
Genome-Wide Study of Conidiation-Related Genes in the Aphid-Obligate Fungal Pathogen Conidiobolus obscurus (Entomophthoromycotina)


Abstract
:1. Introduction
2. Materials and Methods
2.1. Fungal Culture
2.2. Genome Sequencing, Assembly, and Annotation
2.3. RNA Extraction and Transcript Assembly
2.4. Quantification of Transcripts
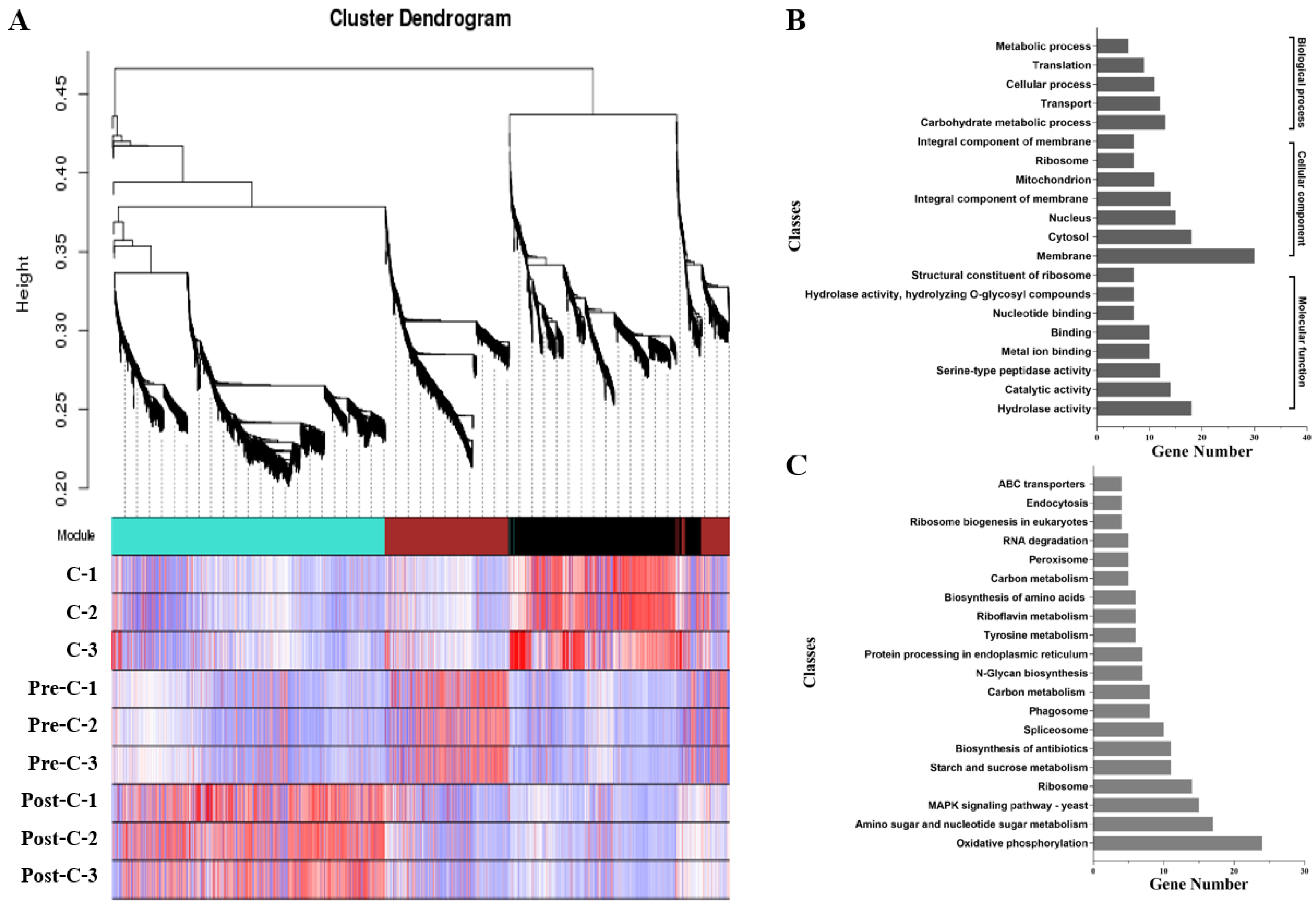
2.5. Co-Expression Network Construction
2.6. Phylogenetic Analysis
2.7. Comparative Genomic Analysis
3. Results
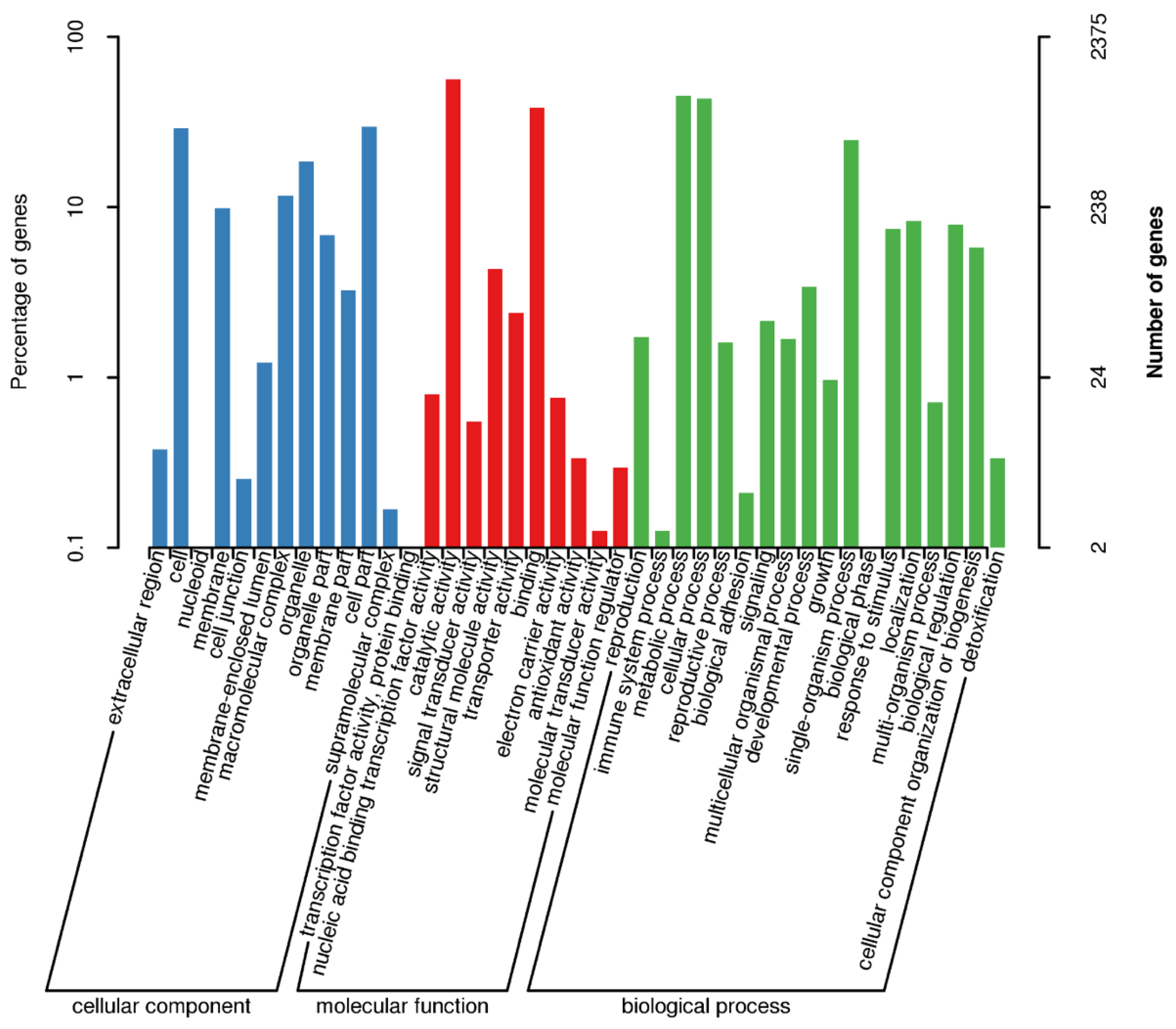
3.1. Global Characteristics of the C. obscurus Genome
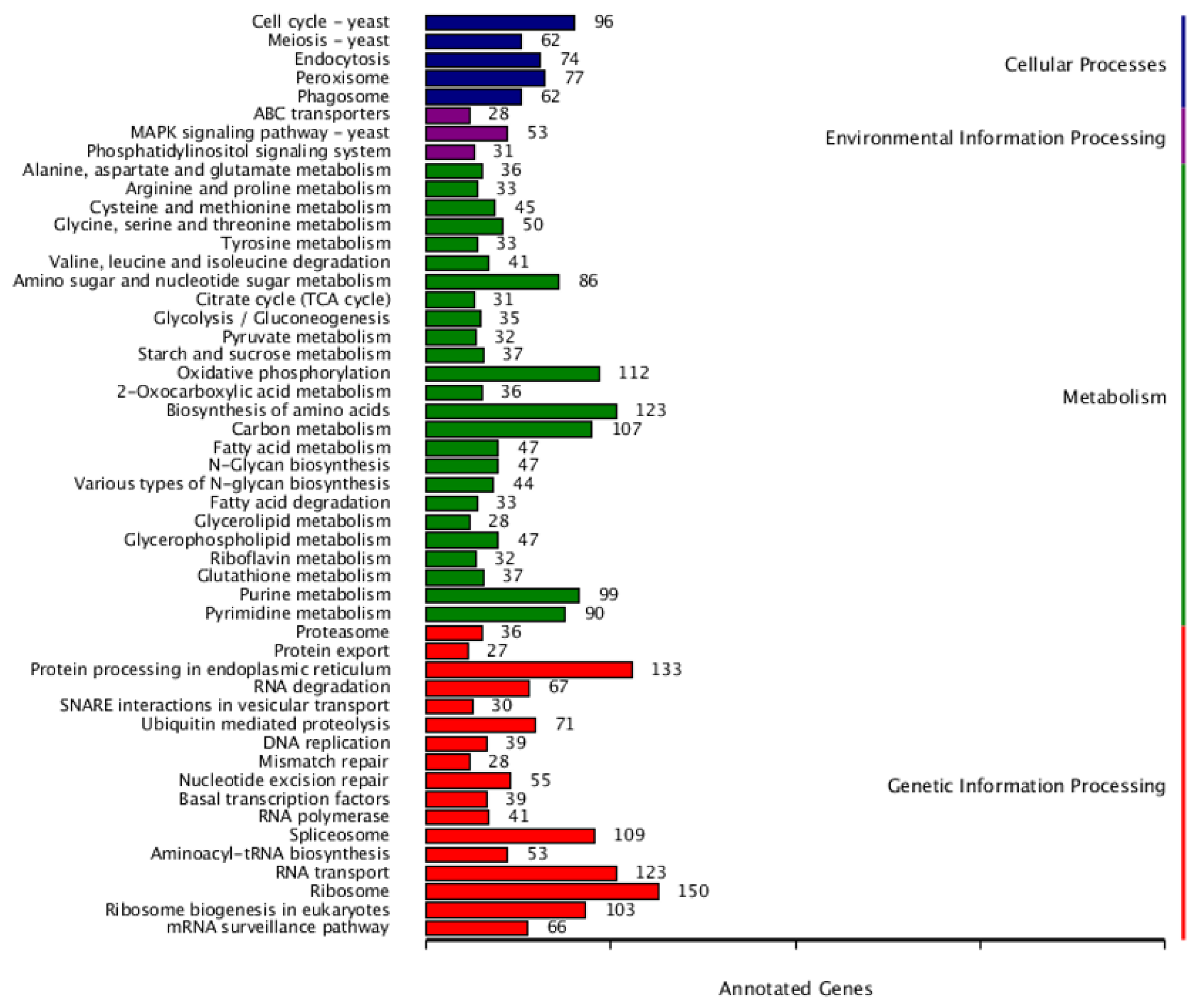
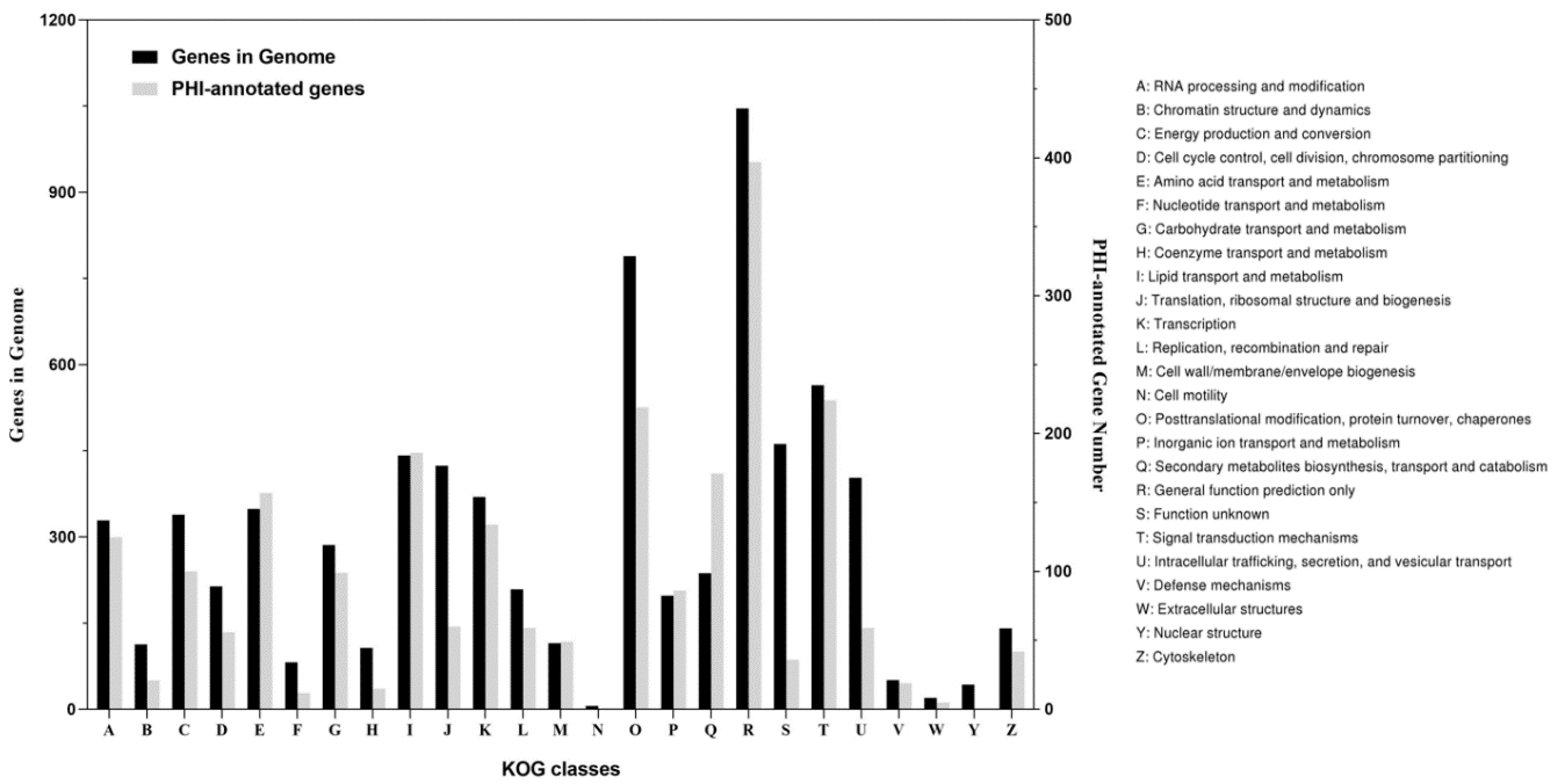
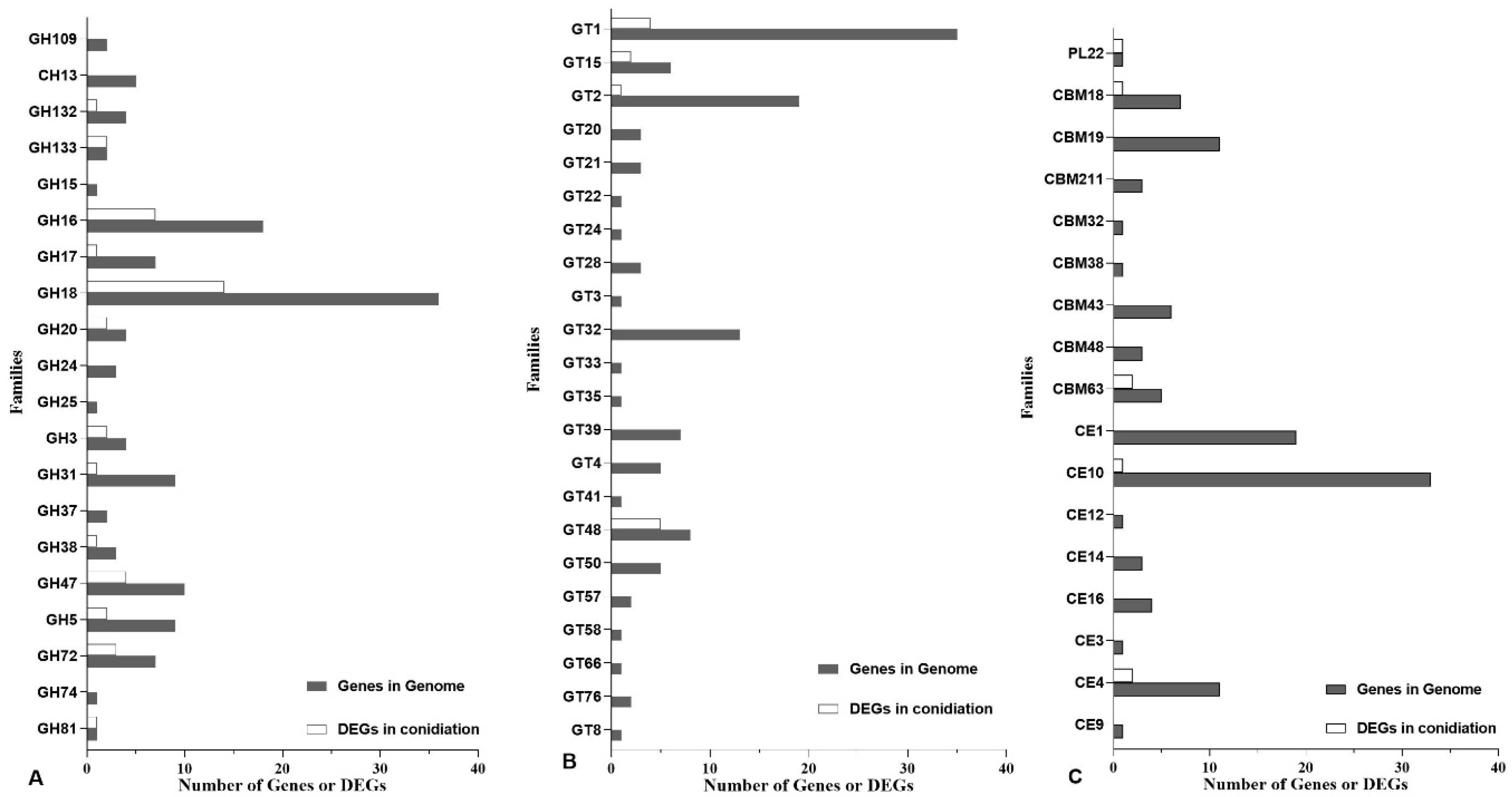
3.2. Annotation in PHI and CAZy Databases
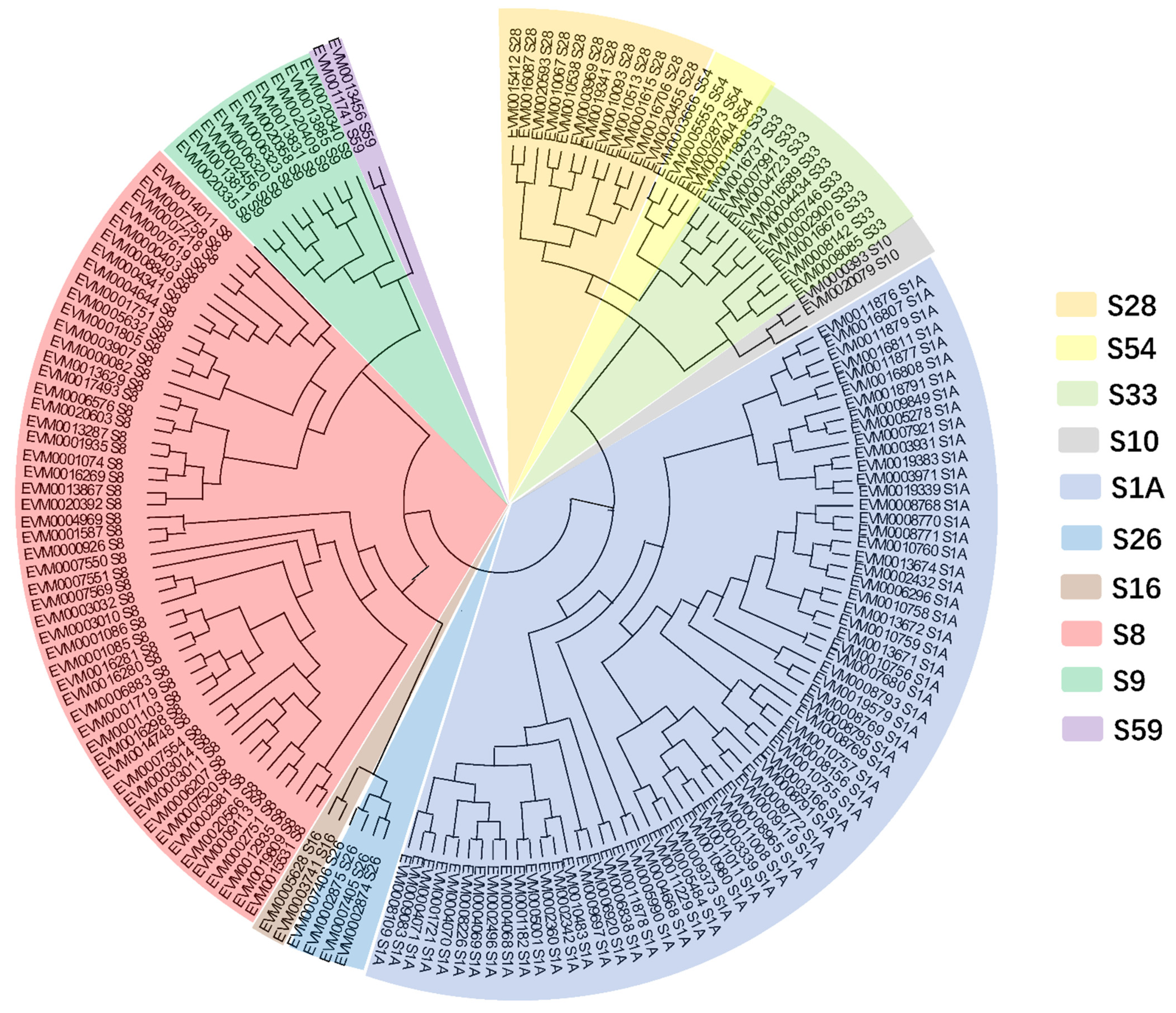
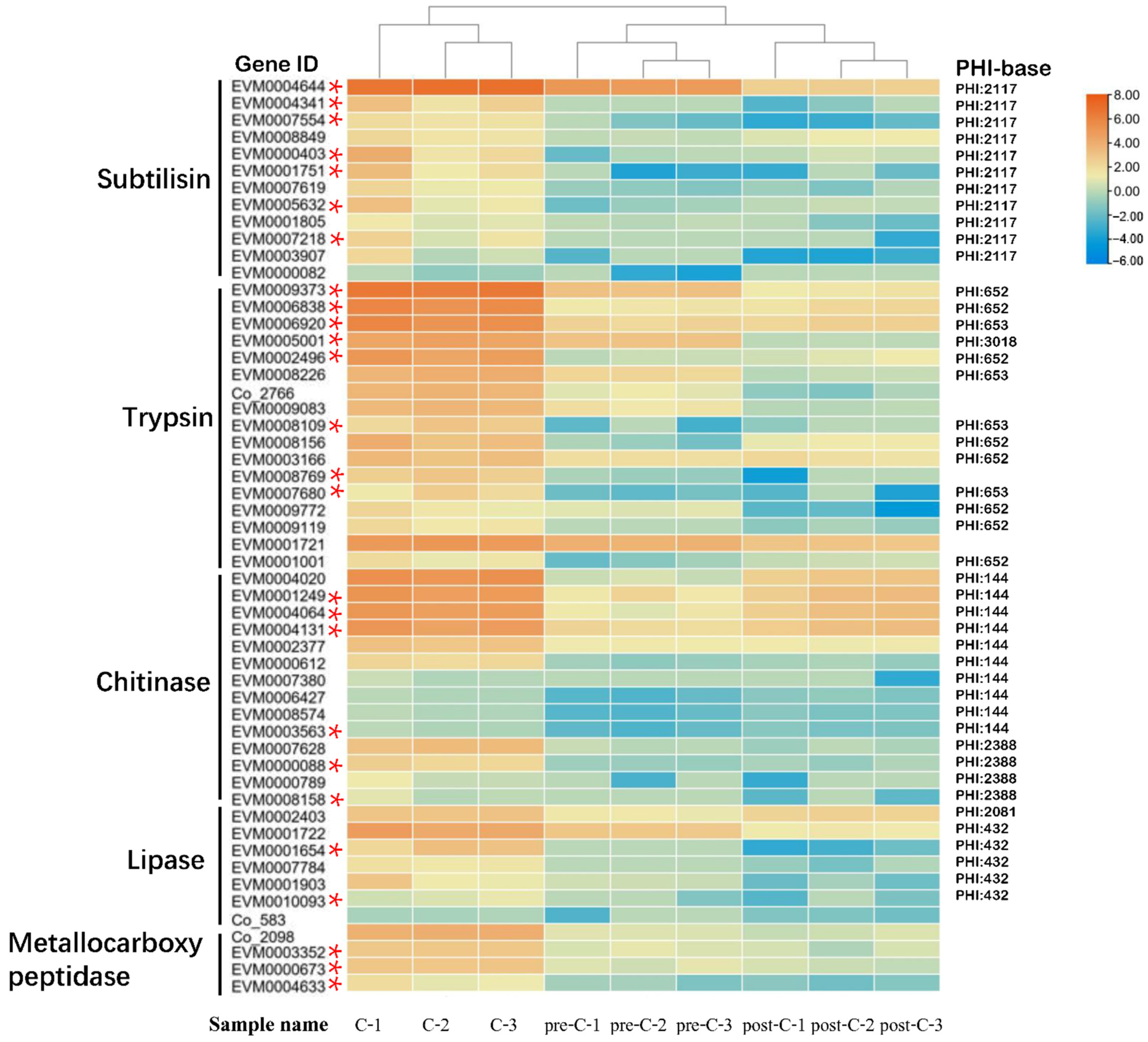
3.3. Abundance of Serine Protease-Encoding Genes in C. obscurus
3.4. Comparative Genomic Analysis
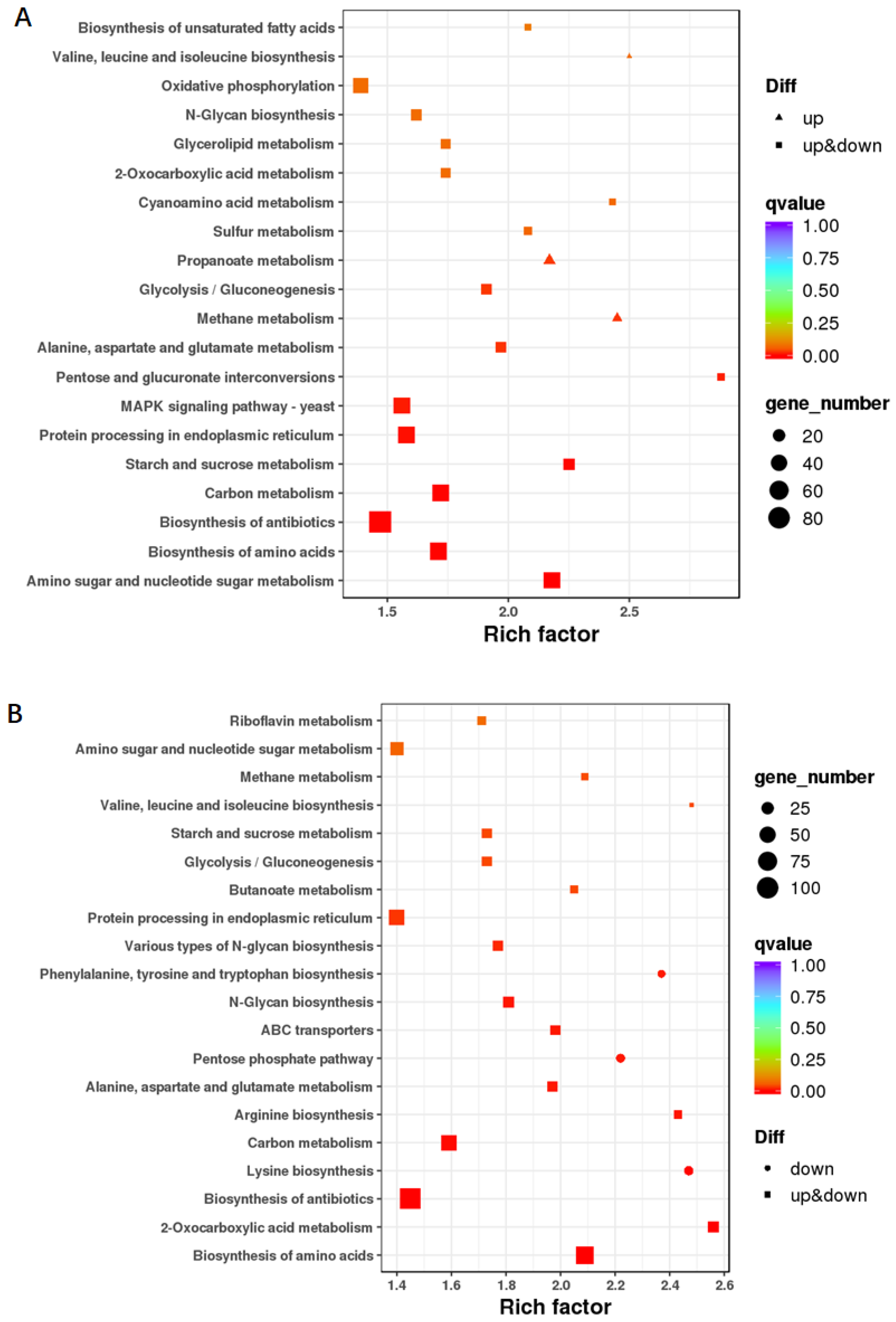
3.5. Gene Differential Expression in the three Conidiation Stages
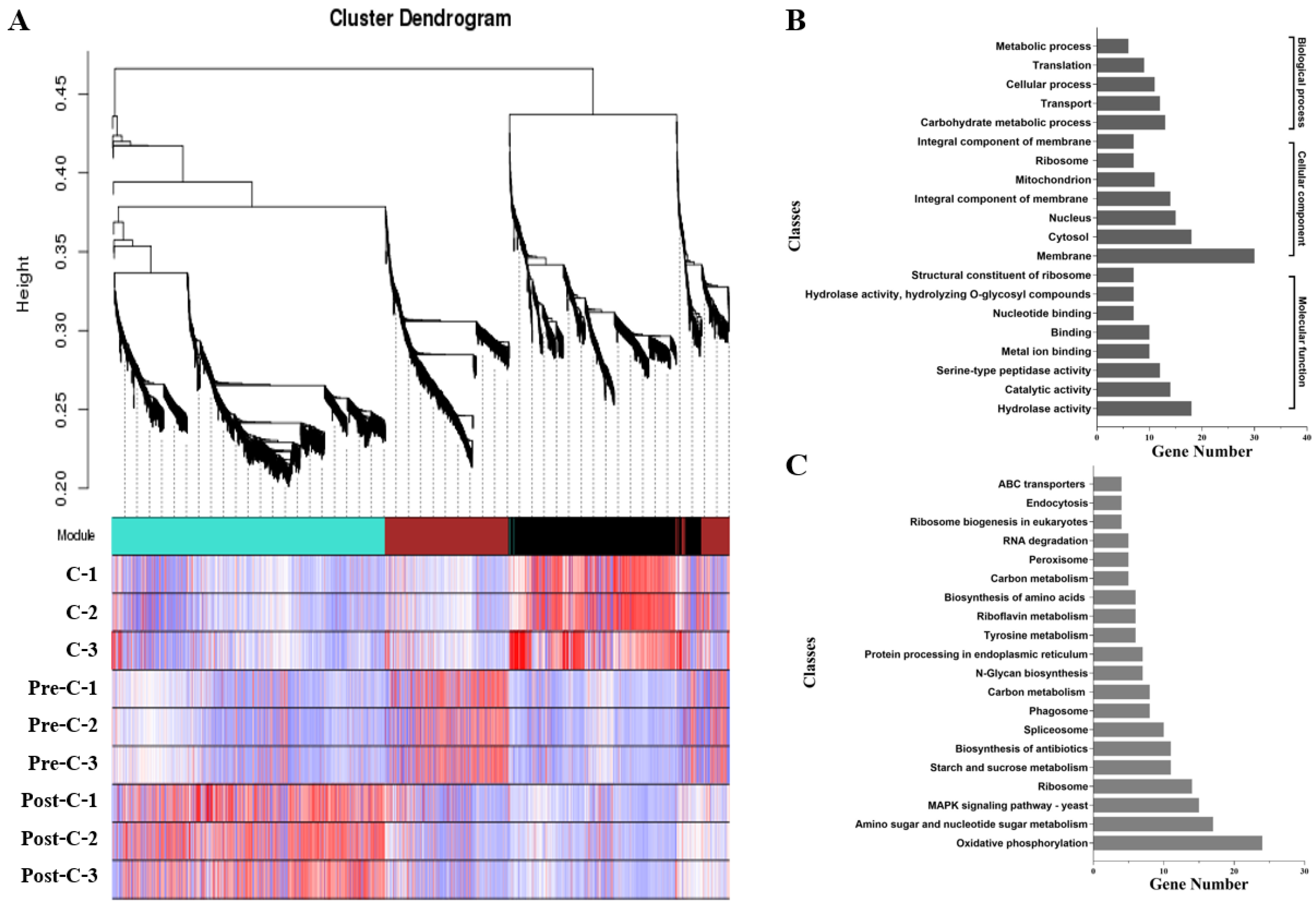
3.6. Co-Expression Network Analysis of Conidiation-Related Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jung, B.; Kim, S.; Lee, J. Microcyle conidiation in filamentous fungi. Mycobiology 2014, 42, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruger-Herreros, C.; Corrochano, L.M. Conidiation in Neurospora crassa: Vegetative reproduction by a model fungus. Int. Microbiol. 2020, 23, 97–105. [Google Scholar] [CrossRef]
- Park, H.S.; Yu, J.H. Genetic control of asexual sporulation in filamentous fungi. Curr. Opin. Microbiol. 2012, 15, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Ball, S.R.; Kwan, A.H.; Sunde, M. Hydrophobin rodlets on the fungal cell wall. Curr. Top. Microbiol. 2019, 425, 29–51. [Google Scholar]
