
The Sporisorium reilianum Effector Vag2 Promotes Head Smut Disease via Suppression of Plant Defense Responses

 , and
, and 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Strains and Growth Conditions of S. reilianum and Maize, Symptom Scoring
2.2. Generation of S. reilianum Gene Deletion and Complementation Strains
2.3. Genomic DNA and RNA Isolation, and qRT-PCR Analysis
2.4. Bimolecular Fluorescence Complementation and Fluorescence Microscopy
2.5. Yeast Two-Hybrid Screening
2.6. Yeast Secretion Trap Assay
2.7. Metabolite Analysis
3. Results
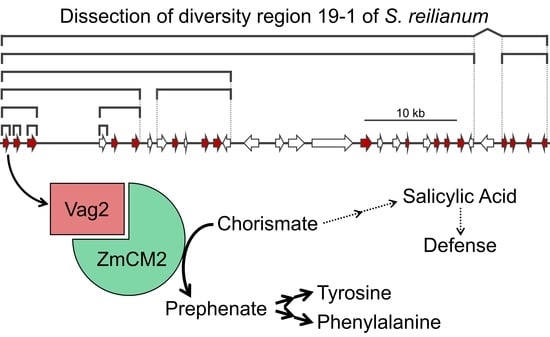
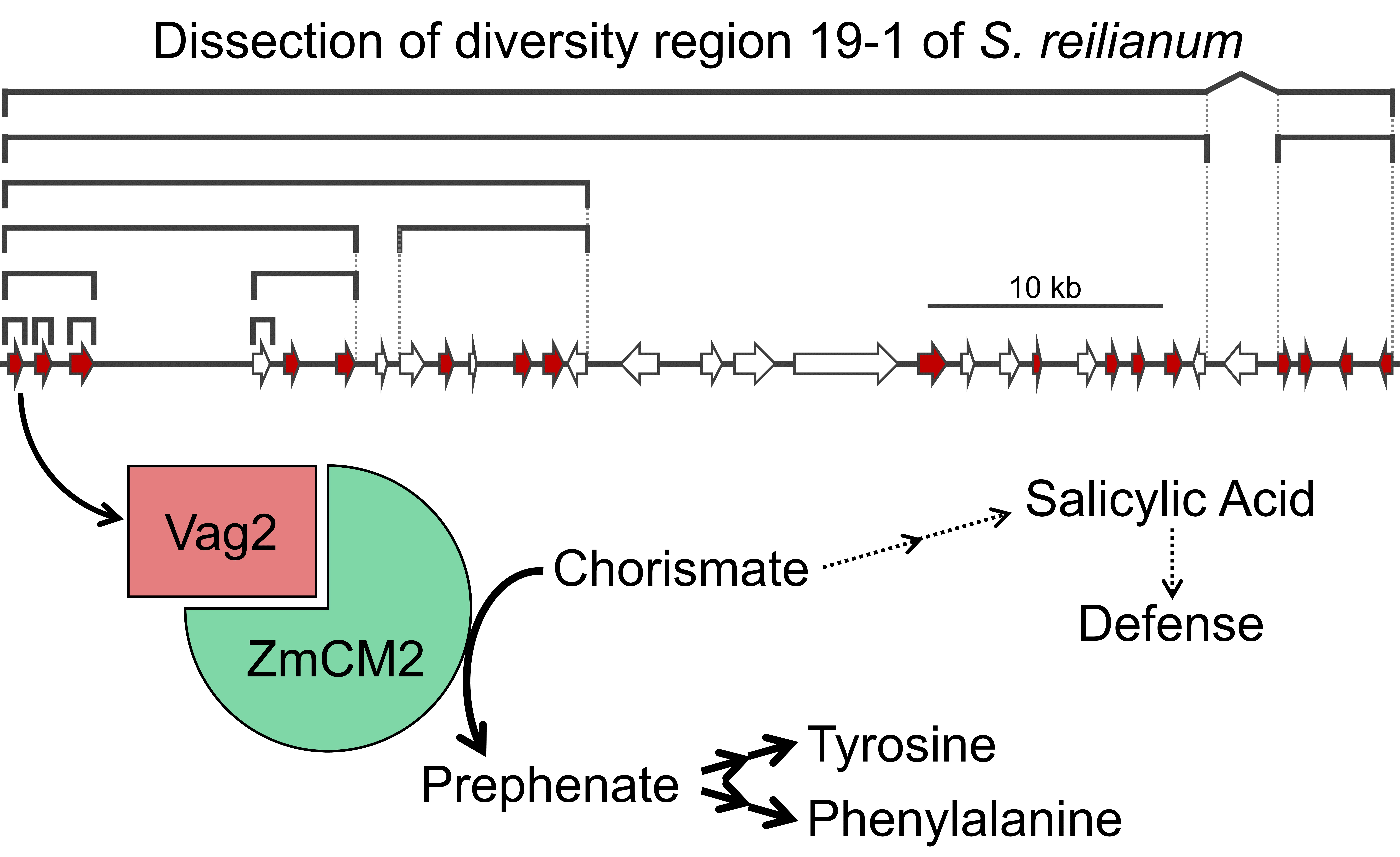
3.1. Diversity Region 19-1 of S. reilianum Contains the Virulence Gene vag2
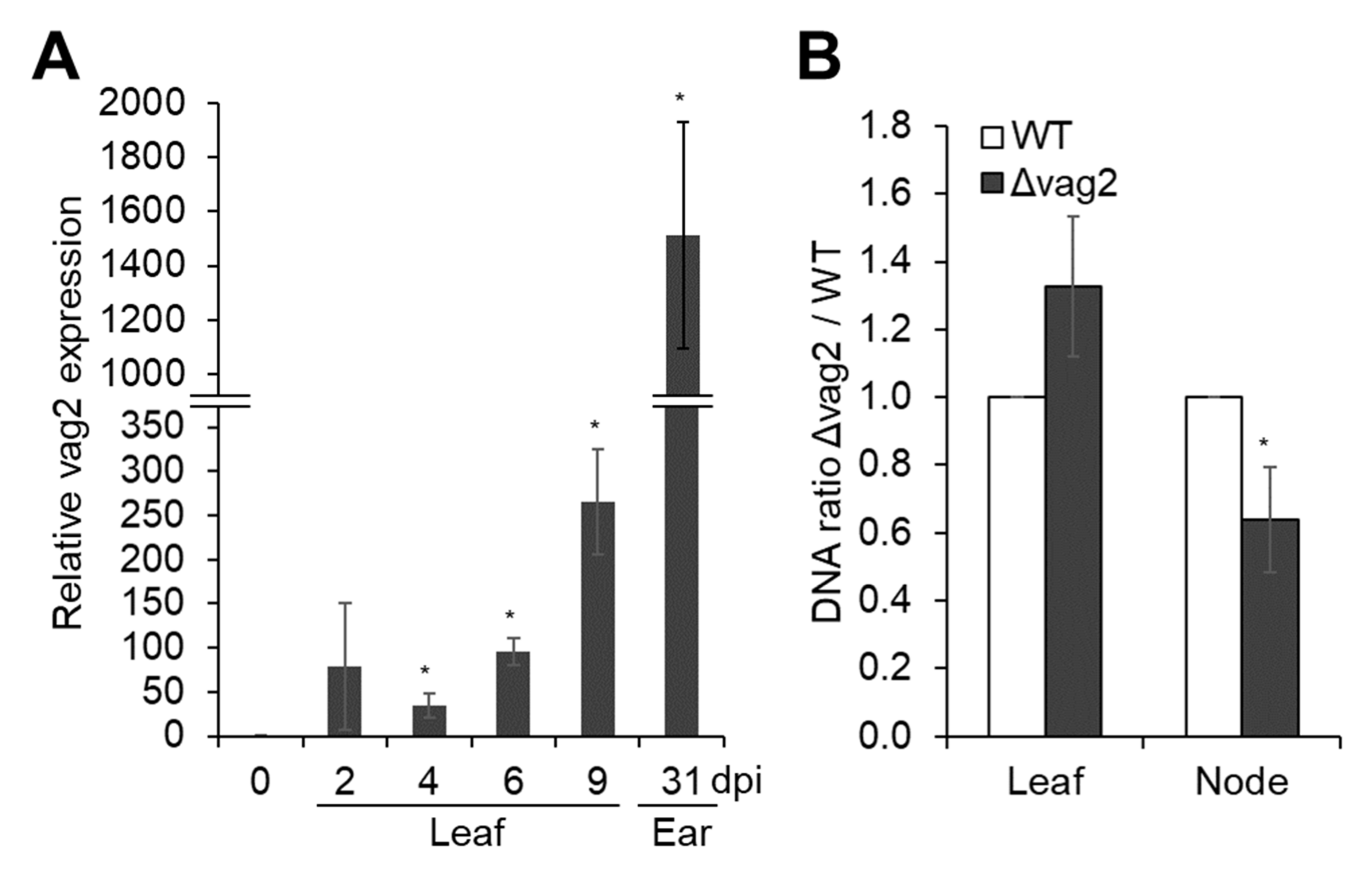
3.2. S. reilianum vag2 Is Transcriptionally Upregulated during Fungal Biotrophic Growth
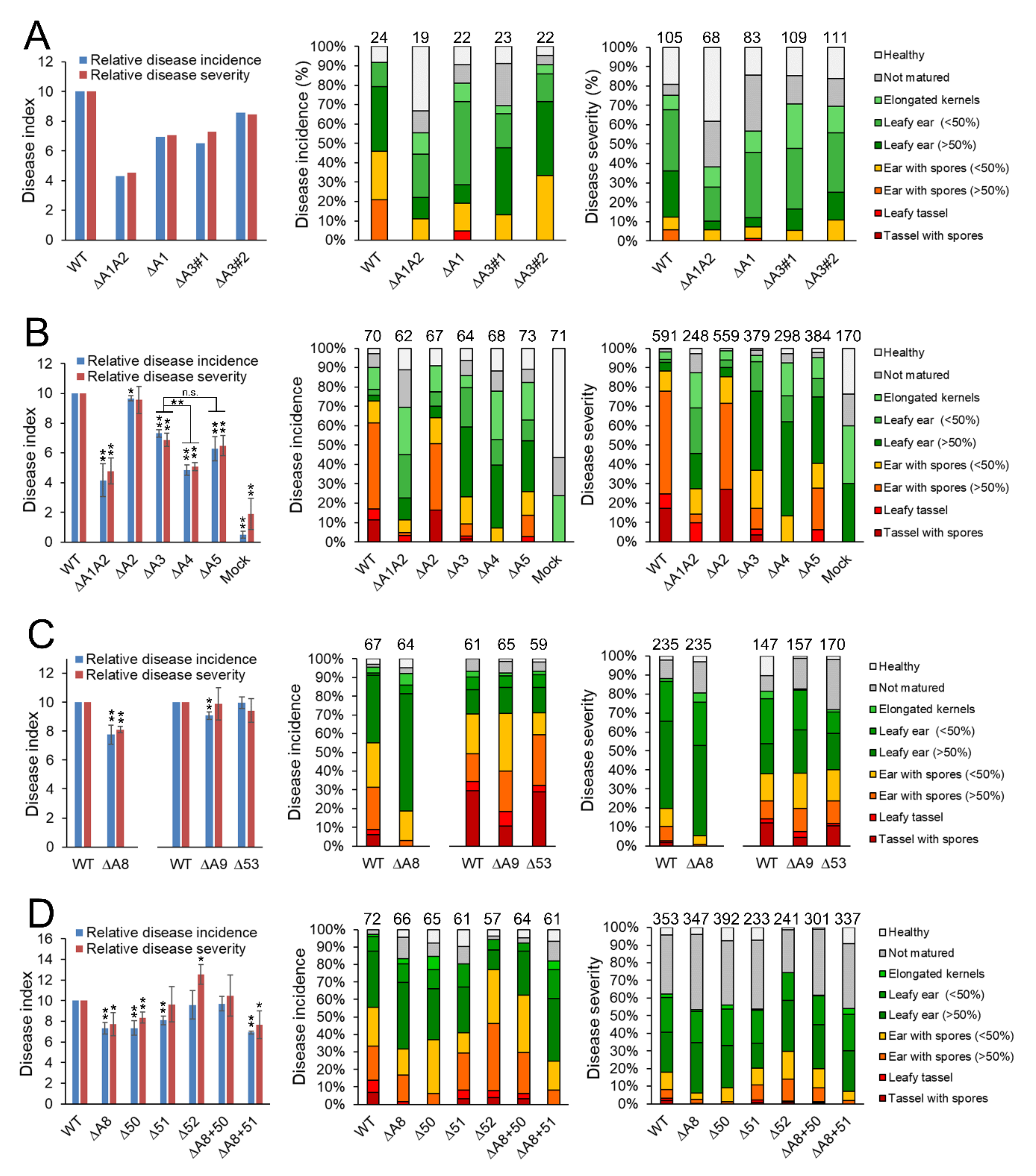
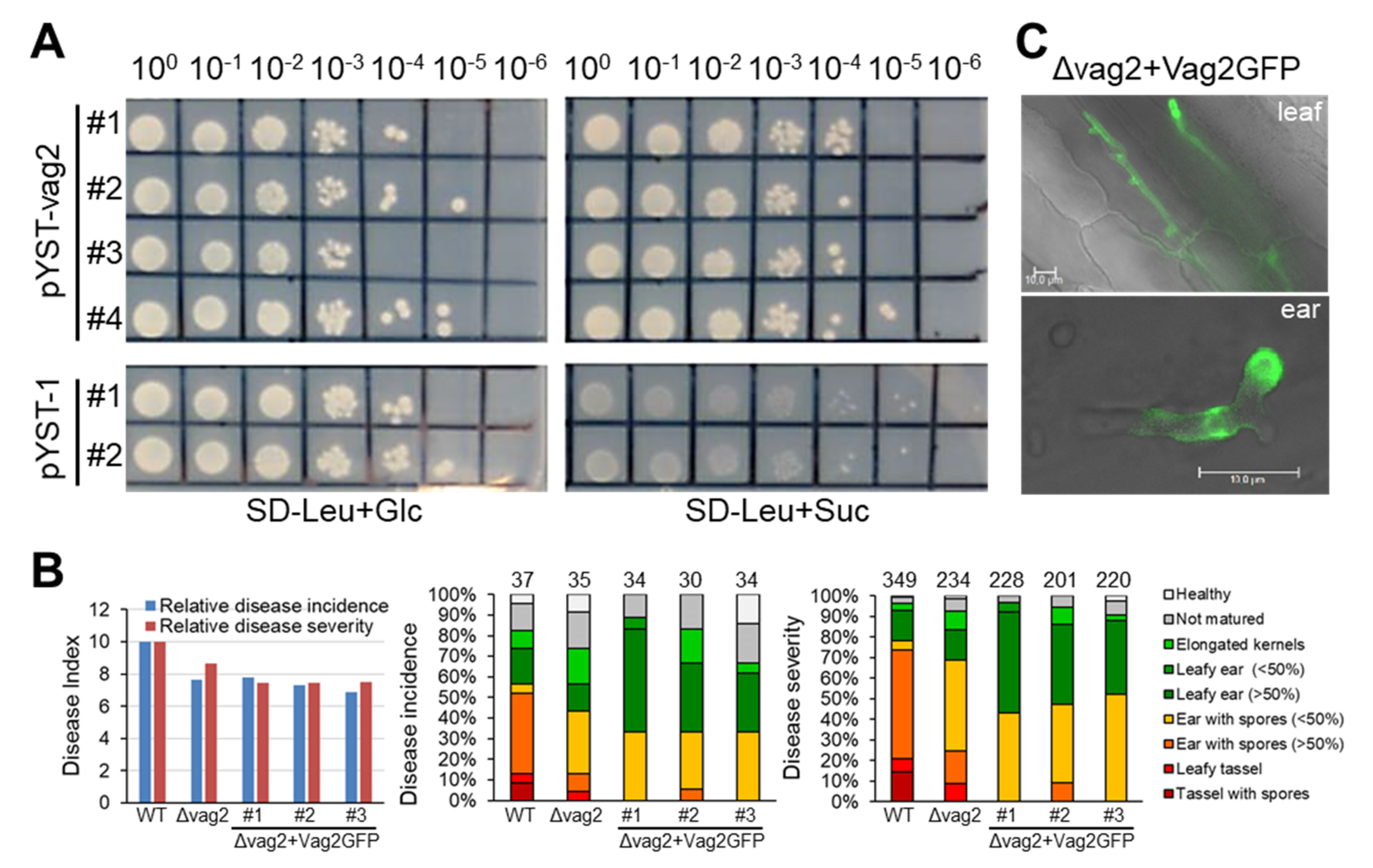
3.3. vag2 Deletion Mutants Show Reduced Systemic Spread in Maize
3.4. Vag2 Has a Functional Secretion Peptide
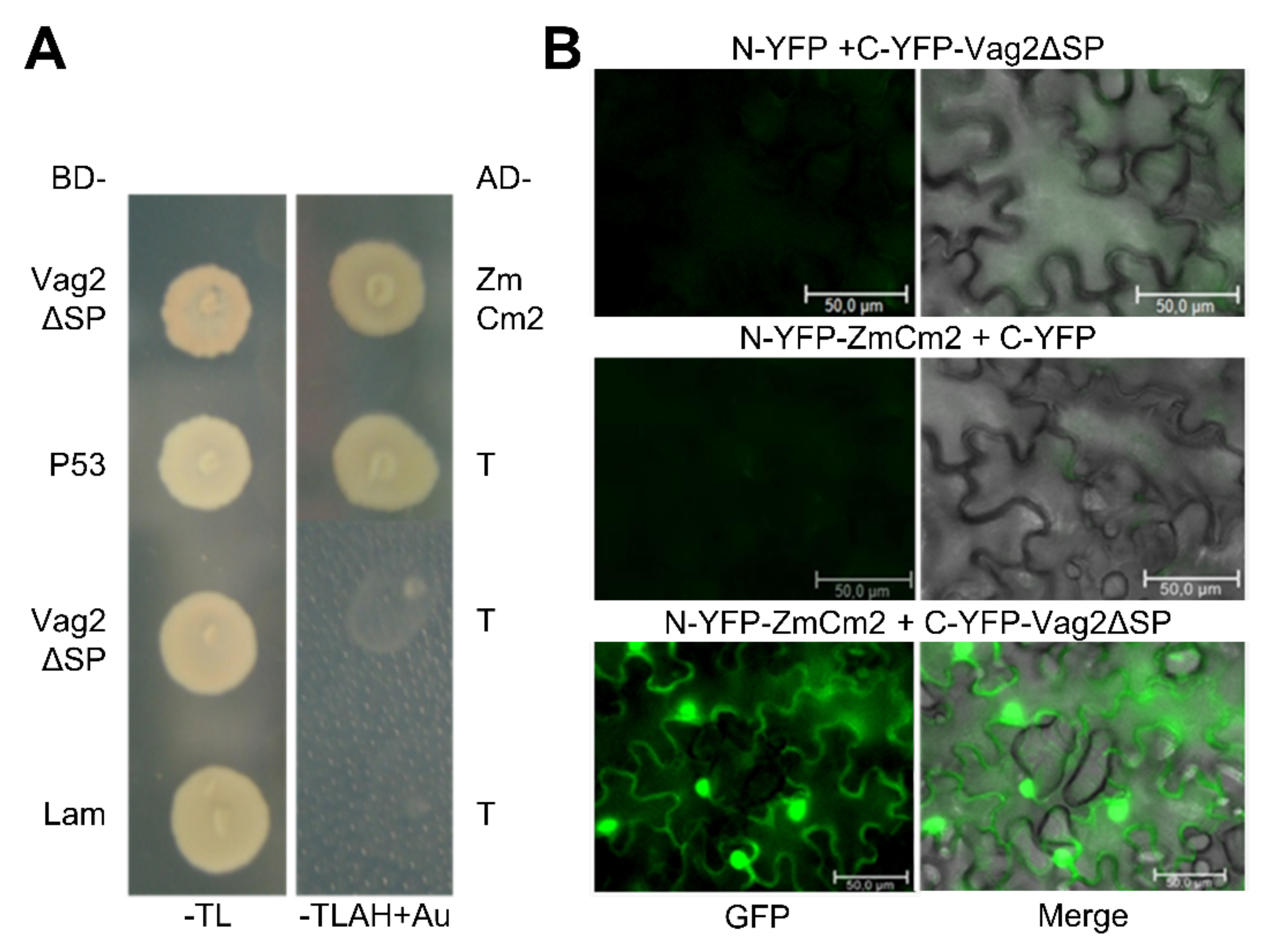
3.5. Interaction Partners of Vag2 Are Involved in Various Plant Processes
3.6. Vag2 Interacts with the Maize Chorismate Mutase 2 (ZmCM2)
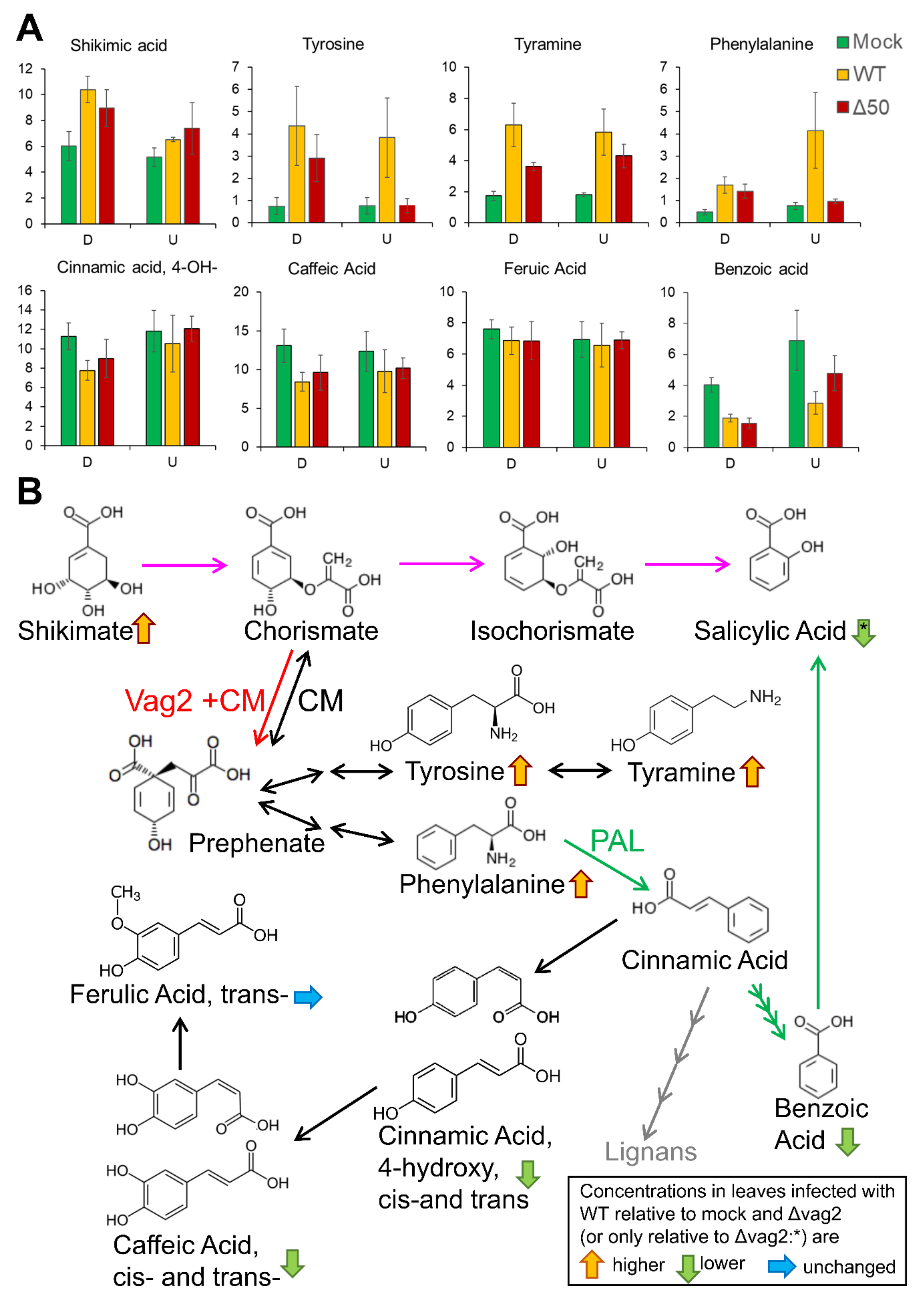
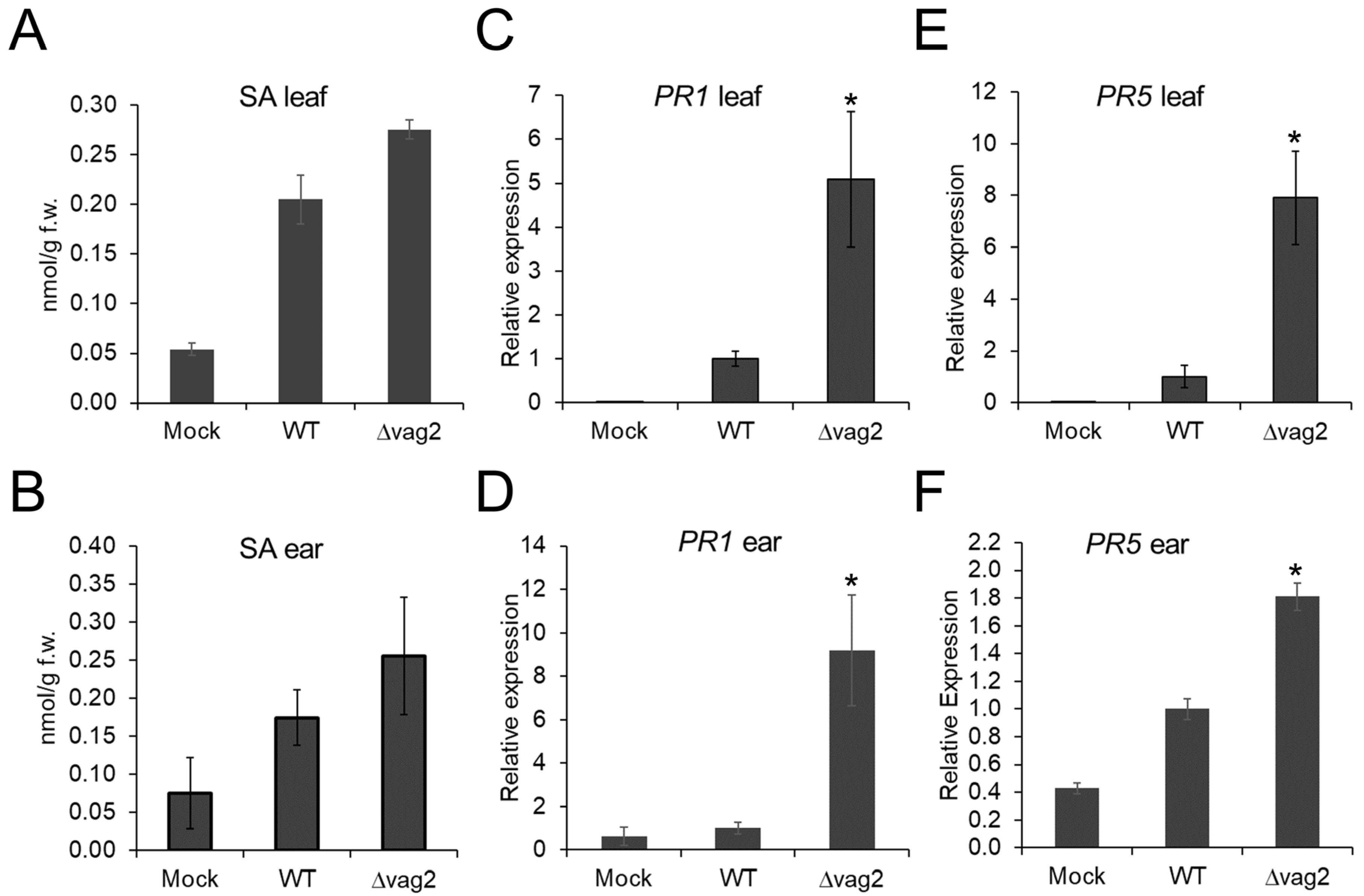
3.7. S. reilianum Δvag2 Deletion Strains Slightly Increase the SA Level in Colonized Tissue and Induce SA-Related Defense Gene Expression in Maize
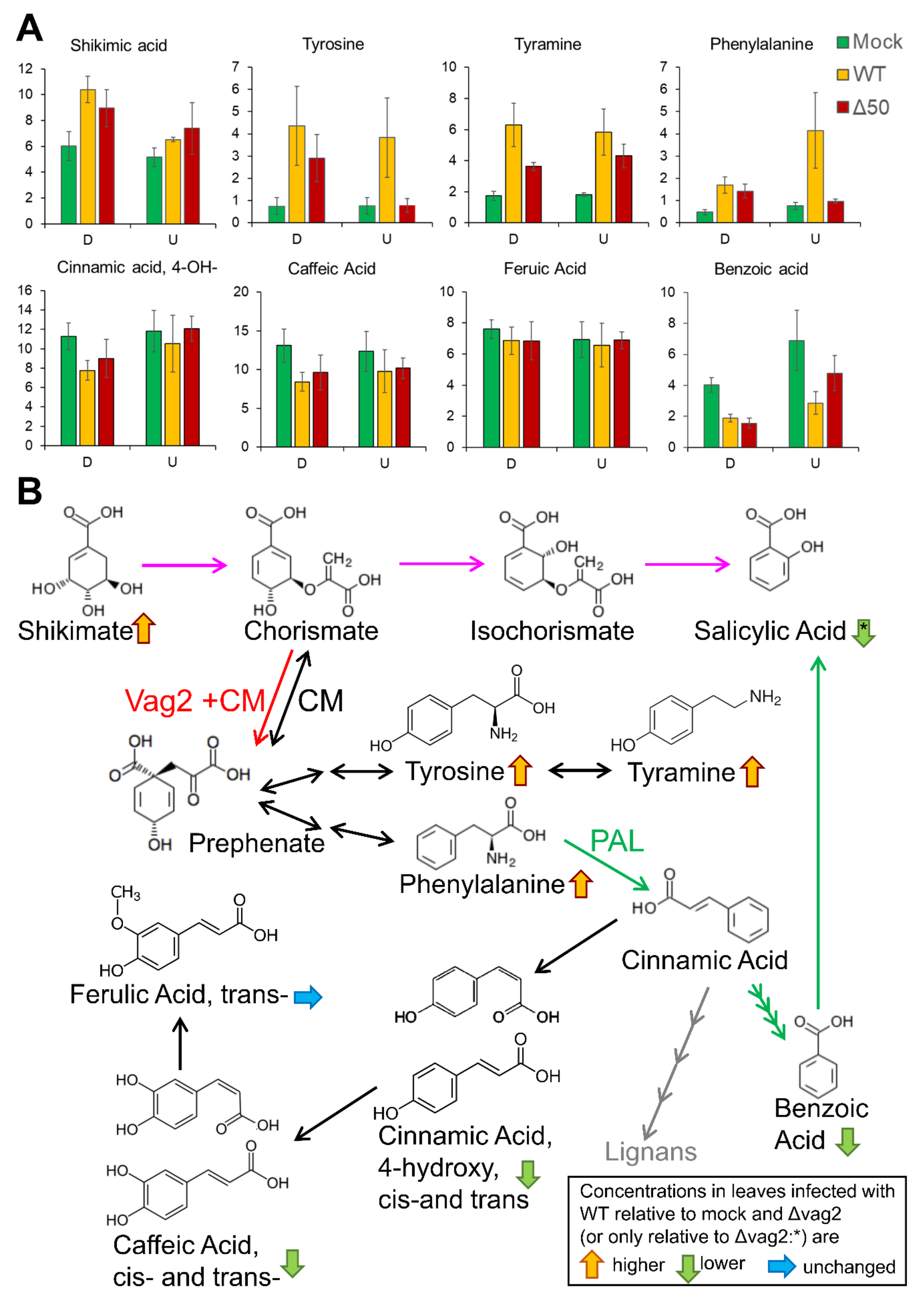
3.8. Metabolite Flux Is Redirected from SA Generation to Aromatic Amino Acid Accumulation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ghareeb, H.; Becker, A.; Iven, T.; Feussner, I.; Schirawski, J. Sporisorium reilianum infection changes inflorescence and branching architectures of maize. Plant Physiol. 2011, 156, 2037–2052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schirawski, J.; Mannhaupt, G.; Münch, K.; Brefort, T.; Schipper, K.; Doehlemann, G.; Di Stasio, M.; Rössel, N.; Mendoza-Mendoza, A.; Pester, D.; et al. Pathogenicity determinants in smut fungi revealed by genome comparison. Science 2010, 330, 1546–1548. [Google Scholar] [CrossRef] [PubMed]
- Matyac, C.A.; Kommedahl, T. Survival of teliospores of Sphacelotheca reiliana in soil. Phytopathology 1986, 76, 487–490. [Google Scholar] [CrossRef]
- Hanna, W.F. Studies in the physiology and cytology of Ustilago zeae and Sorosporium reilianum. Phytopathology 1929, 19, 415–443. [Google Scholar]
- Martinez, C.; Roux, C.; Jauneau, A.; Dargent, R. The biological cycle of Sporisorium reilianum f.sp. zeae: An overview using microscopy. Mycologia 2002, 94, 505–514. [Google Scholar] [CrossRef]
- Schirawski, J.; Heinze, B.; Wagenknecht, M.; Kahmann, R. Mating type loci of Sporisorium reilianum: Novel pattern with three a and multiple b specificities. Eukaryot. Cell 2005, 4, 1317–1327. [Google Scholar] [CrossRef] [Green Version]
- Poloni, A.; Schirawski, J. Host specificity of Sporisorium reilianum is determined by distinct mechanisms in maize and sorghum. Mol. Plant Pathol. 2016, 17, 741–754. [Google Scholar] [CrossRef]
