
Functional Classification and Characterization of the Fungal Glycoside Hydrolase 28 Protein Family


Abstract
1. Introduction
1.1. The GH28 Gene Family Encodes a Number of Different Enzymatic Activities Directed at the Breakdown of the Major Plant Cell Wall Component Pectin
1.2. Structural Analysis of GH28 Enzymes Shows Its Major Characteristics
2. Materials and Methods
2.1. Sequence Mining
2.2. Phylogenetic Reconstruction
2.3. HMMERCTTER Clustering and Classification
2.4. MEME Motifs and Logos
2.5. Structure Modelling and Consurf Analysis
2.6. Generation of Specificity Determining Networks (SDN)
3. Results
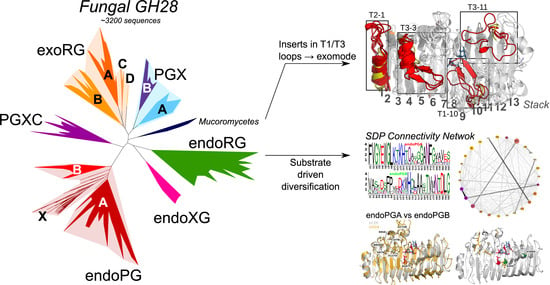
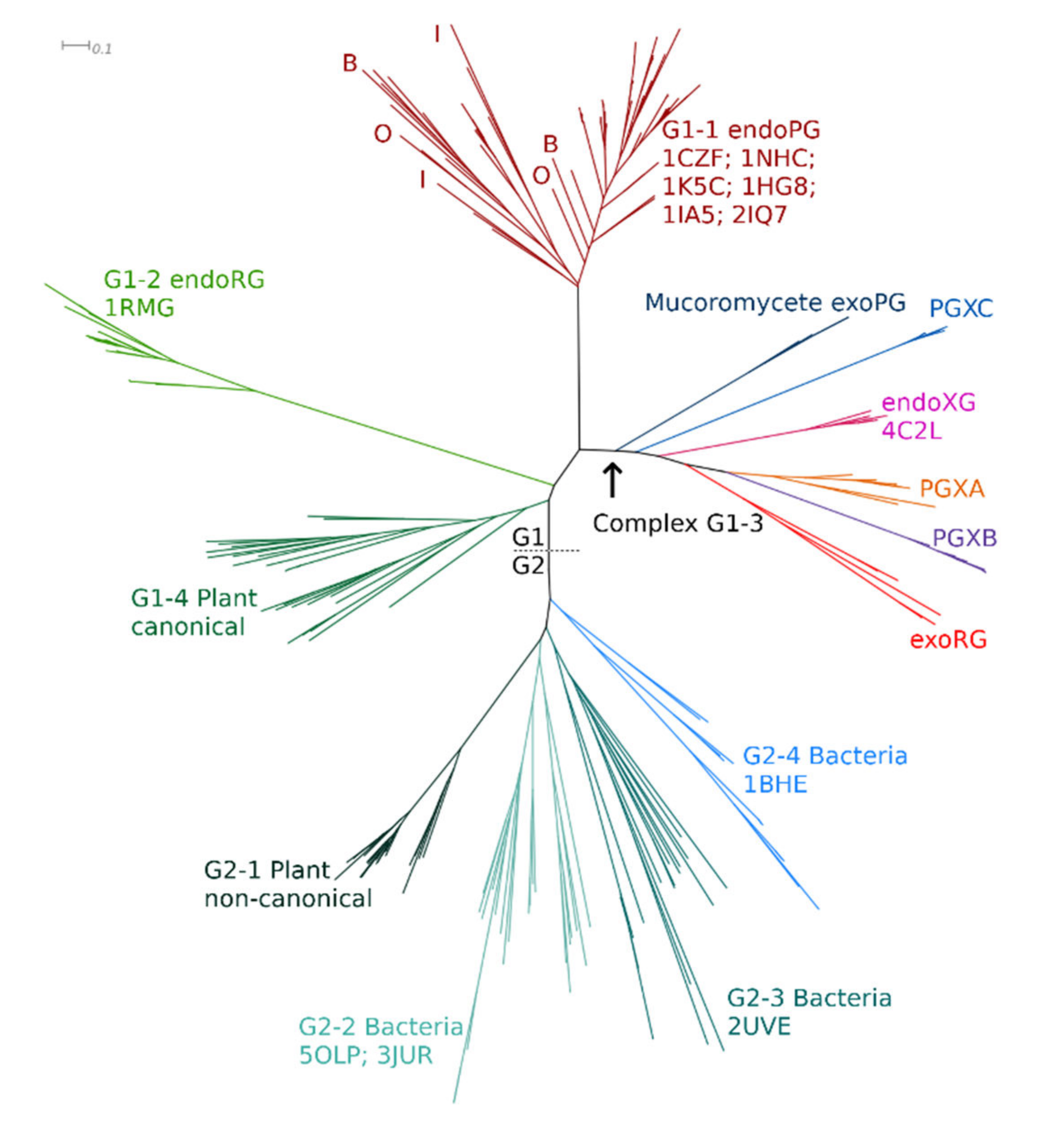
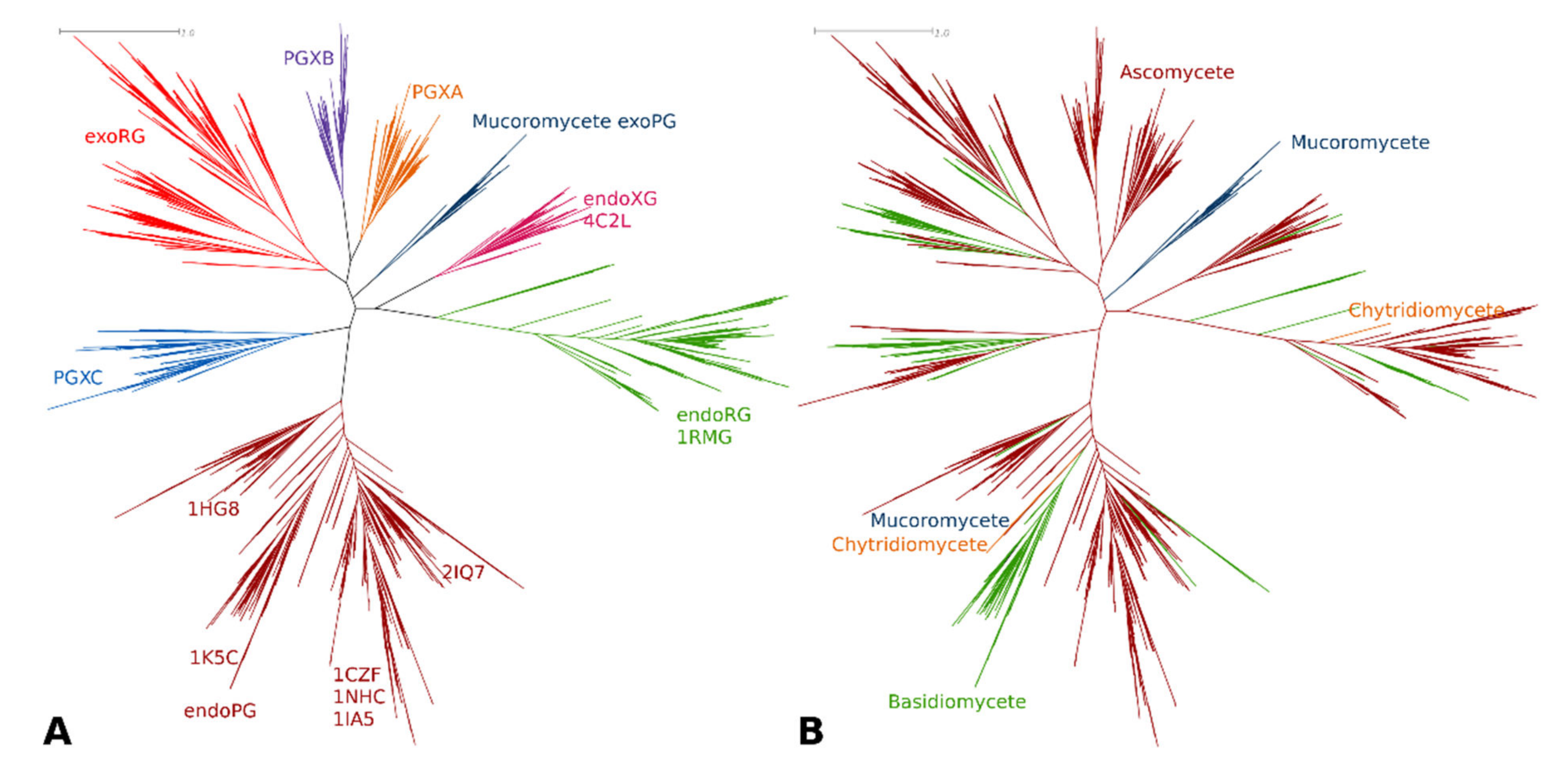
3.1. Phylogenetic Clustering of GH28 Is Explained by Taxonomy and Functional Diversification
3.2. Only Ascomycetes Show the Full Extent of GH28 Diversification
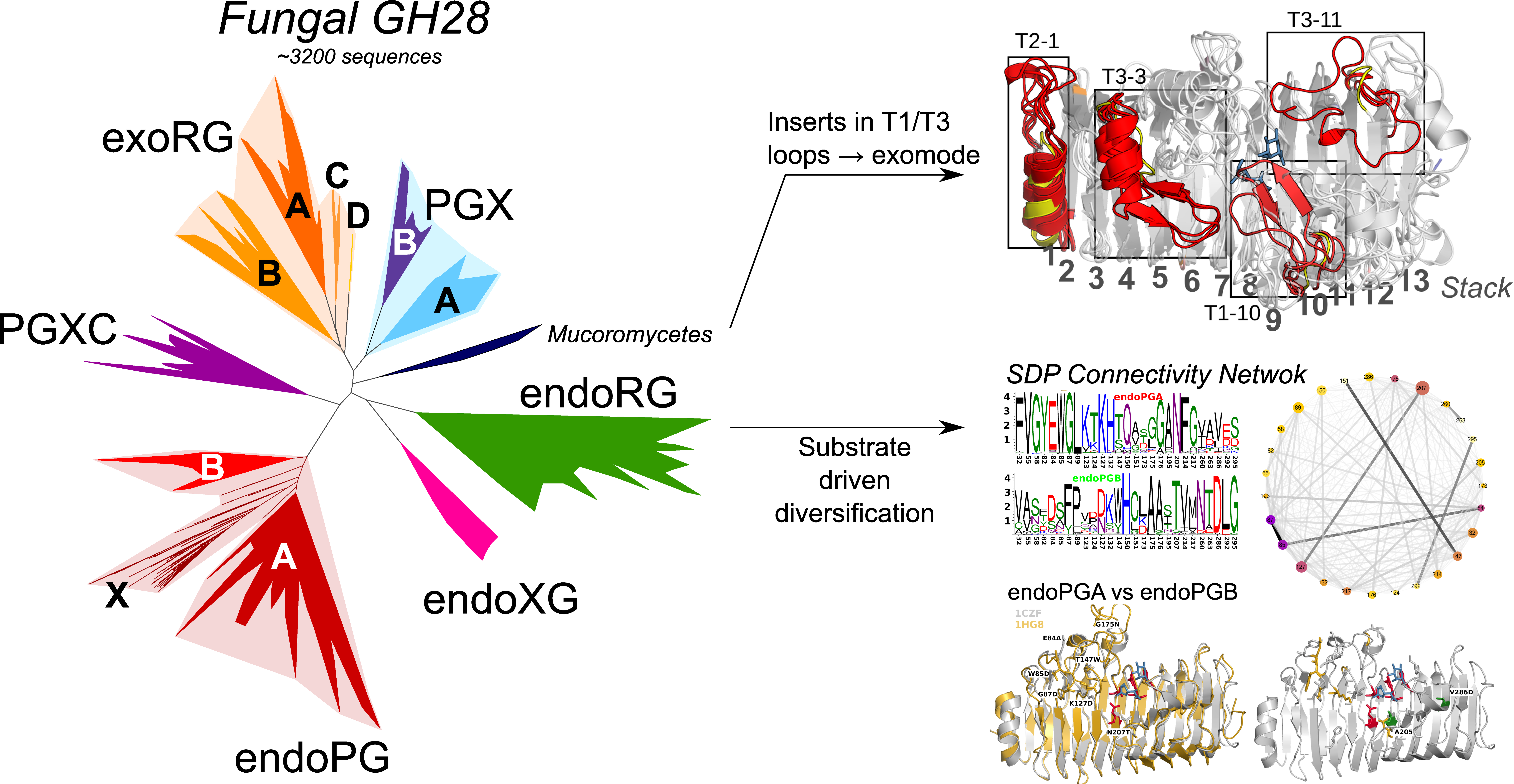
