
Cytochrome P450 Complement May Contribute to Niche Adaptation in Serpula Wood-Decay Fungi


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cultures and Conditions
2.2. Analysis of Predicted Cytochrome P450 Genes
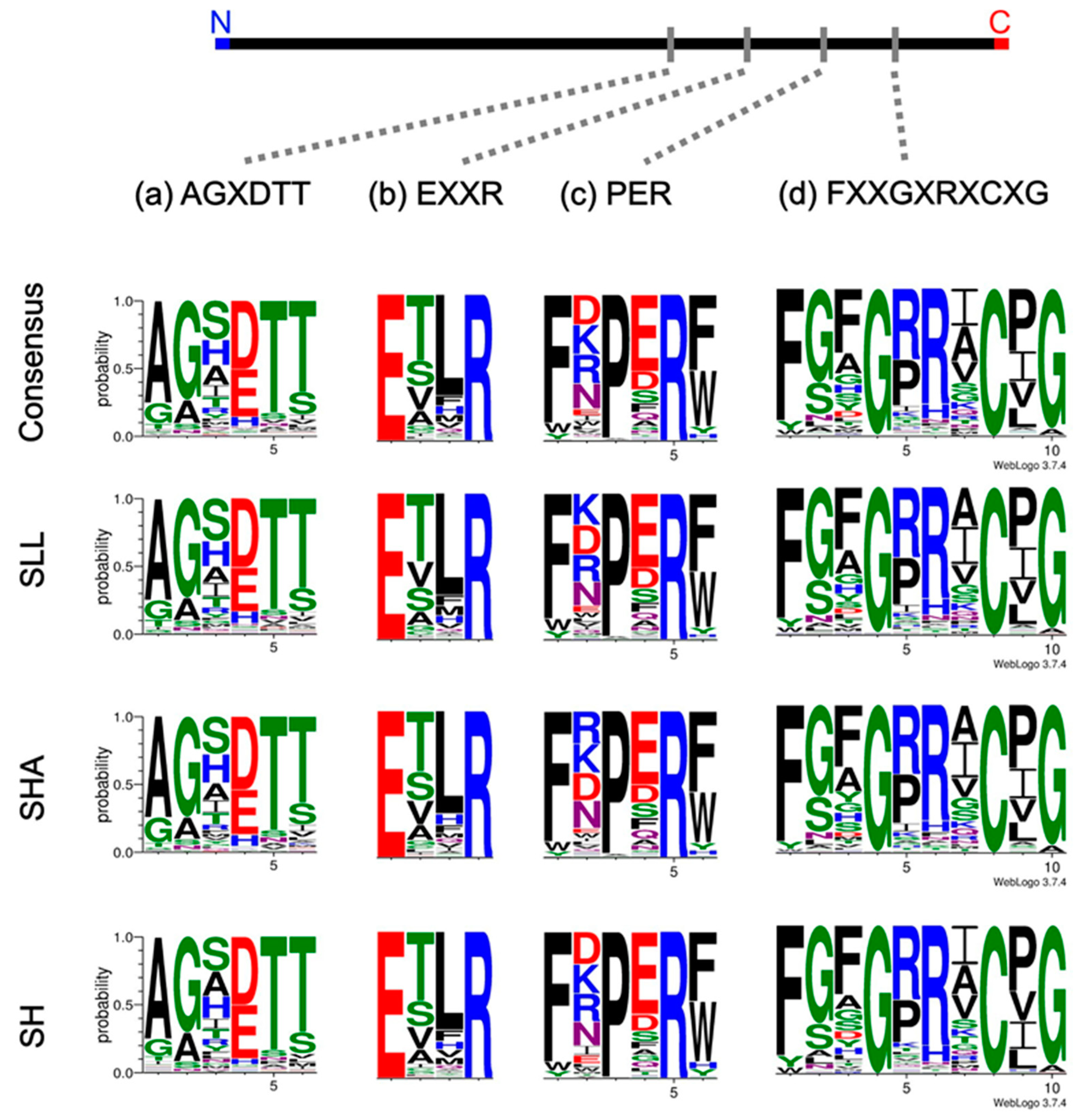
2.3. Generation of Sequence Logo Analysis for Characteristic Motifs
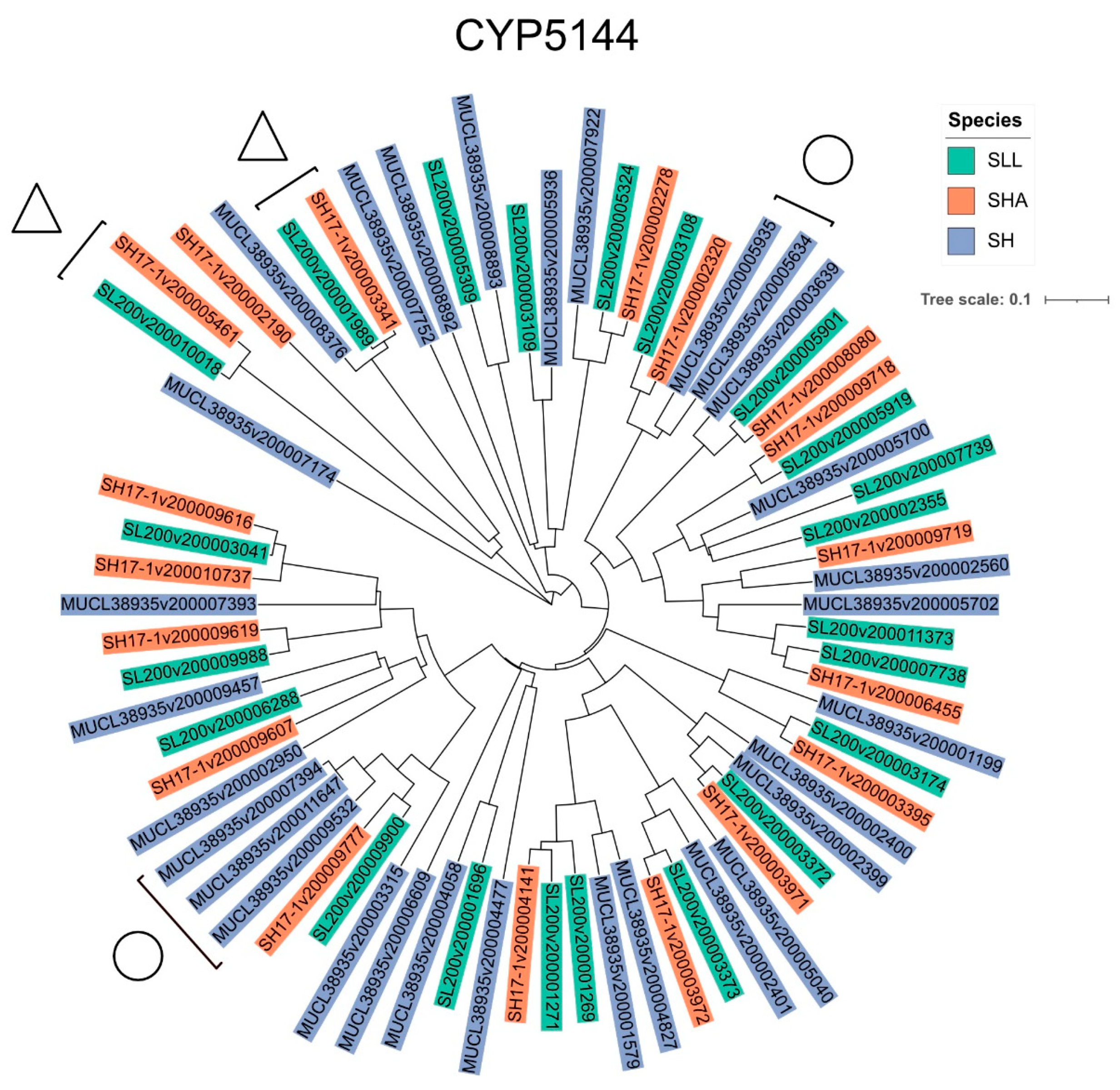
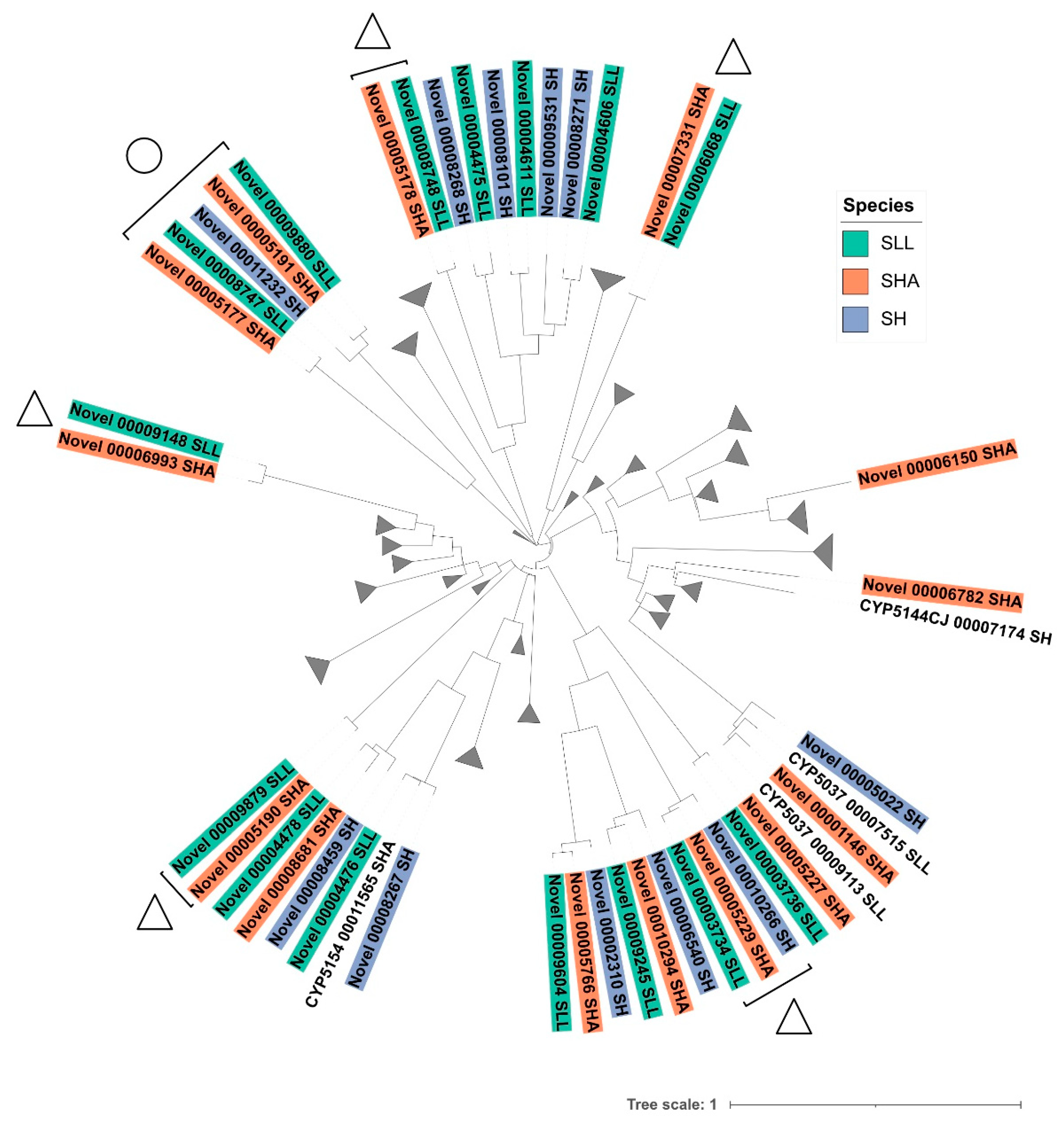
2.4. Generation of Phylogenetic Trees
2.5. Transcriptomic Analysis
3. Results
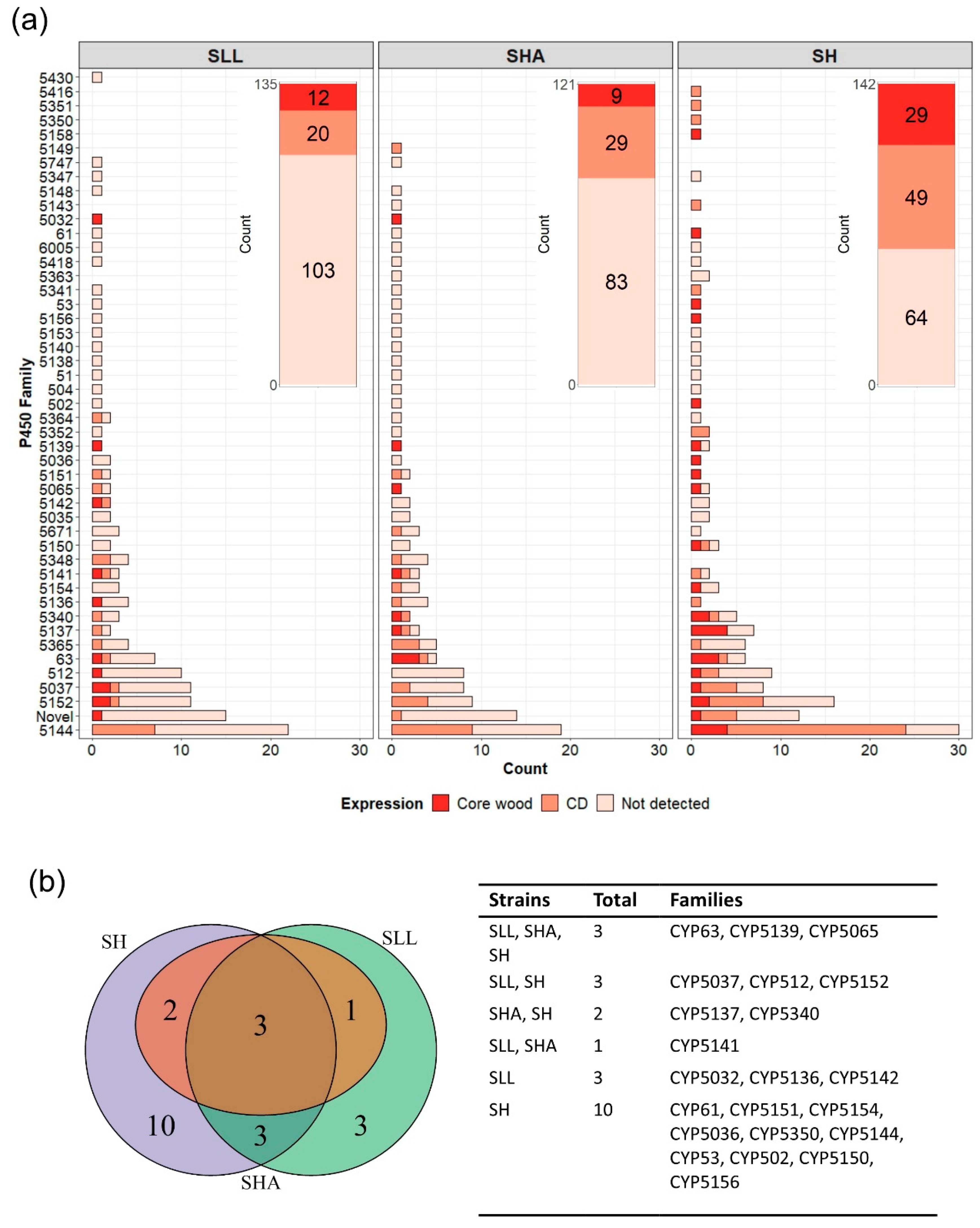
3.1. Identification, Nomenclature and Diversity of P450 Complement
3.2. Evidence of P450 Family Expansion Was Seen in All Strains
3.3. Identification of Novel P450 Sequences Suggested Evidence of Niche Adaptation
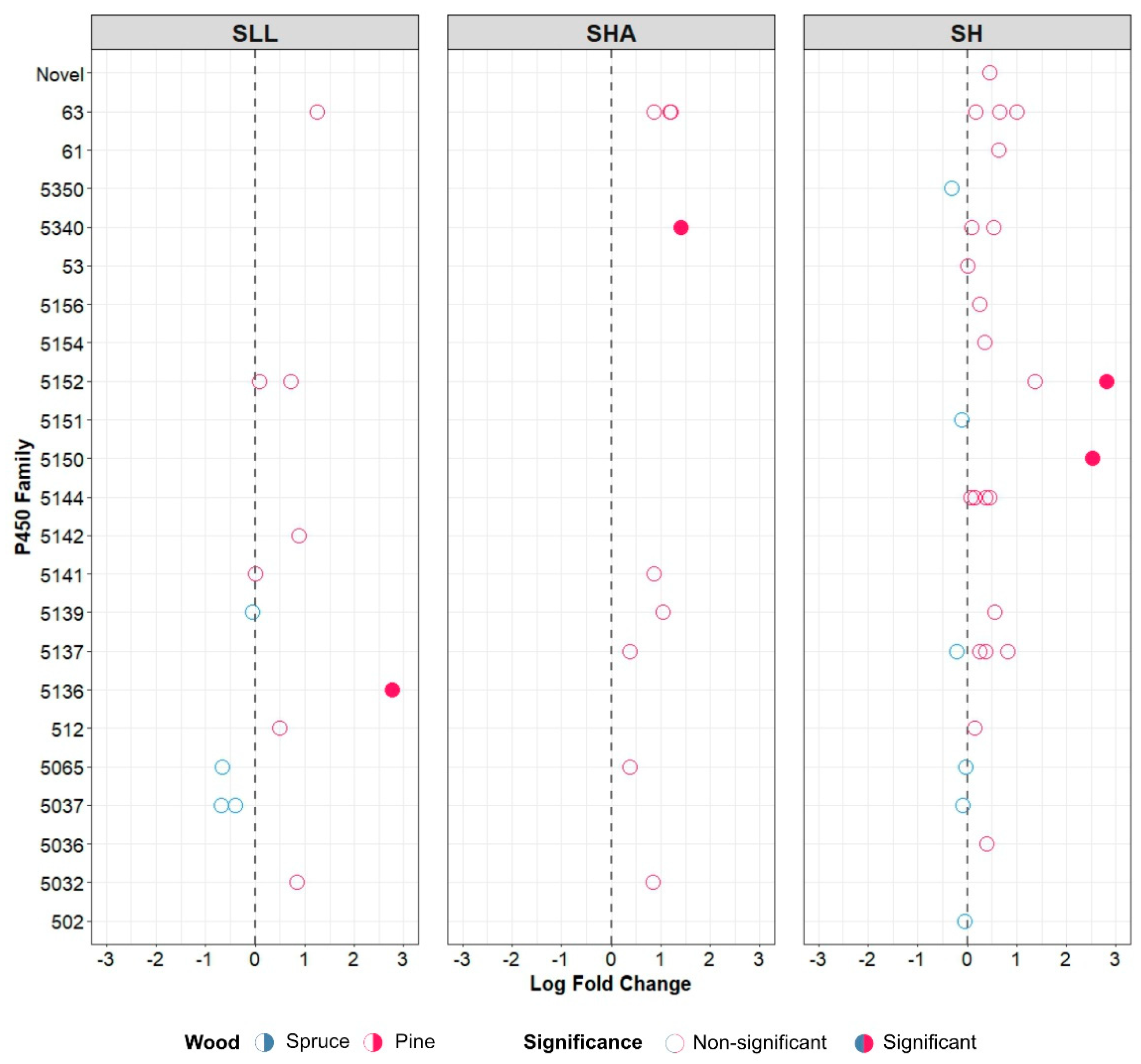
3.4. Differential Expression of Putative P450 Proteins during Growth in Spruce and Pine
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Doddapaneni, H.; Chakraborty, R.; Yadav, J.S. Genome-wide structural and evolutionary analysis of the P450 monooxygenase genes (P450ome) in the white rot fungus Phanerochaete chrysosporium: Evidence for gene duplications and extensive gene clustering. BMC Genom. 2005, 6, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ide, M.; Ichinose, H.; Wariishi, H. Molecular identification and functional characterization of cytochrome P450 monooxygenases from the brown-rot basidiomycete Postia placenta. Arch. Microbiol. 