- Martinez, C.; Roux, C.; Dargent, R. Biotrophic development of Sporisorium reilianum f. sp. zeae in vegetative shoot apex of maize. Phytopathology 1999, 89, 247–253. [Google Scholar] [CrossRef] [Green Version]
- Dangl, J.L.; Jones, J.D. Plant pathogens and integrated defence responses to infection. Nature 2001, 411, 826–833. [Google Scholar] [CrossRef]
- Zipfel, C. Pattern-recognition receptors in plant innate immunity. Curr. Opin. Immunol. 2008, 20, 10–16. [Google Scholar] [CrossRef]
- Zipfel, C. Early molecular events in PAMP-triggered immunity. Curr. Opin. Plant Biol. 2009, 12, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Boller, T.; Felix, G. A renaissance of elicitors: Perception of microbe-associated molecular patterns and danger signals by pattern-recognition receptors. Annu. Rev. Plant Biol. 2009, 60, 379–406. [Google Scholar] [CrossRef] [PubMed]
- Hogenhout, S.A.; Van der Hoorn, R.A.; Terauchi, R.; Kamoun, S. Emerging concepts in effector biology of plant-associated organisms. Mol. Plant Microbe Interact. 2009, 22, 115–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo Presti, L.; Lanver, D.; Schweizer, G.; Tanaka, S.; Liang, L.; Tollot, M.; Zuccaro, A.; Reissmann, S.; Kahmann, R. Fungal effectors and plant susceptibility. Annu. Rev. Plant Biol. 2015, 66, 513–545. [Google Scholar] [CrossRef] [PubMed]
- Thomma, B.P.; Nurnberger, T.; Joosten, M.H. Of PAMPs and effectors: The blurred PTI-ETI dichotomy. Plant Cell 2011, 23, 4–15. [Google Scholar] [CrossRef] [Green Version]
- Bi, G.; Su, M.; Li, N.; Liang, Y.; Dang, S.; Xu, J.; Hu, M.; Wang, J.; Zou, M.; Deng, Y.; et al. The ZAR1 resistosome is a calcium-permeable channel triggering plant immune signaling. Cell 2021, 184, 3528–3541.e12. [Google Scholar] [CrossRef]
- Bari, R.; Jones, J.D. Role of plant hormones in plant defence responses. Plant Mol. Biol. 2009, 69, 473–488. [Google Scholar] [CrossRef]
- Glazebrook, J.; Zook, M.; Mert, F.; Kagan, I.; Rogers, E.E.; Crute, I.R.; Holub, E.B.; Hammerschmidt, R.; Ausubel, F.M. Phytoalexin-deficient mutants of Arabidopsis reveal that PAD4 encodes a regulatory factor and that four PAD genes contribute to downy mildew resistance. Genetics 1997, 146, 381–392. [Google Scholar] [CrossRef]
- Qi, G.; Chen, J.; Chang, M.; Chen, H.; Hall, K.; Korin, J.; Liu, F.; Wang, D.; Fu, Z.Q. Pandemonium breaks out: Disruption of salicylic acid-mediated defense by plant pathogens. Mol. Plant 2018, 11, 1427–1439. [Google Scholar] [CrossRef] [Green Version]
- Ziemann, S.; van der Linde, K.; Lahrmann, U.; Acar, B.; Kaschani, F.; Colby, T.; Kaiser, M.; Ding, Y.; Schmelz, E.; Huffaker, A.; et al. An apoplastic peptide activates salicylic acid signalling in maize. Nat. Plants 2018, 4, 172–180. [Google Scholar] [CrossRef]