- Pell, J.K.; Eilenberg, J.; Hajek, A.E.; Steinkraus, D.C. Biology, Ecology and Pest Management Potential of Entomophthorales; Butt, T.M., Jackson, C., Magan, N., Eds.; CABI Publishing: Wallingford, UK, 2001; pp. 71–132. [Google Scholar]
- Wang, C.; Wang, S. Insect pathogenic fungi: Genomics, molecular interactions, and genetic improvements. Ann. Rev. Entomol. 2017, 62, 73–90. [Google Scholar] [CrossRef] [PubMed]
- Gryganskyi, A.P.; Mullens, B.A.; Gajdeczka, M.T.; Rehner, S.A.; Vilgalys, R.; Hajek, A.E. Hijacked: Co-option of host behavior by entomophthoralean fungi. PLoS Pathog. 2017, 13, e1006274. [Google Scholar] [CrossRef] [Green Version]
- Spatafora, J.W.; Chang, Y.; Benny, G.L. A phylum-level phylogenetic classification of zygomycete fungi based on genome-scale data. Mycologia 2016, 108, 1028–1046. [Google Scholar] [CrossRef] [Green Version]
- Arnesen, J.A.; Małagocka, J.; Gryganskyi, A.; Grigoriev, I.V.; Voigt, K.; Stajich, J.E.; De Fine Licht, H.H. Early diverging insect-pathogenic fungi of the order Entomophthorales possess diverse and unique subtilisin-like serine proteases. G3 2018, 8, 3311–3319. [Google Scholar] [CrossRef] [Green Version]
- Gao, B.J.; Mou, Y.N.; Tong, S.M.; Ying, S.H.; Feng, M.G. Subtilisin-like Pr1 proteases marking the evolution of pathogenicity in a wide-spectrum insect-pathogenic fungus. Virulence 2020, 11, 365–380. [Google Scholar] [CrossRef] [Green Version]
- Xiao, G.; Ying, S.H.; Zheng, P.; Wang, Z.L.; Zhang, S.; Xie, X.Q.; Shang, Y.; St Leger, R.J.; Zhao, G.P.; Wang, C.; et al. Genomic perspectives on the evolution of fungal entomopathogenicity in Beauveria bassiana. Sci. Rep. 2012, 2, 483. [Google Scholar] [CrossRef] [Green Version]