2012, 194, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Carbone, I.; Dean, R.A. The evolutionary history of cytochrome P450 genes in four filamentous Ascomycetes. BMC Evol. Biol. 2007, 7, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, D.E.; Kraševec, N.; Mullins, J.; Nelson, D.R. The CYPome (cytochrome P450 complement) of Aspergillus nidulans. Fungal Genet. Biol. 2009, 46, S53–S61. [Google Scholar] [CrossRef]
- Martinez, D.; Larrondo, L.F.; Putnam, N.; Gelpke, M.D.S.; Huang, K.; Chapman, J.; Helfenbein, K.G.; Ramaiya, P.; Detter, J.C.; Larimer, F.; et al. Genome sequence of the lignocellulose degrading fungus Phanerochaete chrysosporium strain RP78. Nat. Biotechnol. 2004, 22, 695–700. [Google Scholar] [CrossRef]
- Bernhardt, R. Cytochromes P450 as versatile biocatalysts. J. Biotechnol. 2006, 124, 128–145. [Google Scholar] [CrossRef]
- Syed, K.; Nelson, D.R.; Riley, R.; Yadav, J.S. Genomewide annotation and comparative genomics of cytochrome P450 monooxygenases (P450s) in the polypore species Bjerkandera adusta, Ganoderma sp. and Phlebia brevispora. Mycologia 2013, 105, 1445–1455. [Google Scholar] [CrossRef]
- Hori, C.; Ishida, T.; Igarashi, K.; Samejima, M.; Suzuki, H.; Master, E.; Ferreira, P.; Ruiz-Dueñas, F.J.; Held, B.; Canessa, P.; et al. Analysis of the Phlebiopsis gigantea genome, transcriptome and secretome provides insight into its pioneer colonization strategies of wood. PLoS Genet. 2014, 10, e1004759. [Google Scholar] [CrossRef] [Green Version]