- Doehlemann, G.; Wahl, R.; Horst, R.J.; Voll, L.M.; Usadel, B.; Poree, F.; Stitt, M.; Pons-Kühnemann, J.; Sonnewald, U.; Kahmann, R.; et al. Reprogramming a maize plant: Transcriptional and metabolic changes induced by the fungal biotroph Ustilago maydis. Plant J. 2008, 56, 181–195. [Google Scholar] [CrossRef] [PubMed]
- Djamei, A.; Schipper, K.; Rabe, F.; Ghosh, A.; Vincon, V.; Kahnt, J.; Osorio, S.; Tohge, T.; Fernie, A.R.; Feussner, I.; et al. Metabolic priming by a secreted fungal effector. Nature 2011, 478, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Rabe, F.; Ajami-Rashidi, Z.; Doehlemann, G.; Kahmann, R.; Djamei, A. Degradation of the plant defence hormone salicylic acid by the biotrophic fungus Ustilago maydis. Mol. Microbiol. 2013, 89, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Rabe, F.; Seitner, D.; Bauer, L.; Navarrete, F.; Czedik-Eysenberg, A.; Rabanal, F.A.; Djamei, A. Phytohormone sensing in the biotrophic fungus Ustilago maydis—The dual role of the transcription factor Rss1. Mol. Microbiol. 2016, 102, 290–305. [Google Scholar] [CrossRef]
- Kämper, J.; Kahmann, R.; Bölker, M.; Ma, L.J.; Brefort, T.; Saville, B.J.; Banuett, F.; Kronstad, J.W.; Gold, S.E.; Müller, O.; et al. Insights from the genome of the biotrophic fungal plant pathogen Ustilago maydis. Nature 2006, 444, 97–101. [Google Scholar] [CrossRef]
- Laurie, J.D.; Ali, S.; Linning, R.; Mannhaupt, G.; Wong, P.; Güldener, U.; Münsterkötter, M.; Moore, R.; Kahmann, R.; Bakkeren, G.; et al. Genome comparison of barley and maize smut fungi reveals targeted loss of RNA silencing components and species-specific presence of transposable elements. Plant Cell 2012, 24, 1733–1745. [Google Scholar] [CrossRef] [Green Version]
- Que, Y.; Xu, L.; Wu, Q.; Liu, Y.; Ling, H.; Liu, Y.; Zhang, Y.; Guo, J.; Su, Y.; Chen, J.; et al. Genome sequencing of Sporisorium scitamineum provides insights into the pathogenic mechanisms of sugarcane smut. BMC Genom. 2014, 15, 996. [Google Scholar] [CrossRef] [Green Version]
- Sharma, R.; Mishra, B.; Runge, F.; Thines, M. Gene loss rather than gene gain is associated with a host jump from monocots to dicots in the smut fungus Melanopsichium pennsylvanicum. Genome Biol. Evol. 2014, 6, 2034–2049. [Google Scholar] [CrossRef] [Green Version]
- Perlin, M.H.; Amselem, J.; Fontanillas, E.; Toh, S.S.; Chen, Z.; Goldberg, J.; Duplessis, S.; Henrissat, B.; Young, S.; Zeng, Q.; et al. Sex and parasites: Genomic and transcriptomic analysis of Microbotryum lychnidis-dioicae, the biotrophic and plant-castrating anther smut fungus. BMC Genom. 2015, 16, 461. [Google Scholar] [CrossRef] [Green Version]
- Taniguti, L.M.; Schaker, P.D.; Benevenuto, J.; Peters, L.P.; Carvalho, G.; Palhares, A.; Quecine, M.C.; Nunes, F.R.; Kmit, M.C.; Wai, A.; et al. Complete genome sequence of Sporisorium scitamineum and biotrophic interaction transcriptome with Sugarcane. PLoS ONE 2015, 10, e0129318. [Google Scholar] [CrossRef]