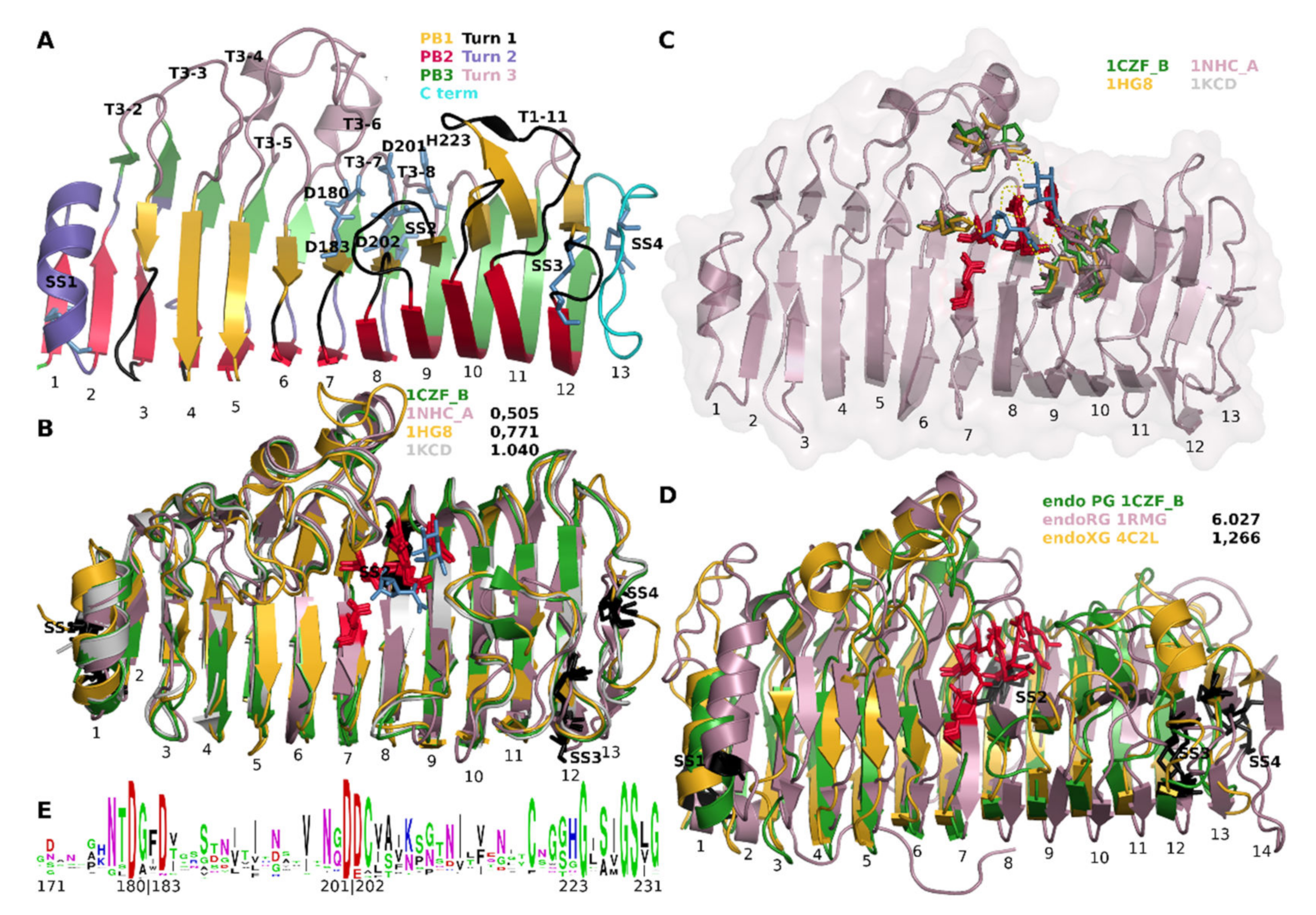
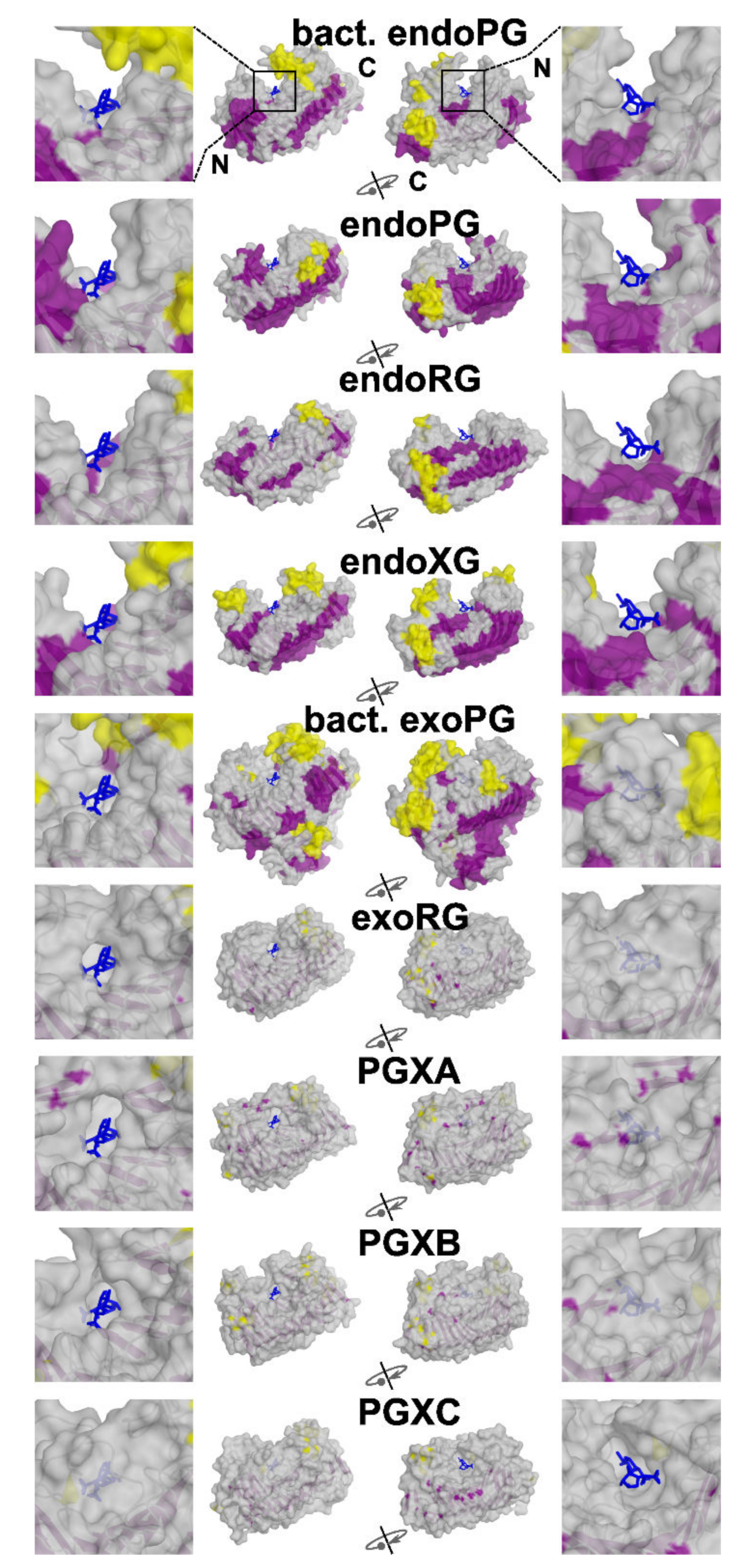
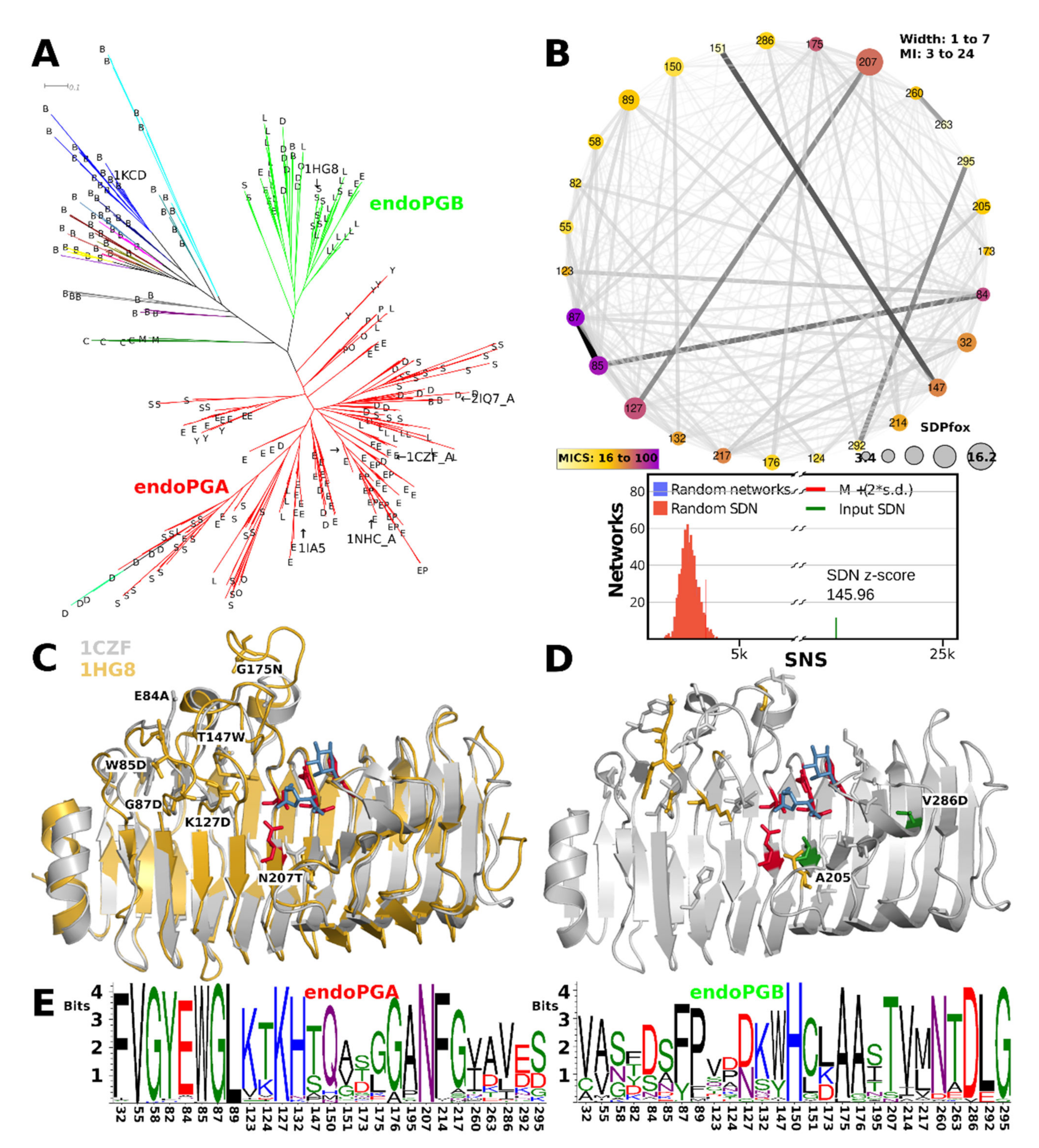
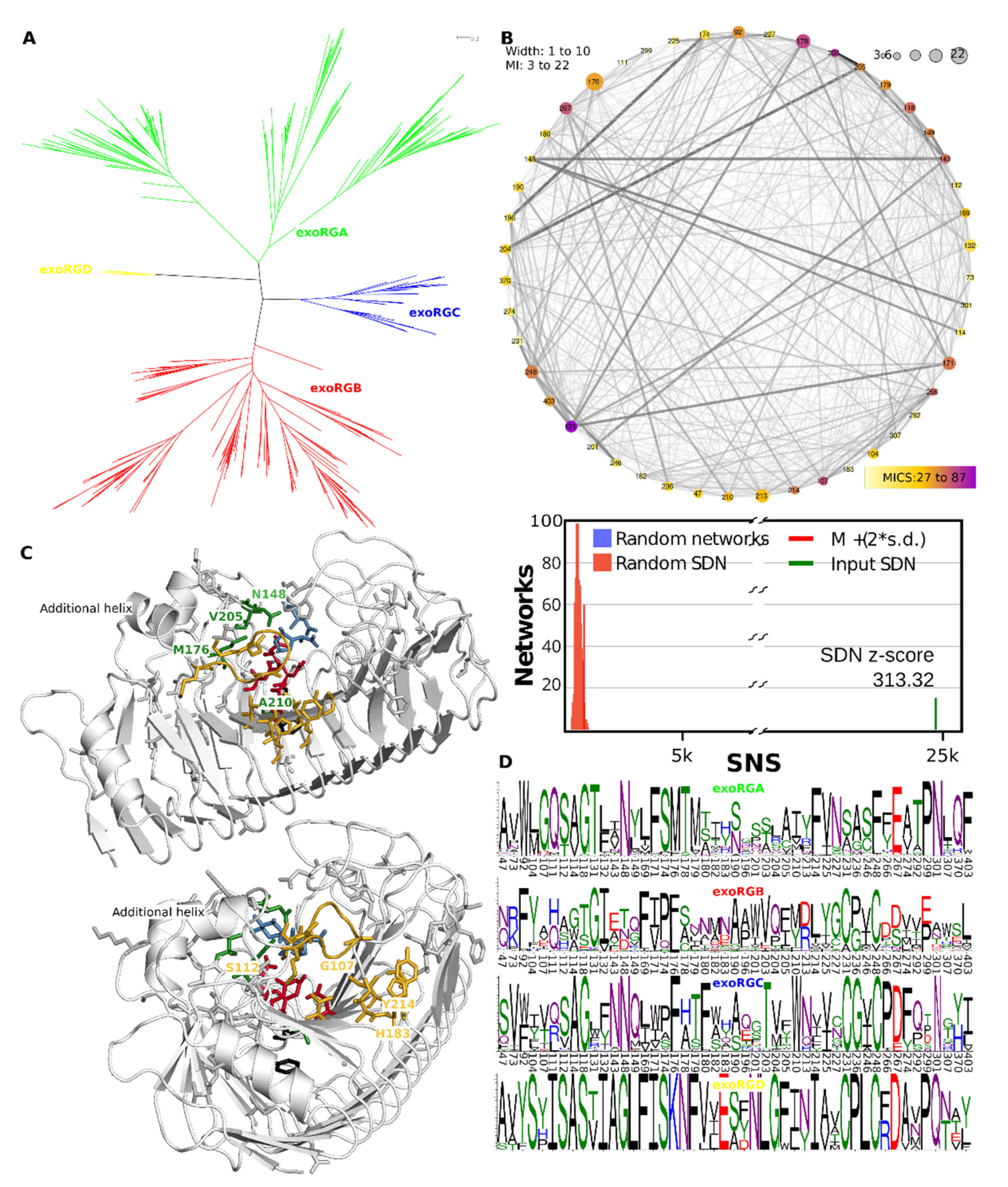
3.3. Functional Diversification in the Ascomycete GH28 Protein Family Can Be Explained by Subfamily Specific Loops That Cover the Binding Groove
3.4. Evolutionary Conservation Analysis
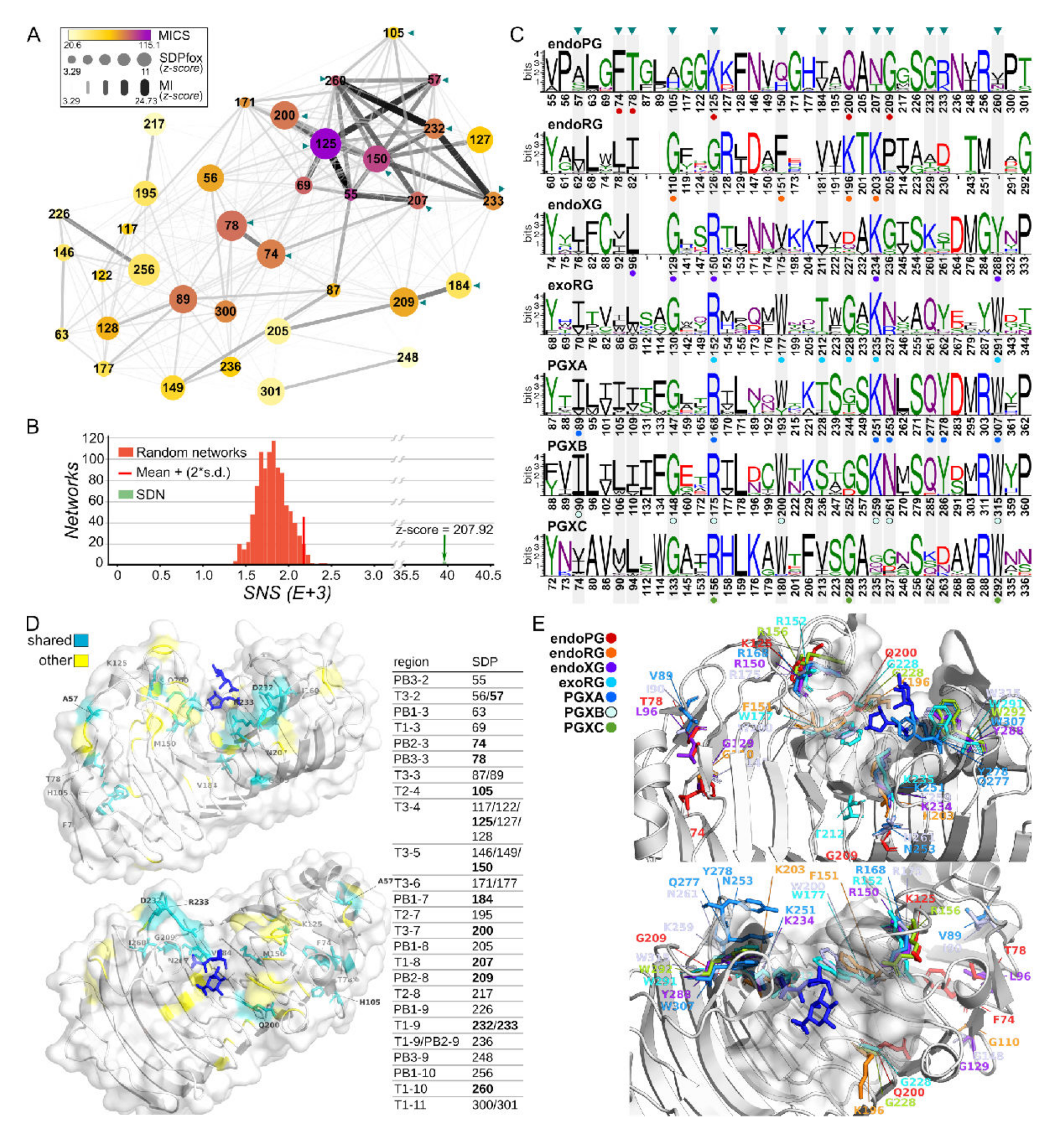
3.5. Superfamily Specificity Determining Positions Show Large Physicochemical Differences in the Binding Groove
3.6. Specificity Determining Positions and Rhamnogalacturonan
3.7. Specificity Determining Positions and Xylogalacturonan
3.8. PGXA and PGXB SDPs Do Not Show Clear Signs of Functional Diversification
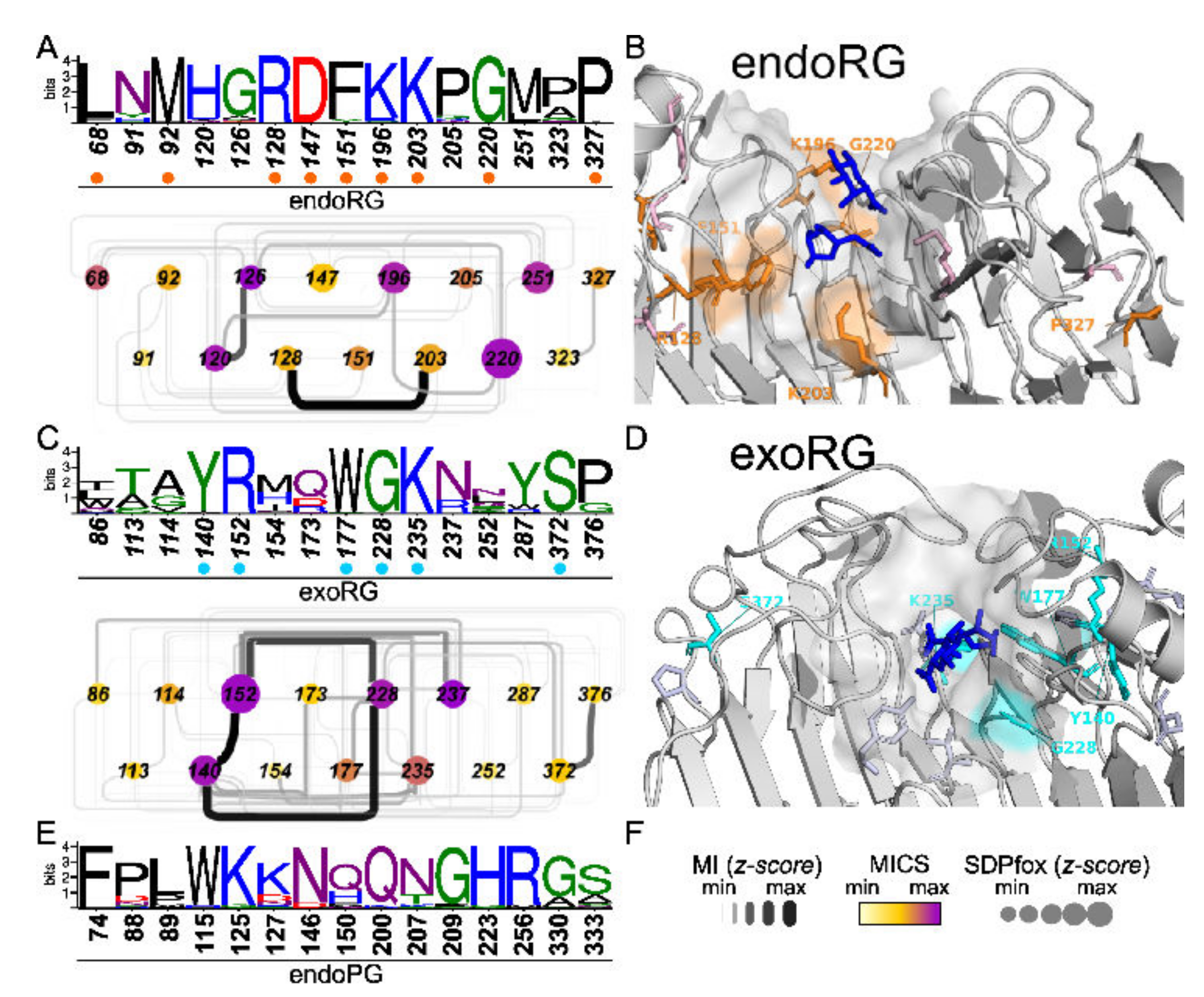
3.9. Ascomycetes endoPG Show Diversification of the T3 Loops
3.10. exoRGs Show Diversification of Site and Close by Subsites
3.11. A Small but Significantly Different endoRG Subfamily Is Detected
4. Discussion