- Gao, Q.; Jin, K.; Ying, S.H.; Zhang, Y.; Xiao, G.; Shang, Y.; Duan, Z.; Hu, X.; Xie, X.Q.; Zhou, G.; et al. Genome sequencing and comparative transcriptomics of the model entomopathogenic fungi Metarhizium anisopliae and M. acridum. PLoS Genet. 2011, 7, e1001264. [Google Scholar] [CrossRef] [Green Version]
- Jackson, M.A.; Dunlap, C.A.; Jaronski, S.T. Ecological considerations in producing and formulating fungal entomopathogens for us in insect biocontrol. BioControl 2010, 55, 129–145. [Google Scholar] [CrossRef]
- Grell, M.N.; Jensen, A.B.; Olsen, P.B.; Eilenberg, J.; Lange, L. Secretome of fungus-infected aphids documents high pathogen activity and weak host response. Fungal Genet. Biol. 2011, 48, 343–352. [Google Scholar]
- Zhou, X.; Wang, D.W.; Zhang, X.; Wang, J.H. The influence of aphid-specific pathogen Conidiobolus obscurus (Entomophthoromycota: Entomophthorales) on the mortality and fecundity of bamboo aphids. J. For. Res. 2014, 19, 388–394. [Google Scholar] [CrossRef]
- Wang, F.; Sethiya, P.; Hu, X.; Guo, S.; Chen, Y.; Li, A.; Tan, K.; Wong, K.H. Transcription in fungal conidia before dormancy produces phenotypically variable conidia that maximize survival in different environments. Nat. Microbiol. 2021, 6, 1066–1081. [Google Scholar] [CrossRef]
- Ye, G.F.; Zhang, L.H.; Zhou, X. Long noncoding RNAs are potentially involved in the degeneration of virulence in an aphid-obligate pathogen, Conidiobolus obscurus (Entomophthoromycotina). Virulence 2021, 21, 1705–1716. [Google Scholar] [CrossRef]
- De Fine Licht, H.H.; Hajek, A.E.; Eilenberg, J.; Jensen, A.B. Utilizing genomics to study entomopathogenicity in the fungal phylum Entomophthoromycota: A review of current genetic resources. Adv. Genet. 2016, 94, 41–65. [Google Scholar] [PubMed]
- Chang, Y.; Wang, S.; Satoshi, S.; Aerts, A.; Choi, C.; Clum, A.; LaButti, K.M.; Lindquist, E.A.; Ngan, C.Y.; Ohm, R.A.; et al. Phylogenomic analyses indicate that early fungi evolved digesting cell walls of algal ancestors of land plants. Genome Biol. Evol. 2015, 7, 1590–1601. [Google Scholar] [CrossRef] [PubMed]