- Rabe, F.; Bosch, J.; Stirnberg, A.; Guse, T.; Bauer, L.; Seitner, D.; Rabanal, F.A.; Czedik-Eysenberg, A.; Uhse, S.; Bindics, J.; et al. A complete toolset for the study of Ustilago bromivora and Brachypodium sp. as a fungal-temperate grass pathosystem. eLife 2016, 5, e20522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutheil, J.Y.; Mannhaupt, G.; Schweizer, G.; Sieber, C.M.K.; Münsterkötter, M.; Güldener, U.; Schirawski, J.; Kahmann, R. A tale of genome compartmentalization: The evolution of virulence clusters in smut fungi. Genome Biol. Evol. 2016, 8, 681–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, Z.; Pan, Y.; Zhang, Y.; Cui, H.; Jin, G.; McHardy, A.C.; Fan, L.; Yu, X. Comparative whole-genome analysis reveals artificial selection effects on Ustilago esculenta genome. DNA Res. 2017, 24, 635–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Courville, K.J.; Frantzeskakis, L.; Gul, S.; Haeger, N.; Kellner, R.; Heßler, N.; Day, B.; Usadel, B.; Gupta, Y.K.; van Esse, H.P.; et al. Smut infection of perennial hosts: The genome and the transcriptome of the Brassicaceae smut fungus Thecaphora thlaspeos reveal functionally conserved and novel effectors. New Phytol. 2019, 222, 1474–1492. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.; Sultana, T.; Kesanakurti, P.; Hambleton, S. Genome sequencing and comparison of five Tilletia species to identify candidate genes for the detection of regulated species infecting wheat. IMA Fungus 2019, 10, 11. [Google Scholar] [CrossRef]
- Doehlemann, G.; van der Linde, K.; Assmann, D.; Schwammbach, D.; Hof, A.; Mohanty, A.; Jackson, D.; Kahmann, R. Pep1, a secreted effector protein of Ustilago maydis, is required for successful invasion of plant cells. PLoS Pathog. 2009, 5, e1000290. [Google Scholar] [CrossRef] [Green Version]
- Hemetsberger, C.; Herrberger, C.; Zechmann, B.; Hillmer, M.; Doehlemann, G. The Ustilago maydis effector Pep1 suppresses plant immunity by inhibition of host peroxidase activity. PLoS Pathog. 2012, 8, e1002684. [Google Scholar] [CrossRef] [Green Version]
- Doehlemann, G.; Reissmann, S.; Assmann, D.; Fleckenstein, M.; Kahmann, R. Two linked genes encoding a secreted effector and a membrane protein are essential for Ustilago maydis-induced tumour formation. Mol. Microbiol. 2011, 81, 751–766. [Google Scholar] [CrossRef]
- Mueller, A.N.; Ziemann, S.; Treitschke, S.; Assmann, D.; Doehlemann, G. Compatibility in the Ustilago maydis-maize interaction requires inhibition of host cysteine proteases by the fungal effector Pit2. PLoS Pathog. 2013, 9, e1003177. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, S.; Brefort, T.; Neidig, N.; Djamei, A.; Kahnt, J.; Vermerris, W.; Koenig, S.; Feussner, K.; Feussner, I.; Kahmann, R. A secreted Ustilago maydis effector promotes virulence by targeting anthocyanin biosynthesis in maize. eLife 2014, 3, e01355. [Google Scholar] [CrossRef]
