Johnson, R.D.; Mace, W.; Ram, A.; Lukito, Y.; Dupont, P.Y.; Johnson, L.J.; Scott, D.B.; et al. Epichloë hybrida, sp. nov., an emerging model system for investigating fungal allopolyploidy. Mycologia 2017, 109, 715–729. [Google Scholar] [CrossRef] [Green Version]
- Winter, D.J.; Ganley, A.R.D.; Young, C.A.; Liachko, I.; Schardl, C.L.; Dupont, P.-Y.; Berry, D.; Ram, A.; Scott, B.; Cox, M.P. Repeat elements organise 3D genome structure and mediate transcription in the filamentous fungus Epichloë festucae. PLoS Genet. 2018, 14, e1007467. [Google Scholar] [CrossRef] [Green Version]
- Treindl, A.D.; Stapley, J.; Winter, D.J.; Cox, M.P.; Leuchtmann, A. Chromosome-level genomes provide insights into genome evolution, organization and size in Epichloë fungi. Genomics 2021, 113, 4267–4275. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.; Berlin, K.; Miller, J.; Bergman, N.; Phillippy, A. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [Green Version]
- Xiao, C.-L.; Chen, Y.; Xie, S.-Q.; Chen, K.-N.; Wang, Y.; Han, Y.; Luo, F.; Xie, Z. MECAT: Fast mapping, error correction, and de novo assembly for single-molecule sequencing reads. Nat. Methods 2017, 14, 1072–1074. [Google Scholar] [CrossRef]
- Chen, Y.; Nie, F.; Xie, S.-Q.; Zheng, Y.-F.; Dai, Q.; Bray, T.; Wang, Y.-X.; Xing, J.-F.; Huang, Z.-J.; Wang, D.-P.; et al. Efficient assembly of nanopore reads via highly accurate and intact error correction. Nat. Commun. 2021, 12, 60. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef] [PubMed]
- Flynn, M.J.; Hubley, R.; Goubert, C.; Rosen, J.; Clark, A.G.; Feschotte, C.; Smit, A.F. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 2020, 117, 9451–9457. [Google Scholar] [CrossRef] [PubMed]
- Testa, A.C.; Oliver, R.P.; Hane, J.K. OcculterCut: A comprehensive survey of AT-rich regions in fungal genomes. Genome Biol. Evol. 2016, 8, 2044–2064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [Green Version]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Oberti, H.; Abreo, E.; Reyno, R.; Feijoo, M.; Murchio, S.; Dalla-Rizza, M. New draft genome sequence of the ergot disease fungus Claviceps paspali. Microbiol. Resour. Announc. 2020, 9, 29. [Google Scholar] [CrossRef]
- Oberti, H.; Spangenberg, G.; Cogan, N.; Reyno, R.; Feijoo, M.; Murchio, S.; Dalla-Rizza, M. Genome-wide analysis of Claviceps paspali: Insights into the secretome of the main species causing ergot disease in Paspalum spp. BMC Genom. 2021, 22, 766. [Google Scholar] [CrossRef]
- Tang, H.; Wang, X.; Bowers, J.E.; Ming, R.; Alam, M.; Paterson, A.H. Unraveling ancient hexaploidy through multiply-aligned angiosperm gene maps. Genome Res. 2008, 18, 1944–1954. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, J.; Hickey, G.; Diekhans, M.; Fiddes, I.T.; Novak, A.M.; Deran, A.; Fang, Q.; Xie, D.; Feng, S.; Stiller, J.; et al. Progressive Cactus is a multiple-genome aligner for the thousand-genome era. Nature 2020, 587, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Hickey, G.; Paten, B.; Earl, D.; Zerbino, D.; Haussler, D. HAL: A hierarchical format for storing and analyzing multiple genome alignments. Bioinformatics 2013, 29, 1341–1342. [Google Scholar] [CrossRef] [PubMed]
- R Development Core Team. A Language and Environment for Statistical Computing. 2022. Available online: https://www.r-project.org (accessed on 1 May 2022).