4.1. Functional Diversification in Fungi Has Resulted in Six Major GH28 Subfamilies
4.2. Many but Not All Identified Specificity Determining Positions Correspond to the Site or a Subsite
4.3. Loops That Cover the Binding Groove Determine Exomode Activity
5. Concluding Remarks and Future Prospects
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McCann, M.C.; Roberts, K. Plant cell wall architecture: The role of pectins. Prog. Biotechnol. 1996, 14C, 91–107. [Google Scholar] [CrossRef]
- Vanitha, T.; Khan, M. Role of Pectin in Food Processing and Food Packaging. In Pectins—Extraction, Purification, Characterization and Applications; Masuelli, M., Ed.; IntechOpen: London, UK, 2019. [Google Scholar] [CrossRef]
- Schomburg, I.; Jeske, L.; Ulbrich, M.; Placzek, S.; Chang, A.; Schomburg, D. The BRENDA enzyme information system—From a database to an expert system. J. Biotechnol. 2017, 261, 194–206. [Google Scholar] [CrossRef] [PubMed]
- Lombard, V.; Golaconda Ramulu, H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, S.; Nagel, C.W. Isolation of an oligogalacturonate hydrolase from a Bacillus specie. Arch. Biochem. Biophys. 1968, 124, 513–520. [Google Scholar] [CrossRef]
- Kanungo, A.; Bag, B.P. Structural insights into the molecular mechanisms of pectinolytic enzymes. J. Proteins Proteom. 2019, 104, 325–344. [Google Scholar] [CrossRef]
- Van Der Vlugt-Bergmans, C.J.B.; Meeuwsen, P.J.A.; Voragen, A.G.J.; Van Ooyen, A.J.J. Endo-Xylogalacturonan Hydrolase, a Novel Pectinolytic Enzyme. Appl. Environ. Microbiol. 2000, 66, 36. [Google Scholar] [CrossRef][Green Version]
- Martens-Uzunova, E.S.; Zandleven, J.S.; Benen, J.A.E.; Awad, H.; Kools, H.J.; Beldman, G.; Voragen, A.G.J.; Berg, J.A.V.D.; Schaap, P.J. A new group of exo-acting family 28 glycoside hydrolases of Aspergillus niger that are involved in pectin degradation. Biochem. J. 2006, 400 Pt 1, 43. [Google Scholar] [CrossRef]
- Ridley, B.L.; O’Neill, M.A.; Mohnen, D. Pectins: Structure, biosynthesis, and oligogalacturonide-related signaling. Phytochemistry 2001, 57, 929–967. [Google Scholar] [CrossRef]
- Van Pouderoyen, G.; Snijder, H.J.; Benen, J.A.E.; Dijkstra, B.W. Structural insights into the processivity of endopolygalacturonase I from Aspergillus niger. FEBS Lett. 2003, 554, 462–466. [Google Scholar] [CrossRef]
- Rozeboom, H.J.; Beldman, G.; Schols, H.A.; Dijkstra, B.W. Crystal structure of endo-xylogalacturonan hydrolase from Aspergillus tubingensis. FEBS J. 2013, 280, 6061–6069. [Google Scholar] [CrossRef] [PubMed]
- Benen, J.A.E.; Kester, H.C.M.; Visser, J. Kinetic characterization of Aspergillus niger N400 endopolygalacturonases I, II and C. Eur. J. Biochem. 1999, 259, 577–585. [Google Scholar] [CrossRef]
- Interpro Classification of Protein Families; Pectate Lyase Like Entry. Available online: https://www.ebi.ac.uk/interpro/entry/InterPro/IPR012334/ (accessed on 3 December 2021).