- Redkar, A.; Hoser, R.; Schilling, L.; Zechmann, B.; Krzymowska, M.; Walbot, V.; Doehlemann, G. A secreted effector protein of Ustilago maydis guides maize leaf cells to form tumors. Plant Cell 2015, 27, 1332–1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutra, D.; Agrawal, N.; Ghareeb, H.; Schirawski, J. Screening of secreted proteins of Sporisorium reilianum f. sp. zeae for cell death suppression in Nicotiana benthamiana. Front. Plant Sci. 2020, 11, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghareeb, H.; Zhao, Y.; Schirawski, J. Sporisorium reilianum possesses a pool of effector proteins that modulate virulence on maize. Mol. Plant Pathol. 2019, 20, 124–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brefort, T.; Tanaka, S.; Neidig, N.; Doehlemann, G.; Vincon, V.; Kahmann, R. Characterization of the largest effector gene cluster of Ustilago maydis. PLoS Pathog. 2014, 10, e1003866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kämper, J. A PCR-based system for highly efficient generation of gene replacement mutants in Ustilago maydis. Mol. Gen. Genom. 2004, 271, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, C.S.; Winston, F. A ten-minute DNA preparation from yeast efficiently releases autonomous plasmids for transformation of Escherichia coli. Gene 1987, 57, 267–272. [Google Scholar] [CrossRef]
- Walter, M.; Chaban, C.; Schütze, K.; Batistic, O.; Weckermann, K.; Näke, C.; Blazevic, D.; Grefen, C.; Schumacher, K.; Oecking, C.; et al. Visualization of protein interactions in living plant cells using bimolecular fluorescence complementation. Plant J. 2004, 40, 428–438. [Google Scholar] [CrossRef]
- Koncz, C.; Schell, J. The promoter of TL-DNA gene 5 controls the tissue-specific expression of chimaeric genes carried by a novel type of Agrobacterium binary vector. Mol. Gen. Genet. 1986, 204, 383–396. [Google Scholar] [CrossRef]
- Ghareeb, H.; Drechsler, F.; Löfke, C.; Teichmann, T.; Schirawski, J. Suppressor of apical dominance1 of Sporisorium reilianum modulates inflorescence branching architecture in maize and Arabidopsis. Plant Physiol. 2015, 169, 2789–2804. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.J.; Kim, B.D.; Rose, J.K. Identification of eukaryotic secreted and cell surface proteins using the yeast secretion trap screen. Nat. Protoc. 2006, 1, 2439–2447. [Google Scholar] [CrossRef]
- Ternes, P.; Feussner, K.; Werner, S.; Lerche, J.; Iven, T.; Heilmann, I.; Riezman, H.; Feussner, I. Disruption of the ceramide synthase LOH1 causes spontaneous cell death in Arabidopsis thaliana. New Phytol. 2011, 192, 841–854. [Google Scholar] [CrossRef] [PubMed]
- Dethloff, F.; Erban, A.; Orf, I.; Alpers, J.; Fehrle, I.; Beine-Golovchuk, O.; Schmidt, S.; Schwachtje, J.; Kopka, J. Profiling methods to identify cold-regulated primary metabolites using gas chromatography coupled to mass spectrometry. Methods Mol. Biol. 2014, 1166, 171–197. [Google Scholar] [CrossRef] [PubMed]
- Kopka, J.; Schauer, N.; Krueger, S.; Birkemeyer, C.; Usadel, B.; Bergmüller, E.; Dörmann, P.; Weckwerth, W.; Gibon, Y.; Stitt, M.; et al. GMD@CSB.DB: The Golm Metabolome Database. Bioinformatics 2005, 21, 1635–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luedemann, A.; Strassburg, K.; Erban, A.; Kopka, J. TagFinder for the quantitative analysis of gas chromatography-mass spectrometry (GC-MS)-based metabolite profiling experiments. Bioinformatics 2008, 24, 732–737. [Google Scholar] [CrossRef] [PubMed]