- Syed, K.; Shale, K.; Pagadala, N.S.; Tuszynski, J. Systematic identification and evolutionary analysis of catalytically versatile cytochrome p450 monooxygenase families enriched in model basidiomycete fungi. PLoS ONE 2014, 9, e86683. [Google Scholar] [CrossRef]
- Schmidt, O. Indoor wood-decay basidiomycetes: Damage, causal fungi, physiology, identification and characterization, prevention and control. Mycol. Progress 2007, 6, 261–279. [Google Scholar] [CrossRef]
- Balasundaram, S.V.; Hess, J.; Durling, M.B.; Moody, S.C.; Thorbek, L.; Progida, C.; LaButti, K.; Aerts, A.; Barry, K.; Grigoriev, I.V.; et al. The fungus that came in from the cold: Dry rot’s pre-adapted ability to invade buildings. ISME J. 2018, 12, 791–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skrede, I.; Murat, C.; Hess, J.; Maurice, S.; Sønstebø, J.H.; Kohler, A.; Barry-Etienne, D.; Eastwood, D.; Högberg, N.; Martin, F.; et al. Contrasting demographic histories revealed in two invasive populations of the dry rot fungus Serpula lacrymans. Mol. Ecol. 2021, 30, 2772–2789. [Google Scholar] [CrossRef] [PubMed]
- Palfreyman, J.W.; Gartland, J.S.; Sturrock, C.J.; Lester, D.; White, N.A.; Low, G.A.; Bech-Andersen, J.; Cooke, D.E. The relationship between ‘wild’ and ‘building’ isolates of the dry rot fungus Serpula lacrymans. FEMS Microbiol. Lett. 2003, 228, 281–286. [Google Scholar] [CrossRef] [Green Version]