- Superfamily Protein HMM Library; Pectate Lyase Like Entry. Available online: https://supfam.mrc-lmb.cam.ac.uk/SUPERFAMILY/cgi-bin/scop.cgi?sunid=51127 (accessed on 3 December 2021).
- Structural Classification of Protein Folds; Pectate Lyase Like Entry. Available online: http://scop.mrc-lmb.cam.ac.uk/term/4002788 (accessed on 3 December 2021).
- Michel, G.; Chantalat, L.; Fanchon, E.; Henrissat, B.; Kloareg, B.; Dideberg, O. The iota-carrageenase of Alteromonas fortis: A beta-helix fold-containing enzyme for the degradation of a highly polyanionic polysaccharide. J. Biol. Chem. 2001, 276, 40202–40209. [Google Scholar] [CrossRef]
- McCarthy, T.W.; Der, J.P.; Honaas, L.A.; de Pamphilis, C.W.; Anderson, C.T. Phylogenetic analysis of pectin-related gene families in Physcomitrella patens and nine other plant species yields evolutionary insights into cell walls. BMC Plant Biol. 2014, 14, 1–14. [Google Scholar] [CrossRef]
- Götesson, A.; Marshall, J.S.; Jones, D.A.; Hardham, A.R. Characterization and evolutionary analysis of a large polygalacturonase gene family in the oomycete plant pathogen Phytophthora cinnamomi. Mol. Plant Microbe Interact. 2002, 15, 907–921. [Google Scholar] [CrossRef]
- Allen, M.L.; Mertens, J.A. Molecular cloning and expression of three polygalacturonase cDNAs from the tarnished plant bug, Lygus lineolaris. J. Insect Sci. 2008, 8, 1–14. [Google Scholar] [CrossRef]
- Jaubert, S.; Laffaire, J.B.; Abad, P.; Rosso, M.N. A polygalacturonase of animal origin isolated from the root-knot nematode Meloidogyne incognita. FEBS Lett. 2002, 522, 109–112. [Google Scholar] [CrossRef]
- Flint, H.J.; Scott, K.P.; Duncan, S.H.; Louis, P.; Forano, E. Microbial degradation of complex carbohydrates in the gut. Gut Microbes 2012, 3, 289. [Google Scholar] [CrossRef]
- Yuan, P.; Meng, K.; Wang, Y.; Luo, H.; Huang, H.; Shi, P.; Bai, Y.; Yang, P.; Yao, B. Abundance and Genetic Diversity of Microbial Polygalacturonase and Pectate Lyase in the Sheep Rumen Ecosystem. PLoS ONE 2012, 7, 7. [Google Scholar] [CrossRef]
- Ten Have, A.; Mulder, W.; Visser, J.; Van Kan, J.A.L. The endopolygalacturonase gene Bcpg1 is required to full virulence of Botrytis cinerea. Mol. Plant Microbe Interact. 1998, 11, 1009–1016. [Google Scholar] [CrossRef]
- Shieh, M.T.; Brown, R.L.; Whitehead, M.P.; Cary, J.W.; Cotty, P.J.; Cleveland, T.E.; Dean, R.A. Molecular genetic evidence for the involvement of a specific polygalacturonase, P2c, in the invasion and spread of Aspergillus flavus in cotton bolls. Appl. Environ. Microbiol. 1997, 63, 3548. [Google Scholar] [CrossRef]
- Kars, I.; Krooshof, G.H.; Wagemakers, L.; Joosten, R.; Benen, J.A.E.; Van Kan, J.A.L. Necrotizing activity of five Botrytis cinerea endopolygalacturonases produced in Pichia pastoris. Plant J. 2005, 43, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Rong, W.; He, C. Two Xanthomonas extracellular polygalacturonases, PghAxc and PghBxc, are regulated by type III secretion regulators HrpX and HrpG and are required for virulence. Mol. Plant Microbe Interact. 2008, 21, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Martens-Uzunova, E.S.; Schaap, P.J. An evolutionary conserved d-galacturonic acid metabolic pathway operates across filamentous fungi capable of pectin degradation. Fungal Genet. Biol. 2008, 45, 1449–1457. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Thiewes, H.; van Kan, J.A.L. The d-galacturonic acid catabolic pathway in Botrytis cinerea. Fungal Genet. Biol. 2011, 48, 990–997. [Google Scholar] [CrossRef]
- Petersen, T.N.; Kauppinen, S.; Larsen, S. The crystal structure of rhamnogalacturonase A from Aspergillus aculeatus: A right-handed parallel beta helix. Structure 1997, 5, 533–544. [Google Scholar] [CrossRef]
- Van Santen, Y.; Benen, J.A.E.; Schröter, K.H.; Kalk, K.H.; Armand, S.; Visser, J.; Dijkstra, B.W. 1.68-A crystal structure of endopolygalacturonase II from Aspergillus niger and identification of active site residues by site-directed mutagenesis. J. Biol. Chem. 1999, 274, 30474–30480. [Google Scholar] [CrossRef]
- Prosite Documentation Polygalacturonase Active Site. Available online: https://prosite.expasy.org/PDOC00415 (accessed on 4 December 2021).