- Strehmel, N.; Hummel, J.; Erban, A.; Strassburg, K.; Kopka, J. Retention index thresholds for compound matching in GC-MS metabolite profiling. J. Chromatogr. B Analyt. Technol. Biomed Life Sci. 2008, 87, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Monaco, M.K.; Sen, T.Z.; Dharmawardhana, P.D.; Ren, L.; Schaeffer, M.; Naithani, S.; Amarasinghe, V.; Thomason, J.; Harper, L.; Gardiner, J.; et al. Maize metabolic network construction and transcriptome analysis. Plant Genome 2013, 6, plantgenome2012.09.0025. [Google Scholar] [CrossRef] [Green Version]
- Wildermuth, M.C.; Dewdney, J.; Wu, G.; Ausubel, F.M. Isochorismate synthase is required to synthesize salicylic acid for plant defence. Nature 2001, 414, 562–565. [Google Scholar] [CrossRef]
- Dempsey, D.A.; Vlot, A.C.; Wildermuth, M.C.; Klessig, D.F. Salicylic acid biosynthesis and metabolism. Arab. Book 2011, 9, e0156. [Google Scholar] [CrossRef] [Green Version]
- Eberhard, J.; Ehrler, T.T.; Epple, P.; Felix, G.; Raesecke, H.R.; Amrhein, N.; Schmid, J. Cytosolic and plastidic chorismate mutase isozymes from Arabidopsis thaliana: Molecular characterization and enzymatic properties. Plant J. 1996, 10, 815–821. [Google Scholar] [CrossRef]
- Parthasarathy, A.; Cross, P.J.; Dobson, R.; Adams, L.E.; Savka, M.A.; Hudson, A.O. A three-ring circus: Metabolism of the three proteogenic aromatic amino acids and their role in the health of plants and animals. Front. Mol. Biosci. 2018, 5, 29. [Google Scholar] [CrossRef] [PubMed]
- Potter, S.; Moreland, D.E.; Kreuz, K.; Ward, E. Induction of cytochrome P450 genes by ethanol in maize. Drug Metab. Drug Interact. 1995, 12, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Rösler, J.; Krekel, F.; Amrhein, N.; Schmid, J. Maize phenylalanine ammonia-lyase has tyrosine ammonia-lyase activity. Plant Physiol. 1997, 113, 175–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, R.A.; Barros, J. Lignin biosynthesis: Old roads revisited and new roads explored. Open Biol. 2019, 9, 190215. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, N.; Reissmann, S.; Schipper, K.; Gonzalez, C.; Assmann, D.; Glatter, T.; Moretti, M.; Ma, L.S.; Rexer, K.H.; Snetselaar, K.; et al. A cell surface-exposed protein complex with an essential virulence function in Ustilago maydis. Nat. Microbiol. 2021, 6, 722–730. [Google Scholar] [CrossRef]
- Alcântara, A.; Bosch, J.; Nazari, F.; Hoffmann, G.; Gallei, M.; Uhse, S.; Darino, M.A.; Olukayode, T.; Reumann, D.; Baggaley, L.; et al. Systematic Y2H screening reveals extensive effector-complex formation. Front. Plant Sci. 2019, 10, 1437. [Google Scholar] [CrossRef] [Green Version]
- Ali, S.; Laurie, J.D.; Linning, R.; Cervantes-Chávez, J.A.; Gaudet, D.; Bakkeren, G. An immunity-triggering effector from the Barley smut fungus Ustilago hordei resides in an Ustilaginaceae-specific cluster bearing signs of transposable element-assisted evolution. PLoS Pathog. 2014, 10, e1004223. [Google Scholar] [CrossRef]