- Vanden Wymelenberg, A.; Gaskell, J.; Mozuch, M.; BonDurant, S.S.; Sabat, G.; Ralph, J.; Skyba, O.; Mansfield, S.D.; Blanchette, R.A.; Grigoriev, I.V.; et al. Significant alteration of gene expression in wood decay fungi Postia placenta and Phanerochaete chrysosporium by plant species. Appl. Environ. Microbiol. 2011, 77, 4499–4507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hess, J.; Balasundaram, S.V.; Bakkemo, R.I.; Drula, E.; Henrissat, B.; Högberg, N.; Eastwood, D.; Skrede, I. Niche differentiation and evolution of the wood decay machinery in the invasive fungus Serpula lacrymans. ISME J. 2021, 15, 592–604. [Google Scholar] [CrossRef]
- Syed, K.; Mashele, S.S. Comparative analysis of P450 signature motifs EXXR and CXG in the large and diverse kingdom of fungi: Identification of evolutionarily conserved amino acid patterns characteristic of P450 family. PLoS ONE 2014, 9, e95616. [Google Scholar] [CrossRef] [Green Version]
- Nelson, D.R. Cytochrome P450 diversity in the tree of life. Biochem. Biophys. Acta 2017, 1866, 141–154. [Google Scholar] [CrossRef]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [Green Version]
- Okonechnikov, K.; Golosova, O.; Fursov, M.; Team, U. Unipro ugene: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Rzhetsky, A.; Nei, M. A simple method for estimating and testing minimum-evolution trees. Mol. Biol. Evol. 1992, 9, 945–967. [Google Scholar]