- Sieger, G.M.; Ziegler, W.M.; Klein, D.X.; Sokol, H. Proline: The Distribution, Frequency, Positioning, and Common Functional Roles of Proline and Polyproline Sequences in the Human Proteome. PLoS ONE 2013, 8, e53785. [Google Scholar] [CrossRef]
- Federici, L.; Caprari, C.; Mattei, B.; Savino, C.; Di Matteo, A.; De Lorenzo, G.; Cervone, F.; Tsernoglou, D. Structural requirements of endopolygalacturonase for the interaction with PGIP (polygalacturonase-inhibiting protein). Proc. Natl. Acad. Sci. USA 2001, 98, 13425–13430. [Google Scholar] [CrossRef]
- Shimizu, T.; Nakatsu, T.; Miyairi, K.; Okuno, T.; Kato, H. Active-site architecture of endopolygalacturonase I from Stereum purpureum revealed by crystal structures in native and ligand-bound forms at atomic resolution. Biochemistry 2002, 41, 6651–6659. [Google Scholar] [CrossRef]
- McClendon, J.H. The active site of yeast endo-polygalacturonase contains seven subsites. Phytochemistry 1979, 18, 765–769. [Google Scholar] [CrossRef]
- Pagès, S.; Heijne, W.H.M.; Kester, H.C.M.; Visser, J.; Benen, J.A.E. Subsite mapping of Aspergillus niger endopolygalacturonase II by site-directed mutagenesis. J. Biol. Chem. 2000, 275, 29348–29353. [Google Scholar] [CrossRef]
- Kester, H.C.M.; Magaud, D.; Roy, C.; Anker, D.; Doutheau, A.; Shevchik, V.; Cotte-Pattat, N.; Benen, J.A.E.; Visser, J. Performance of selected microbial pectinases on synthetic monomethyl-esterified di- and trigalacturonates. J. Biol. Chem. 1999, 274, 37053–37059. [Google Scholar] [CrossRef][Green Version]
- Massa, C.; Clausen, M.H.; Stojan, J.; Lamba, D.; Campa, C. Study of the mode of action of a polygalacturonase from the phytopathogen Burkholderia cepacia. Biochem. J. 2007, 407 Pt 2, 207. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7, Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Deorowicz, S.; Debudaj-Grabysz, A.; Gudys, A. FAMSA: Fast and accurate multiple sequence alignment of huge protein families. Sci. Rep. 2016, 6, 33964. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Felsenstein, J. Confidence Limits on Phylogenies: An Approach Using the Bootstrap. Evolution 1985, 39, 783. [Google Scholar] [CrossRef]
- Lemoine, F.; Domelevo Entfellner, J.B.; Wilkinson, E.; Correia, D.; Dávila Felipe, M.; De Oliveira, T.; Gascuel, O. Renewing Felsenstein’s phylogenetic bootstrap in the era of big data. Nature 2018, 556, 452–456. [Google Scholar] [CrossRef]
- Pagnuco, I.A.; Revuelta, M.V.; Bondino, H.G.; Brun, M.; Ten Have, A. HMMER cut-off threshold tool (HMMERCTTER): Supervised classification of superfamily protein sequences with a reliable cut-off threshold. PLoS ONE 2018, 13, e0193757. [Google Scholar] [CrossRef]
- Lichtarge, O.; Sowa, M.E. Evolutionary Predictions of Binding Surfaces and Interactions. Curr. Opin. Struct. Biol. 2002, 12, 21–27. Available online: http://www.ncbi.nlm.nih.gov/pubmed/11839485 (accessed on 28 June 2017). [CrossRef]
- Wilkins, A.; Erdin, S.; Lua, R.; Lichtarge, O. Evolutionary trace for prediction and redesign of protein functional sites. Methods Mol. Biol. 2012, 819, 29–42. [Google Scholar] [CrossRef]
- Mazin, P.V.; Gelfand, M.S.; Mironov, A.A.; Rakhmaninova, A.B.; Rubinov, A.R.; Russell, R.B.; Kalinina, O.V. An automated stochastic approach to the identification of the protein specificity determinants and functional subfamilies. Algorithms Mol. Biol. 2010, 5, 29. [Google Scholar] [CrossRef]
- Simonetti, F.L.; Teppa, E.; Chernomoretz, A.; Nielsen, M.; Marino Buslje, C. MISTIC: Mutual information server to infer coevolution. Nucleic Acids Res. 2013, 41, W8–W14. [Google Scholar] [CrossRef]
- Pereira, J.; Simpkin, A.J.; Hartmann, M.D.; Rigden, D.J.; Keegan, R.M.; Lupas, A.N. High-accuracy protein structure prediction in CASP14. Proteins Struct. Funct. Bioinform. 2021, 89, 1687–1699. [Google Scholar] [CrossRef]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef]
- Pfam Glyco_hydro_28 (PF00295) Hidden Markov Model Profile. Available online: https://pfam.xfam.org/family/PF00295/hmm (accessed on 6 December 2021).
- Eddy, S.R. Accelerated Profile HMM Searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef]
- HmmerWeb Version 2.41.2. Available online: https://www.ebi.ac.uk/Tools/hmmer/search/hmmsearch (accessed on 30 November 2021).
- Bairoch, A.; Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000, 28, 45–48. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W. Gapped BLAST and PSI-BLAST: A New Generation of Protein Database Search Programs. Nucleic Acids Res. 1997, 25, 3389–3402. Available online: http://www.ncbi.nlm.nih.gov/pubmed/9254694 (accessed on 22 June 2017). [CrossRef]
- Sequence Download Site Pfam Glyco_hydro_28 (PF00295): Glyco_hydro_28 (PF00295). Available online: https://pfam.xfam.org/family/Glyco_hydro_28#tabview=tab7 (accessed on 1 November 2021).
- Apweiler, R.; Bairoch, A.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M.; et al. The Uniprot Consortium The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef] [PubMed]
- Larsson, A. AliView: A fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 2014, 30, 3276–3278. [Google Scholar] [CrossRef] [PubMed]
- Junier, T.; Pagni, M. Dotlet: Diagonal plots in a Web browser. Bioinformatics 2000, 16, 178–179. [Google Scholar] [CrossRef] [PubMed]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Criscuolo, A.; Gribaldo, S. BMGE (Block Mapping and Gathering with Entropy): A new software for selection of phylogenetic informative regions from multiple sequence alignments. BMC Evol. Biol. 2010, 10, 210. [Google Scholar] [CrossRef]
- Gouy, M.; Guindon, S.; Gascuel, O. SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 2010, 27, 221–224. [Google Scholar] [CrossRef]
- Huson, D.H.; Scornavacca, C. Dendroscope 3: An interactive tool for rooted phylogenetic trees and networks. Syst. Biol. 2012, 61, 1061–1067. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL): An online tool for phylogenetic tree display and annotation. Bioinformatics 2007, 23, 127–128. [Google Scholar] [CrossRef]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME Suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef]
- MEME Server. Available online: https://meme-suite.org/meme/tools/meme (accessed on 29 December 2021).
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef]
- ColabFold: AlphaFold2 Using MMseqs2. Available online: https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/AlphaFold2.ipynb (accessed on 3 December 2021).
- Ashkenazy, H.; Abadi, S.; Martz, E.; Chay, O.; Mayrose, I.; Pupko, T.; Ben-Tal, N. ConSurf 2016: An improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016, 44, W344–W350. [Google Scholar] [CrossRef]
- The Consurf Conservation Server. Available online: https://consurf.tau.ac.il/ (accessed on 3 December 2021).