- Ngwenya, M.L.; Chen, W.; Basson, A.K.; Shandu, H.S.; Yu, J.H.; Nelson, D.R.; Syed, K. Blooming of unusual cytochrome P450s by tandem duplication in the pathogenic fungus Conidiobolus coronatus. Int. J. Mol. Sci. 2018, 19, 1711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Nie, Y.; Yu, D.; Xie, X.; Qin, L.; Yang, Y.; Huang, B. Genome-wide study of saprotrophy-related genes in the basal fungus Conidiobolus heterosporus. Appl. Microbiol. Biotechnol. 2020, 104, 6261–6272. [Google Scholar] [CrossRef]
- Zhou, X.; Feng, M.G.; Huang, Z.H. Effects of cryopreservation at −80 °C on the formulation and pathogenicity of the obligate aphid pathogen Pandora nouryi. Pol. J. Microbiol. 2014, 63, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Olsen, H.E.; Paten, B. The Oxford Nanopore MinION: Delivery of nanopore sequencing to the genomics community. Genome Biol. 2016, 17, 239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.Q.; Chen, F.Z.; Gao, F.; Li, F.; Liu, K.; You, L.; Hua, C.; Yang, F.; Liu, W.; Peng, C.; et al. CNSA: A data repository for archiving omics data. Database 2020, 2020, baaa055. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruan, J.; Li, H. Fast and accurate long-read assembly with wtdbg2. Nat. Methods 2020, 17, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [Green Version]
- Tarailo-Graovac, M.; Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 2009, Chapter 4, unit 4.10. [Google Scholar] [CrossRef]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Nawrocki, E.P.; Eddy, S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 2013, 29, 2933–2935. [Google Scholar] [CrossRef] [Green Version]