- Zuckerkandl, E.; Pauling, L. Evolutionary divergence and convergence in proteins. In Evolving Genes and Proteins; Bryson, V., Vogel, H., Eds.; Elsevier: Amsterdam, The Netherlands, 1965; pp. 97–166. [Google Scholar]
- Nei, M.; Kumar, S. Molecular Evolution and Phylogenetics; Oxford University Press: Oxford, UK, 2000. [Google Scholar]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [PubMed]
- Letunic, I.; Bork, P. Interactive tree of life (itol): An online tool for phylogenetic tree display and annotation. Bioinformatics 2007, 23, 127–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A bioconductor pack-age for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, D.R. Cytochrome P450 nomenclature, 2004. In Cytochrome P450 Protocols, 2nd ed.; Phillips, I., Shephard, E., Eds.; Humana Press: Totowa, NJ, USA, 2006; pp. 1–10. [Google Scholar]
- Carlsen, T.; Engh, I.B.; Decock, C.; Rajchenberg, M.; Kauserud, H. Multiple cryptic species with divergent substrate affinities in the Serpula himantioides species complex. Fungal Biol. 2011, 115, 54–61. [Google Scholar] [CrossRef]
- Lysøe, E.; Harris, L.J.; Walkowiak, S.; Subramaniam, R.; Divon, H.H.; Riiser, E.S.; Llorens, C.; Gabaldón, T.; Kistler, H.C.; Jonkers, W.; et al. The genome of the generalist plant pathogen Fusarium avenaceum is enriched with genes involved in redox, signaling and secondary metabolism. PLoS ONE 2014, 9, e112703. [Google Scholar] [CrossRef] [Green Version]