- Craven, K.D.; Hsiau, P.T.W.; Leuchtmann, A.; Hollin, W.; Schardl, C.L. Multigene phylogeny of Epichloë species, fungal symbionts of grasses. Ann. Mo. Bot. Gard. 2001, 88, 14–34. [Google Scholar] [CrossRef]
- Gladyshev, E. Repeat-induced point mutation and other genome defense mechanisms in Fungi. Microbiol. Spectr. 2017, 5, 0042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schardl, C.L.; Young, C.A.; Hesse, U.; Amyotte, S.G.; Andreeva, K.; Calie, P.J.; Fleetwood, D.J.; Haws, D.C.; Moore, N.; Oeser, B.; et al. Plant-symbiotic fungi as chemical engineers: Multi-genome analysis of the Clavicipitaceae reveals dynamics of alkaloid loci. PLoS Genet. 2013, 9, e1003323. [Google Scholar] [CrossRef] [Green Version]
- Badet, T.; Fouché, S.; Hartmann, F.E.; Zala, M.; Croll, D. Machine-learning predicts genomic determinants of meiosis-driven structural variation in a eukaryotic pathogen. Nat. Commun. 2021, 12, 3551. [Google Scholar] [CrossRef]
- Catanach, A.; Crowhurst, R.; Deng, C.; David, C.; Bernatchez, L.; Wellenreuther, M. The genomic pool of standing structural variation outnumbers single nucleotide polymorphism by threefold in the marine teleost Chrysophrys auratus. Mol. Ecol. 2019, 28, 1210–1223. [Google Scholar] [CrossRef]
- Hane, J.K.; Rouxel, T.; Howlett, B.J.; Kema, G.H.J.; Goodwin, S.B.; Oliver, R.P. A novel mode of chromosomal evolution peculiar to filamentous Ascomycete fungi. Genome Biol. 2011, 12, R45. [Google Scholar] [CrossRef] [Green Version]
- Dunham, M.J.; Badrane, H.; Ferea, T.; Adams, J.; Brown, P.O.; Rosenzweig, F.; Botstein, D. Characteristic genome rearrangements in experimental evolution of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 2002, 99, 16144–16149. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Peterson, T. Genome rearrangements by nonlinear transposons in maize. Genetics 1999, 153, 1403–1410. [Google Scholar] [CrossRef]
- Kuldau, G.A.; Tsai, H.F.; Schardl, C.L. Genome sizes of Epichloë species and anamorphic hybrids. Mycologia 1999, 91, 776–782. [Google Scholar] [CrossRef]
- Clay, K.; Schardl, C.L. Evolutionary origins and ecological consequences of endophyte symbiosis with grasses. Am. Nat. 2002, 160, S99–S127. [Google Scholar] [CrossRef] [PubMed]
- Ambrose, K.V.; Koppenhöfer, A.M.; Belanger, F.C. Horizontal gene transfer of a bacterial insect toxin gene into the Epichloë fungal symbionts of grasses. Sci. Rep. 2014, 4, 5562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, E.L.; Robbins, T.P.; Bureau, T.E.; Kermicle, J.; Dellaporta, S.L. Transposon-mediated chromosomal rearrangements and gene duplications in the formation of the maize R-r complex. EMBO J. 1995, 14, 2350. [Google Scholar] [CrossRef]
- Xuan, Y.H.; Piao, H.L.; Je, B.I.; Park, S.J.; Park, S.H.; Huang, J.; Zhang, J.B.; Peterson, T.; Han, C.D. Transposon Ac/Ds-induced chromosomal rearrangements at the rice OsRLG5 locus. Nucleic Acids Res. 2011, 39, e149. [Google Scholar] [CrossRef]