- Stanke, M.; Waack, S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 2003, 2, ii215–ii225. [Google Scholar] [CrossRef] [Green Version]
- Keilwagen, J.; Wenk, M.; Erickson, J.L.; Schattat, M.H.; Grau, J.; Hartung, F. Using intron position conservation for homology-based gene prediction. Nucleic Acids Res. 2016, 44, e89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [PubMed]
- Kim, D.; Landmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential genes and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowite 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Stoeckert, C.J., Jr.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [Green Version]
- Bowman, S.M.; Free, S.J. The structure and synthesis of the fungal cell wall. BioEssays 2006, 28, 799–808. [Google Scholar] [CrossRef]
- Utsugi, T.; Minemura, M.; Hirata, A.; Abe, M.; Watanabe, D.; Ohya, Y. Movement of yeast 1,3-β-glucan synthase is essential for uniform cell wall synthesis. Genes Cells 2002, 7, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Saha, S.; Anilkumar, A.A.; Mayor, S. GPI-anchored protein organization and dynamics at the cell surface. J. Lipid Res. 2016, 57, 159–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajek, A.E.; Eastburn, C.C. Attachment and germination of Entomophaga maimaiga conidia on host and non-host larval cuticle. J. Invertebr. Pathol. 2003, 82, 12–22. [Google Scholar] [CrossRef]
- Małagocka, J.; Grell, M.N.; Lange, L.; Eilenberg, J.; Jensen, A.B. Transcriptome of an entomophthoralean fungus (Pandora formicae) shows molecular machinery adjusted for successful host exploitation and transmission. J. Invertebr. Pathol. 2015, 128, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.H.; Zhou, X.; Guo, K.; Zhang, X.Q.; Lin, H.P.; Montalva, C. Transcriptomic insight into pathogenicity-associated factors of Conidiobolus obscurus, an obligate aphid-pathogenic fungus belonging to Entomopthoromycota. Pest Manag. Sci. 2018, 74, 1677–1686. [Google Scholar] [CrossRef] [PubMed]