- Valette, N.; Perrot, T.; Sormani, R.; Gelhaye, E.; Morel-Rouhier, M. Antifungal activities of wood extractives. Fungal Biol. Rev. 2017, 31, 113–123. [Google Scholar] [CrossRef]
- Schultz, T.P.; Nicholas, D.D.; Preston, A.F. A brief review of the past, present and future of wood preservation. Pest. Manag. Sci. 2007, 63, 784–788. [Google Scholar] [CrossRef]
- Watkinson, S.; Eastwood, D. Serpula lacrymans, wood and buildings. In Advances in Applied Microbiology; Academic Press: Cambridge, MA, USA, 2012; Volume 78, pp. 121–149. [Google Scholar]
- Giovannoni, S.; Cameron Thrash, J.; Temperton, B. Implications of streamlining theory for microbial ecology. ISME J. 2014, 8, 1553–1565. [Google Scholar] [CrossRef] [Green Version]
- MacDonald, J.; Doering, M.; Canam, T.; Gong, Y.; Guttman, D.S.; Campbell, M.M.; Master, E.R. Transcriptomic responses of the softwood-degrading white-rot fungus Phanerochaete carnosa during growth on coniferous and deciduous wood. Appl. Environ. Microbiol. 2011, 77, 3211–3218. [Google Scholar] [CrossRef] [Green Version]
- Savluchinske-Feio, S.; Nunes, L.; Pereira, P.T.; Silva, A.M.; Roseiro, J.C.; Gigante, B.; Curto, M.J.M. Activity of dehydroabietic acid derivatives against wood contaminant fungi. J. Microbiol. Meth. 2007, 70, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Chigu, N.L.; Hirosue, S.; Nakamura, C.; Teramoto, H.; Ichinose, H.; Wariishi, H. Cytochrome P450 monooxygenases involved in anthracene metabolism by the white-rot basidiomycete Phanerochaete chrysosporium. Appl. Microbiol. Biotechnol. 2010, 87, 1907–1916. [Google Scholar] [CrossRef] [PubMed]
- Hirosue, S.; Tazaki, M.; Hiratsuka, N.; Yanai, S.; Kabumoto, H.; Shinkyo, R.; Arisawa, A.; Sakaki, T.; Tsunekawa, H.; Johdo, O.; et al. Insight into functional diversity of cytochrome p450 in the white-rot basidiomycete Phanerochaete chrysosporium: Involvement of versatile monooxygenase. Biochem. Biophys. Res. Commun. 2011, 407, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Yap, H.-Y.Y.; Chooi, Y.-H.; Firdaus-Raih, M.; Fung, S.-Y.; Ng, S.-T.; Tan, C.-S.; Tan, N.-H. The genome of the tiger milk mushroom, Lignosus rhinocerotis, provides insights into the genetic basis of its medicinal properties. BMC Genom. 2014, 15, 635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, J.; Doddapaneni, H.; Subramanian, V. P450ome of the white rot fungus Phanerochaete chrysosporium: Structure, evolution and regulation of expression of genomic p450 clusters. Biochem. Soc. Trans. 2006, 34, 1165–1169. [Google Scholar] [CrossRef]
- Harju, A.M.; Venäläinen, M.; Anttonen, S.; Viitanen, H.; Kainulainen, P.; Saranpää, P.; Vapaavuori, E. Chemical factors affecting the brown-rot decay resistance of scots pine heartwood. Trees 2003, 17, 263–268. [Google Scholar] [CrossRef]
