
Characteristics of the Genome, Transcriptome and Ganoderic Acid of the Medicinal Fungus Ganoderma lingzhi
 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Fungal Strains and Culture Conditions
2.2. Genome Sequencing, Assembly, and Annotation
2.3. Identification of CAZymes and Mating Type (MAT) Genes
2.4. Ganoderic Acid Extraction and Mass Spectrometry Analysis
2.5. RNA-Seq
3. Results and Discussion
3.1. The Genome Characteristics of G. lingzhi
3.2. Gene Annotation
3.3. GO and KEGG Analysis
3.4. Identification of CAZymes
3.5. Mating Gene Loci in G. lingzhi
3.6. The Pathway of Ganoderic Acid Biosynthesis
3.7. Identification of GAs by Mass Spectrometry
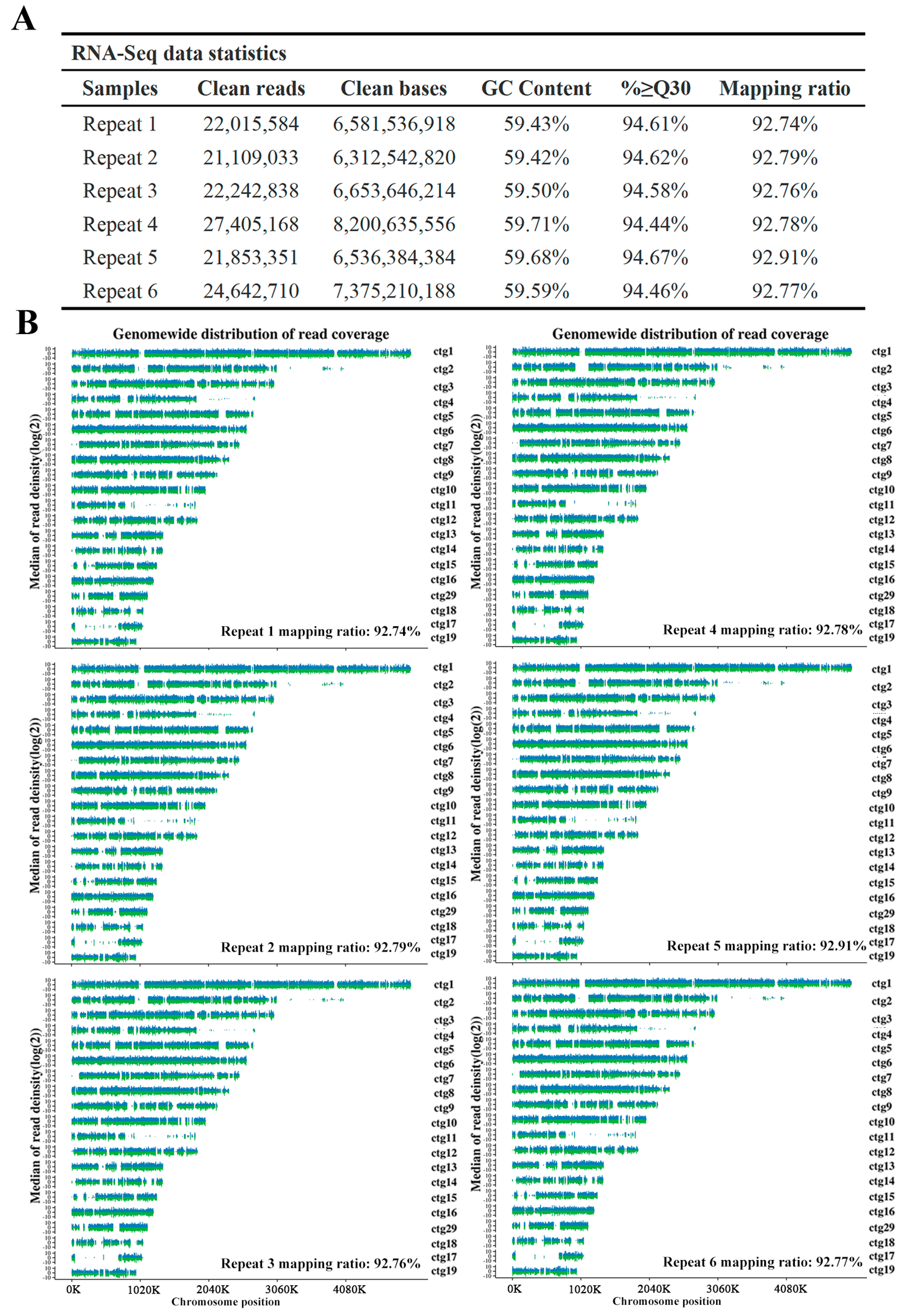
3.8. RNA-Seq Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kladar, N.V.; Gavarić, N.S.; Božin, B.N. Ganoderma: Insights into anticancer effects. Eur. J. Cancer Prev. 2016, 25, 462–471. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Wu, S.H.; Dai, Y.C. Species clarification of the prize medicinal Ganoderma mushroom “Lingzhi”. Fungal Divers. 2012, 56, 49–62. [Google Scholar] [CrossRef]
- Ren, A.; Shi, L.; Zhu, J.; Yu, H.S.; Jiang, A.L.; Zheng, H.H.; Zhao, M.W. Shedding light on the mechanisms underlying the environmental regulation of secondary metabolite ganoderic acid in Ganoderma lucidum using physiological and genetic methods. Fungal Genet. Biol. 2019, 128, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.Y.; Tian, D.N.; Liu, Y.Z.; Li, H.; Zhu, J.L.; Li, M.; Xin, M.H.; Xia, J. Review of the molecular mechanisms of Ganoderma lucidum triterpenoids: Ganoderic acids A, C2, D, F, DM, X and Y. Eur. J. Med. Chem. 2019, 174, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.H.; Wu, H.Y.; Wu, K.M.; Liu, T.T.; Liou, R.F.; Tsai, S.F.; Shiao, M.S.; Ho, L.T.; Tzean, S.S.; Yang, U.C. Generation and analysis of the expressed sequence tags from the mycelium of Ganoderma lucidum. PLoS ONE 2013, 8, e61127. [Google Scholar] [CrossRef]