- Montenegro Alonso, A.P.; Ali, S.; Song, X.; Linning, R.; Bakkeren, G. UhAVR1, an HR-triggering avirulence effector of Ustilago hordei, is secreted via the ER-golgi pathway, localizes to the cytosol of barley cells during in planta-expression, and contributes to virulence early in infection. J. Fungi 2020, 6, 178. [Google Scholar] [CrossRef]
- Rekhter, D.; Lüdke, D.; Ding, Y.; Feussner, K.; Zienkiewicz, K.; Lipka, V.; Wiermer, M.; Zhang, Y.; Feussner, I. Isochorismate-derived biosynthesis of the plant stress hormone salicylic acid. Science 2019, 365, 498–502. [Google Scholar] [CrossRef]
- Torrens-Spence, M.P.; Bobokalonova, A.; Carballo, V.; Glinkerman, C.M.; Pluskal, T.; Shen, A.; Weng, J.K. PBS3 and EPS1 complete salicylic acid biosynthesis from isochorismate in Arabidopsis. Mol. Plant 2019, 12, 1577–1586. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Interaction Partner | Frequency | Gene ID | Description |
|---|---|---|---|
| Metabolism | |||
| IP1 | 24 | GRMZM2G179454 | Zea mays chorismate mutase (ZmCm2) |
| IP2 | 11 | GRMZM2G027663 | Putative ThiC superfamily protein |
| IP3 | 10 | GRMZM2G006329 | Zea mays Enzyme: pleckstrin homology (PH) domain-containing protein |
| IP4 | 6 | GRMZM2G135588 | Putative citrate synthase family protein |
| IP5 | 4 | GRMZM2G081585 | Chloroplastic iron-superoxide dismutase (sodB) |
| IP6 | 3 | AC208571.4_FG001 | Hypothetical protein containing a haloacid dehalogenase-like hydrolase family domain / NHL repeat domain |
| IP7 | 3 | GRMZM2G300862 | Aspartate kinase |
| IP8 | 3 | GRMZM2G014788 | Unknown protein containing Carboxypeptidase regulatory-like domain |
| IP9 | 2 | GRMZM2G135588 | Aspartate kinase homoserine dehydrogenase 2 (akh2) |
| IP10 | 2 | GRMZM2G049538 | Terpene synthase1 |
| IP11 | 1 | GRMZM2G151934 | Zea mays protein DA1-related 2-like |
| IP12 | 1 | GRMZM2G043198 | Pyruvate dehydrogenase 2 (pdh2) |
| IP13 | 1 | GRMZM2G121612 | Starch synthase |
| IP14 | 1 | GRMZM2G118806 | Uncharacterized protein with proteolysis and peptidase activity |
| IP15 | 1 | GRMZM2G088689 | 2-Oxoisovalerate dehydrogenase (acylating) |
| IP16 | 1 | GRMZM2G448142 | Putative NADH-quinone oxidoreductase subunit K |
| Transcription/DNA binding | |||
| IP17 | 7 | GRMZM2G100246 | Unknown protein containing NOT2,3,5 domain |
| IP18 | 4 | GRMZM2G440943 | Helicase/SANT-associated, DNA binding protein |
| IP19 | 2 | GRMZM2G351304 | Uncharacterized protein containing chromosome segregation protein SMC domain |
| IP20 | 2 | AC225308.2_FG005 | Putative homeodomain-like transcription factor superfamily protein |
| IP21 | 1 | GRMZM2G133016 | MYB DNA-binding domain superfamily protein |
| IP22 | 1 | GRMZM5G876621 | Zea mays putative RING zinc finger domain superfamily protein |
| IP23 | 1 | GRMZM2G377369 | Uncharacterized protein containing DNA binding site |
| IP24 | 1 | AC226373.2 | Zink finger C-x8-C-x5-C-x3-H type family protein |