- PyMOL. PyMOL: The PyMOL Molecular Graphics System Version 2.0. Schrödinger, LLC.: New York, NY, USA, 2015. [Google Scholar]
- Kirsch, R.; Gramzow, L.; Theißen, G.; Siegfried, B.D.; ffrench-Constant, R.H.; Heckel, D.G.; Pauchet, Y. Horizontal gene transfer and functional diversification of plant cell wall degrading polygalacturonases: Key events in the evolution of herbivory in beetles. Insect Biochem. Mol. Biol. 2014, 52, 33–50. [Google Scholar] [CrossRef]
- Pickersgill, R.; Smith, D.; Worboys, K.; Jenkins, J. Crystal Structure of Polygalacturonase from Erwinia carotovora ssp. carotovora. J. Biol. Chem. 1998, 273, 24660–24664. [Google Scholar] [CrossRef]
- Abbott, D.W.; Boraston, A.B. The structural basis for exopolygalacturonase activity in a family 28 glycoside hydrolase. J. Mol. Biol. 2007, 368, 1215–1222. [Google Scholar] [CrossRef]
- Glaser, F.; Pupko, T.; Paz, I.; Bell, R.E.; Bechor-Shental, D.; Martz, E.; Ben-Tal, N. ConSurf: Identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics 2003, 19, 163–164. [Google Scholar] [CrossRef]
- Cho, S.W.; Lee, S.; Shin, W. The X-ray structure of Aspergillus aculeatus polygalacturonase and a modeled structure of the polygalacturonase-octagalacturonate complex. J. Mol. Biol. 2001, 311, 863–878. [Google Scholar] [CrossRef]
- Nei, M.; Gu, X.; Sitnikova, T. Evolution by the birth-and-death process in multigene families of the vertebrate immune system. Proc. Natl. Acad. Sci. USA 1997, 94, 7799–7806. [Google Scholar] [CrossRef]
- Bondino, H.G.; Valle, E.M.; ten Have, A. Evolution and functional diversification of the small heat shock protein/α-crystallin family in higher plants. Planta 2012, 235, 1299–1313. [Google Scholar] [CrossRef]
- Young, M.A.; Larson, D.E.; Sun, C.W.; George, D.R.; Ding, L.; Miller, C.A.; Lin, L.; Pawlik, K.M.; Chen, K.; Fan, X.; et al. Background mutations in parental cells account for most of the genetic heterogeneity of induced pluripotent stem cells. Cell Stem Cell 2012, 10, 570–582. [Google Scholar] [CrossRef]
- Wagner, A. Neutralism and selectionism: A network-based reconciliation. Nat. Rev. Genet. 2008, 9, 965–974. [Google Scholar] [CrossRef]
- André-Leroux, G.; Tessier, D.; Bonnin, E. Endopolygalacturonases reveal molecular features for processivity pattern and tolerance towards acetylated pectin. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2008, 1794, 5–13. [Google Scholar] [CrossRef]
- Ralet, M.C.; Crépeau, M.J.; Bonnin, E. Evidence for a blockwise distribution of acetyl groups onto homogalacturonans from a commercial sugar beet (Beta vulgaris) pectin. Phytochemistry 2008, 69, 1903–1909. [Google Scholar] [CrossRef]
- Tu, T.; Pan, X.; Meng, K.; Luo, H.; Ma, R.; Wang, Y.; Yao, B. Substitution of a non-active-site residue located on the T3 loop increased the catalytic efficiency of endo-polygalacturonases. Process Biochem. 2016, 51, 1230–1238. [Google Scholar] [CrossRef]
- Tu, T.; Meng, K.; Luo, H.; Turunen, O.; Zhang, L.; Cheng, Y.; Su, X.; Ma, R.; Shi, P.; Wang, Y.; et al. New Insights into the Role of T3 Loop in Determining Catalytic Efficiency of GH28 Endo-Polygalacturonases. PLoS ONE 2015, 10, e0135413. [Google Scholar] [CrossRef]
- Yang, Y.; Yu, Y.; Liang, Y.; Anderson, C.T.; Cao, J. A profusion of molecular scissors for pectins: Classification, expression, and functions of plant polygalacturonases. Front. Plant Sci. 2018, 9, 1208. [Google Scholar] [CrossRef]
- Bustamante, J.P.; Radusky, L.; Boechi, L.; Estrin, D.A.; ten Have, A.; Martí, M.A. Evolutionary and Functional Relationships in the Truncated Hemoglobin Family. PLoS Comput. Biol. 2016, 12, e1004701. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| All | endoPG | endoRG | endoXG | PGXA | PGXB | PGXC | Mucoro | exoRG | N | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Dothideomycete | 298 | 109 | 0.37 | 36 | 0.12 | 7 | 0.02 | 51 | 0.17 | 14 | 0.05 | 8 | 0.03 | 0 | NA | 73 | 0.24 | 298 |
| Eurotiomycete | 1150 | 394 | 0.34 | 246 | 0.21 | 81 | 0.07 | 100 | 0.09 | 81 | 0.07 | 54 | 0.05 | 0 | NA | 194 | 0.17 | 1150 |
| Leotiomycete | 429 | 140 | 0.33 | 60 | 0.14 | 54 | 0.13 | 59 | 0.14 | 25 | 0.06 | 19 | 0.04 | 0 | NA | 72 | 0.17 | 429 |
| Neolectomycetes | 2 | 2 | NA | 0 | NA | 0 | NA | 0 | NA | 0 | NA | 0 | NA | 0 | NA | 0 | NA | 2 |
| Orbiliomycetes | 30 | 12 | NA | 0 | NA | 4 | NA | 9 | NA | 3 | NA | 0 | NA | 0 | NA | 2 | NA | 30 |
| Pezizomycetes | 15 | 6 | NA | 4 | NA | 0 | NA | 2 | NA | 3 | NA | 0 | NA | 0 | NA | 0 | NA | 15 |
| Saccharomycetes | 13 | 13 | NA | 0 | NA | 0 | NA | 0 | NA | 0 | NA | 0 | NA | 0 | NA | 0 | NA | 13 |
| Sordariomycetes | 787 | 241 | 0.31 | 78 | 0.10 | 35 | 0.04 | 117 | 0.15 | 137 | 0.17 | 44 | 0.06 | 0 | NA | 135 | 0.17 | 787 |
| Fungi 14919 | 12 | 3 | NA | 1 | NA | 2 | NA | 2 | NA | 1 | NA | 0 | NA | 0 | NA | 3 | NA | 12 |
| ∑Ascomycetes | 2736 | 920 | 0.34 | 425 | 0.16 | 183 | 0.07 | 340 | 0.12 | 264 | 0.10 | 125 | 0.05 | 0 | NA | 479 | 0.18 | 2736 |
| Basidio | 499 | 208 | 0.42 | 59 | 0.12 | 8 | 0.02 | 3 | 0.01 | 0 | 0 | 100 | 0.20 | 0 | NA | 121 | 0.24 | 499 |
| Mucoro | 75 | 2 | NA | 1 | NA | 0 | NA | 0 | NA | 0 | NA | 0 | NA | 72 | NA | 0 | NA | 75 |
| Chytridio | 9 | 6 | NA | 1 | NA | 0 | NA | 0 | NA | 2 | NA | 0 | NA | 0 | NA | 0 | NA | 9 |
| ∑ | 3319 | 1136 | 0.34 | 486 | 0.15 | 191 | 0.06 | 343 | 0.1 | 266 | 0.08 | 225 | 0.07 | 72 | NA | 600 | 0.18 | 3319 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Villarreal, F.; Stocchi, N.; ten Have, A. Functional Classification and Characterization of the Fungal Glycoside Hydrolase 28 Protein Family. J. Fungi 2022, 8, 217. https://doi.org/10.3390/jof8030217
Villarreal F, Stocchi N, ten Have A. Functional Classification and Characterization of the Fungal Glycoside Hydrolase 28 Protein Family. Journal of Fungi. 2022; 8(3):217. https://doi.org/10.3390/jof8030217
Chicago/Turabian StyleVillarreal, Fernando, Nicolás Stocchi, and Arjen ten Have. 2022. "Functional Classification and Characterization of the Fungal Glycoside Hydrolase 28 Protein Family" Journal of Fungi 8, no. 3: 217. https://doi.org/10.3390/jof8030217
APA StyleVillarreal, F., Stocchi, N., & ten Have, A. (2022). Functional Classification and Characterization of the Fungal Glycoside Hydrolase 28 Protein Family. Journal of Fungi, 8(3), 217. https://doi.org/10.3390/jof8030217