- Berthelot, C.; Muffato, M.; Abecassis, J.; Crollius, H.R. The 3D organization of chromatin explains evolutionary fragile genomic regions. Cell Rep. 2015, 10, 1913–1924. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Sun, M.; Zhang, Y.; Song, Z.; Zhang, S.; Zhang, Q.; Xu, J.R.; Liu, H. Extensive chromosomal rearrangements and rapid evolution of novel effector superfamilies contribute to host adaptation and speciation in the basal ascomycetous fungi. Mol. Plant Pathol. 2020, 21, 330–348. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
| Species | Strain | Sequencing Technology | Host Species | Geographical Origin | Sexual Reproduction | NCBI BioProject Number |
|---|---|---|---|---|---|---|
| E. amarillans | NFe708 | PacBio RSII/Illumina | Elymus canadensis | Iowa, USA | Observed | PRJNA532945 |
| E. baconii | E357 | MinION | Calamagrostis villosa | Switzerland | Observed | PRJNA842087 |
| E. brachyelytri | E1124 | MinION | Brachyelytrum erectum | New York, USA | Observed | PRJNA841758 |
| E. bromicola | NFe1 | MinION/Illumina | Hordeum bogdanii | Kazakhstan | Not observed | PRJNA842763 |
| E. clarkii | 1605_22 | PacBio RSII/Illumina | Holcus lanatus | Switzerland | Observed | PRJNA533212 |
| E. elymi | NFe728 | MinION | Elymus virginicus | Oklahoma, USA | Observed | PRJNA839151 |
| E. elymi | NFe732 | PacBio RSII/Illumina | Elymus canadensis | Oklahoma, USA | Not observed | PRJNA532946 |
| E. festucae | E437 | PacBio RSII/Illumina | Festuca pulchella | Japan | Observed | PRJNA842088 |
| E. festucae | Fl1 | PacBio RSII/Illumina | Festuca longifolia | United Kingdom | Observed | PRJNA431450 |
| E. gansuensis | e7080 | MinION | Achnatherum inebrians | China | Not observed | PRJNA842754 |
| E. glyceriae | E2772 | MinION | Glyceria striata | New York, USA | Observed | PRJNA841696 |
| E. poae | Pn1703_3 | PacBio Sequel II/Illumina | Poa nemoralis | France | Observed | PRJNA843480 |
| E. scottii | DSM112488 | MinION | Melica uniflora | Germany | Observed | PRJNA756890 |
| E. typhina | Pt1807_3 | PacBio Sequel II/Illumina | Poa trivialis | Spain | Observed | PRJNA844123 |
| E. typhina | 1756 | PacBio RSII/Illumina | Dactylis glomerata | Switzerland | Observed | PRJNA533210 |
| Species | Strain | Chromosomes | Genome Length | Number of Genes | Repeat Regions (%) | AT-Rich Regions (%) | Number of AT-Rich Blocks | Mean Length of AT-Rich Blocks |
|---|---|---|---|---|---|---|---|---|
| E. amarillans | NFe708 | 7 | 38,224,169 | 7451 | 68 | 41 | 351 | 44,429 |
| E. baconii | E357 | 7 | 39,250,507 | 7486 | 67 | 39 | 528 | 29,265 |
| E. brachyelytri | E1124 | 7 | 44,747,857 | 7335 | 72 | 49 | 777 | 28,132 |
| E. bromicola | NFe1 | 7 | 46,201,037 | 7615 | 72 | 49 | 808 | 27,892 |
| E. clarkii | 1605_22 | 7 | 45,646,793 | 7772 | 72 | 49 | 650 | 34,135 |
| E. elymi | NFe728 | 7 | 34,206,040 | 7424 | 64 | 32 | 340 | 32,533 |
| E. elymi | NFe732 | 7 | 33,820,330 | 8324 | 63 | 32 | 348 | 30,920 |
| E. festucae | E437 | 7 | 33,219,473 | 7764 | 61 | 28 | 395 | 23,630 |
| E. festucae | Fl1 | 7 | 35,023,690 | 7822 | 63 | 32 | 435 | 25,714 |
| E. gansuensis | e7080 | 7 | 39,568,882 | 7601 | 63 | 37 | 428 | 33,994 |
| E. glyceriae | E2772 | 7 | 43,207,551 | 7267 | 71 | 47 | 496 | 40,681 |
| E. poae | Pn1703_3 | 7 | 34,185,484 | 7420 | 63 | 32 | 396 | 27,983 |
| E. scottii | DSM112488 | 7 | 37,342,247 | 6486 | 62 | 37 | 363 | 29,549 |
| E. typhina | Pt1807_3 | 7 | 39,303,283 | 7305 | 68 | 41 | 513 | 31,484 |