| IP25 | 1 | GRMZM2G340749 | General negative regulator of transcription |
| Protein processes | |||
| IP26 | 5 | GRMZM2G027282 | Proteasome 26S subunit 6A (RPT5a) |
| IP27 | 3 | GRMZM2G168119 | Putative HSP20-like chaperone domain family protein |
| IP28 | 2 | GRMZM2G134980 | Putative dnaJ chaperone family protein |
| IP29 | 2 | GRMZM2G006781 | Conserved oligomeric Golgi complex subunit 8 |
| IP30 | 1 | GRMZM2G551402 | Unknown protein containing a ubiquitin carboxyl-terminal hydrolase domain |
| IP31 | 1 | GRMZM2G012631 | HSP protein (HSP90-2) |
| IP32 | 1 | GRMZM2G137495 | DnaJ domain or J-domain. DnaJ/Hsp40 (heat shock protein 40) |
| IP33 | 1 | GRMZM2G162968 | Chaperone protein ClpB2 |
| IP34 | 1 | GRMZM2G154312 | Co-chaperone protein SBA1 |
| Signaling | |||
| IP35 | 4 | GRMZM2G038982 | Uncharacterized protein containing a STKc_MAP3K-like domain |
| IP36 | 3 | GRMZM2G126946 | Zea mays putative calcium-dependent lipid-binding (CaLB domain) family protein |
| IP37 | 1 | GRMZM2G152877 | Uncharacterized protein containing F-box-like and Cupin-like domain domain |
| IP38 | 1 | GRMZM2G326472 | Uncharacterized protein containing a STKc_MAP3K-like domain |
| Nuclear processes | |||
| IP39 | 2 | GRMZM2G159028 | RNA binding protein |
| IP40 | 1 | GRMZM2G111014 | Unknown protein containing DNA gyrase subunit |
| IP41 | 1 | GRMZM2G588223 | Hypothetical protein ZEAMMB73 containing double-stranded RNA binding motif |
| IP42 | 1 | GRMZM2G030128 | DNA repair-recombination protein (rad50) |
| IP43 | 1 | AC205703.4_FG010 | Hypothetical protein ZEAMMB73_142911/ATPase involved in DNA replication, recombination, and repair |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Y.; Agrawal, N.; Ghareeb, H.; Habib, M.T.; Dickmeis, S.; Schwachtje, J.; Iven, T.E.; Kopka, J.; Feussner, I.; Schirawski, J. The Sporisorium reilianum Effector Vag2 Promotes Head Smut Disease via Suppression of Plant Defense Responses. J. Fungi 2022, 8, 498. https://doi.org/10.3390/jof8050498
Zhao Y, Agrawal N, Ghareeb H, Habib MT, Dickmeis S, Schwachtje J, Iven TE, Kopka J, Feussner I, Schirawski J. The Sporisorium reilianum Effector Vag2 Promotes Head Smut Disease via Suppression of Plant Defense Responses. Journal of Fungi. 2022; 8(5):498. https://doi.org/10.3390/jof8050498
Chicago/Turabian StyleZhao, Yulei, Nisha Agrawal, Hassan Ghareeb, Mohammad Tanbir Habib, Sascha Dickmeis, Jens Schwachtje, Tim E. Iven, Joachim Kopka, Ivo Feussner, and Jan Schirawski. 2022. "The Sporisorium reilianum Effector Vag2 Promotes Head Smut Disease via Suppression of Plant Defense Responses" Journal of Fungi 8, no. 5: 498. https://doi.org/10.3390/jof8050498
APA StyleZhao, Y., Agrawal, N., Ghareeb, H., Habib, M. T., Dickmeis, S., Schwachtje, J., Iven, T. E., Kopka, J., Feussner, I., & Schirawski, J. (2022). The Sporisorium reilianum Effector Vag2 Promotes Head Smut Disease via Suppression of Plant Defense Responses. Journal of Fungi, 8(5), 498. https://doi.org/10.3390/jof8050498