- Muszewska, A.; Stepniewska-Dziubinska, M.M.; Steczkiewicz, K.; Pawlowska, J.; Dziedzic, A.; Ginalski, K. Fungal lifestyle reflected in serine protease repertoire. Sci. Rep. 2017, 7, 9147. [Google Scholar] [CrossRef] [Green Version]
- Bagga, S.; Gang, H.; Screen, S.E.; St. Leger, R.J. Reconstructing the diversification of subtilisins in the pathogenic fungus Metarhizium anisopliae. Gene 2004, 324, 159–169. [Google Scholar] [CrossRef]
- Alharbi, A.; Rabadi, S.M.; Alqahtani, M.; Marghani, D.; Worden, M.; Ma, Z.; Malik, M.; Bakshi, C.S. Role of peroxiredoxin of the AhpC/TSA family in antioxidant defense mechanisms of Francisella tularensis. PLoS ONE 2019, 14, e0213699. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Liu, J.; Liu, N.; Kuhn, L.A.; Garavito, R.M.; Ferguson-Miller, S. Translocator protein 18 kDa (TSPO): An old protein with new functions? Biochemistry 2016, 55, 2821–2831. [Google Scholar] [CrossRef] [Green Version]
- Braga, G.U.; Rangel, D.E.; Fernandes, É.K.; Flint, S.D.; Roberts, D.W. Molecular and physiological effects of environmental UV radiation on fungal conidia. Curr. Genet. 2015, 61, 405–425. [Google Scholar] [CrossRef]
- Vallim, M.A.; Miller, K.Y.; Miller, B.L. Aspergillus SteA (sterile12-like) is a homeodomain-C2/H2-Zn2+ finger transcription factor required for sexual reproduction. Mol. Microbiol. 2000, 36, 290–301. [Google Scholar] [CrossRef]