- Willför, S.; Hemming, J.; Reunanen, M.; Holmbom, B. Phenolic and lipophilic extractives in scots pine knots and stemwood. Holzforshung 2003, 57, 359–372. [Google Scholar] [CrossRef]
- Skrede, I.; Engh, I.B.; Binder, M.; Carlsen, T.; Kauserud, H.; Bendiksby, M. Evolutionary history of Serpulaceae (Basidiomycota): Molecular phylogeny, historical biogeography and evidence for a single transition of nutritional mode. BMC Evol. Biol. 2011, 11, 230. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cowan, A.; Skrede, I.; Moody, S.C. Cytochrome P450 Complement May Contribute to Niche Adaptation in Serpula Wood-Decay Fungi. J. Fungi 2022, 8, 283. https://doi.org/10.3390/jof8030283
Cowan A, Skrede I, Moody SC. Cytochrome P450 Complement May Contribute to Niche Adaptation in Serpula Wood-Decay Fungi. Journal of Fungi. 2022; 8(3):283. https://doi.org/10.3390/jof8030283
Chicago/Turabian StyleCowan, Andrew, Inger Skrede, and Suzy Clare Moody. 2022. "Cytochrome P450 Complement May Contribute to Niche Adaptation in Serpula Wood-Decay Fungi" Journal of Fungi 8, no. 3: 283. https://doi.org/10.3390/jof8030283
APA StyleCowan, A., Skrede, I., & Moody, S. C. (2022). Cytochrome P450 Complement May Contribute to Niche Adaptation in Serpula Wood-Decay Fungi. Journal of Fungi, 8(3), 283. https://doi.org/10.3390/jof8030283