| E. typhina | 1756 | 7 | 33,870,766 | 7539 | 64 | 32 | 503 | 27,726 |
| Genome | Terminal Branch Length | Substitutions | Transitions | Transversions | Insertions | Deletions | Inversions | Duplications | Transpositions |
|---|---|---|---|---|---|---|---|---|---|
| E. amarillans | 0.009439 | 413,037 | 278,990 | 134,047 | 7030 | 2924 | 0 | 16,594 | 137 |
| E. baconii | 0.01839 | 608,309 | 320,946 | 287,363 | 10,798 | 962 | 1 | 14,582 | 50 |
| E. brachyelytri | 0.034203 | 772,918 | 465,715 | 307,203 | 11,648 | 3,713 | 0 | 53,611 | 193 |
| E. bromicola | 0.016056 | 794,167 | 464,710 | 329,457 | 13,291 | 2,241 | 2 | 20,666 | 59 |
| E. clarkii | 0.006319 | 493,578 | 296,234 | 197,344 | 14,452 | 1,137 | 1 | 60,699 | 312 |
| E. festucae (E437) | 0.001888 | 307,728 | 173,466 | 134,262 | 6016 | 421 | 1 | 13,191 | 102 |
| E. festucae (Fl1) | 0.001466 | 234,349 | 139,930 | 94,419 | 7410 | 403 | 1 | 29,260 | 172 |
| E. gansuensis | 0.055092 | 1,497,975 | 921,662 | 576,313 | 20,821 | 4835 | 1 | 25,934 | 116 |
| E. glyceriae | 0.023931 | 710,405 | 475,456 | 234,949 | 7697 | 4900 | 0 | 27,682 | 166 |
| E. elymi (NFe728) | 0.000835 | 81,944 | 55,421 | 26,523 | 3946 | 243 | 0 | 5937 | 68 |
| E. elymi (NFe732) | 0.000892 | 156,109 | 99,732 | 56,377 | 3891 | 248 | 3 | 6421 | 80 |
| E. poae | 0.009131 | 500,740 | 283,538 | 217,202 | 7292 | 1025 | 0 | 11,467 | 46 |
| E. scottii | 0.024456 | 649,936 | 408,925 | 241,011 | 11,654 | 4056 | 3 | 25,089 | 166 |
| E. typhina (Pt1807) | 0.010108 | 594,494 | 353,680 | 240,814 | 11,336 | 2080 | 1 | 42,243 | 242 |
| E. typhina (1756) | 0.009271 | 455,536 | 264,745 | 190,791 | 9370 | 932 | 1 | 24,706 | 202 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quenu, M.; Treindl, A.D.; Lee, K.; Takemoto, D.; Thünen, T.; Ashrafi, S.; Winter, D.; Ganley, A.R.D.; Leuchtmann, A.; Young, C.A.; et al. Telomere-to-Telomere Genome Sequences across a Single Genus Reveal Highly Variable Chromosome Rearrangement Rates but Absolute Stasis of Chromosome Number. J. Fungi 2022, 8, 670. https://doi.org/10.3390/jof8070670
Quenu M, Treindl AD, Lee K, Takemoto D, Thünen T, Ashrafi S, Winter D, Ganley ARD, Leuchtmann A, Young CA, et al. Telomere-to-Telomere Genome Sequences across a Single Genus Reveal Highly Variable Chromosome Rearrangement Rates but Absolute Stasis of Chromosome Number. Journal of Fungi. 2022; 8(7):670. https://doi.org/10.3390/jof8070670
Chicago/Turabian StyleQuenu, Mathieu, Artemis D. Treindl, Kate Lee, Daigo Takemoto, Torsten Thünen, Samad Ashrafi, David Winter, Austen R. D. Ganley, Adrian Leuchtmann, Carolyn A. Young, and et al. 2022. "Telomere-to-Telomere Genome Sequences across a Single Genus Reveal Highly Variable Chromosome Rearrangement Rates but Absolute Stasis of Chromosome Number" Journal of Fungi 8, no. 7: 670. https://doi.org/10.3390/jof8070670
APA StyleQuenu, M., Treindl, A. D., Lee, K., Takemoto, D., Thünen, T., Ashrafi, S., Winter, D., Ganley, A. R. D., Leuchtmann, A., Young, C. A., & Cox, M. P. (2022). Telomere-to-Telomere Genome Sequences across a Single Genus Reveal Highly Variable Chromosome Rearrangement Rates but Absolute Stasis of Chromosome Number. Journal of Fungi, 8(7), 670. https://doi.org/10.3390/jof8070670