- Gao, P.; Li, M.; Jin, K.; Xia, Y. The homeobox gene MaH1 governs microcycle conidiation for increased conidial yield by mediating transcription of conidiation pattern shift-related genes in Metarhizium acridum. Appl. Microbiol. Biotechnol. 2019, 103, 2251–2262. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Xu, C.; Qian, Y.; Liu, R.; Zhang, Q.; Zeng, G.; Zhang, X.; Zhao, H.; Fang, W. MAPK cascade-mediated regulation of pathogenicity, conidiation and tolerance to abiotic stresses in the entomopathogenic fungus Metarhizium robertsii. Environ. Microbiol. 2016, 18, 1048–1062. [Google Scholar] [CrossRef] [PubMed]
- Tatebayashi, K.; Yamamoto, K.; Tanaka, K.; Tomida, T.; Maruoka, T.; Kasukawa, E.; Saito, H. Adaptor functions of Cdc42, Ste50, and Sho1 in the yeast osmoregulatory HOG MAPK pathway. EMBO J. 2006, 25, 3033–3044. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | C. obscurus |
|---|---|
| Genome size (Mb) | 37.6 |
| Coverage (×) | 318 |
| Sequence number | 1,145,404 |
| Scaffold number | 167 |
| Scaffold N50 length (bp) | 1,104,530 |
| Scaffold N90 length (bp) | 75,751 |
| G+C content (%) | 26.46 |
| Protein-encoding genes | 10,262 |
| Average number of exons per genes | 4.7 |
| Repeat sequences (bp) | 6,558,671 |
| tRNA number, family number | 526, 45 |
| rRNA number, family number | 8, 2 |
| Other ncRNA number, family number | 49, 20 |
| Internal ID | Pfam Annotation | FPKM a | FC b | |
|---|---|---|---|---|
| C | Pre-C or Post-C | |||
| Up-regulated in C vs. pre-C | ||||
| EVM0006541 | Polysaccharide deacetylase | 220.03 | 0.06 | 9.69 |
| EVM0001654 | Lipase (class 3) | 19.39 | 0 | 9.07 |
| EVM0006408 | Cell surface GPI-anchored protein | 277.16 | 0.47 | 8.94 |
| EVM0003421 | Cytochrome P450 | 12.09 | 0 | 8.77 |
| EVM0008805 | Glycosyl hydrolases family 16 | 10.70 | 0 | 8.66 |
| EVM0002345 | TspO/MBR family | 291.43 | 1.14 | 7.82 |
| EVM0004341 | Subtilase family | 14.41 | 0 | 7.80 |
| Co _242 | Homeobox domain | 18.18 | 0 | 7.70 |
| EVM0009119 | Trypsin | 5.80 | 0 | 7.41 |
| EVM0008624 | AhpC/TSA antioxidant enzyme | 24.49 | 0.13 | 7.09 |
| Up-regulated in C vs. post-C | ||||
| EVM0009712 | TspO/MBR family | 289.08 | 0.04 | 11.57 |
| EVM0002345 | TspO/MBR family | 291.43 | 0.05 | 11.40 |
| EVM0003678 | TspO/MBR family | 411.48 | 0.20 | 10.92 |
| EVM0007081 | TspO/MBR family | 305.94 | 0.14 | 10.73 |
| EVM0005679 | TspO/MBR family | 295.98 | 0.18 | 10.42 |
| EVM0001538 | TspO/MBR family | 783.86 | 0.71 | 10.21 |
| EVM0001670 | Kunitz/Bovine pancreatic trypsin inhibitor | 28.37 | 0 | 9.65 |
| EVM0010258 | Transcription elongation factor SPT6 | 442.33 | 0.62 | 9.59 |
| EVM0003314 | Glycosyl hydrolases family 16 | 21.02 | 0 | 9.52 |
| EVM0009807 | Kunitz/Bovine pancreatic trypsin inhibitor | 19.57 | 0 | 9.34 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Yang, T.; Yu, W.; Wang, X.; Zhou, X.; Zhou, X. Genome-Wide Study of Conidiation-Related Genes in the Aphid-Obligate Fungal Pathogen Conidiobolus obscurus (Entomophthoromycotina). J. Fungi 2022, 8, 389. https://doi.org/10.3390/jof8040389
Zhang L, Yang T, Yu W, Wang X, Zhou X, Zhou X. Genome-Wide Study of Conidiation-Related Genes in the Aphid-Obligate Fungal Pathogen Conidiobolus obscurus (Entomophthoromycotina). Journal of Fungi. 2022; 8(4):389. https://doi.org/10.3390/jof8040389
Chicago/Turabian StyleZhang, Lvhao, Tian Yang, Wangyin Yu, Xiaojun Wang, Xiang Zhou, and Xudong Zhou. 2022. "Genome-Wide Study of Conidiation-Related Genes in the Aphid-Obligate Fungal Pathogen Conidiobolus obscurus (Entomophthoromycotina)" Journal of Fungi 8, no. 4: 389. https://doi.org/10.3390/jof8040389
APA StyleZhang, L., Yang, T., Yu, W., Wang, X., Zhou, X., & Zhou, X. (2022). Genome-Wide Study of Conidiation-Related Genes in the Aphid-Obligate Fungal Pathogen Conidiobolus obscurus (Entomophthoromycotina). Journal of Fungi, 8(4), 389. https://doi.org/10.3390/jof8040389