- Liu, D.B.; Gong, J.; Dai, W.K.; Kang, X.C.; Huang, Z.; Zhang, H.M.; Liu, W.; Liu, L.; Ma, J.P.; Xia, Z.L.; et al. The Genome of Ganderma lucidum provide insights into triterpense biosynthesis and wood degradation. PLoS ONE 2012, 7, e36146. [Google Scholar] [CrossRef]
- Tian, Y.Z.; Wang, Z.F.; Liu, Y.D.; Zhang, G.Z.; Li, G. The whole-genome sequencing and analysis of a Ganoderma lucidum strain provide insights into the genetic basis of its high triterpene content. Genomics 2021, 113, 840–849. [Google Scholar] [CrossRef]
- Chen, S.L.; Xu, J.; Liu, C.; Zhu, Y.J.; Nelson, D.R.; Zhou, S.G.; Li, C.F.; Wang, L.Z.; Guo, X.; Sun, Y.Z.; et al. Genome sequence of the model medicinal mushroom Ganoderma lucidum. Nat Commun. 2012, 3, 913. [Google Scholar] [CrossRef]
- Liu, Y.-N.; Wu, F.-Y.; Tian, R.-Y.; Shi, Y.-X.; Xu, Z.-Q.; Liu, J.-Y.; Huang, J.; Xue, F.-F.; Liu, G.-Q. The Regulatory and Transcriptional Landscape Associated with Triterpenoid and Lipid Metabolisms by the bHLH-Zip Transcription Factor SREBP in the Medicinal Fungus Ganoderma lingzhi. Available online: https://doi.org/10.21203/rs.3.rs-1244597/v1 (accessed on 5 August 2022). [CrossRef]
- Liu, Y.N.; Tong, T.; Zhang, R.R.; Liu, L.M.; Shi, M.L.; Ma, Y.C.; Liu, G.Q. Interdependent nitric oxide and hydrogen peroxide independently regulate the coix seed oil-induced triterpene acid accumulation in Ganoderma lingzhi. Mycologia 2019, 111, 529–540. [Google Scholar] [CrossRef]
- Chin, C.S.; Peluso, P.; Sedlazeck, F.J.; Nattestad, M.; Concepcion, G.T.; Clum, A.; Dunn, C.; O’Malley, R.; Figueroa-Balderas, R.; Morales-Cruz, A.; et al. Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods 2016, 13, 1050–1054. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Earl, A.M. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef] [PubMed]
- Simao, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Gong, W.B.; Wang, Y.H.; Xie, C.L.; Zhou, Y.J.; Zhu, Z.H.; Peng, Y.D. Whole genome sequence of an edible and medicinal mushroom, Hericium erinaceus (Basidiomycota, Fungi). Genomics 2020, 112, 2393–2399. [Google Scholar] [CrossRef]
- Han, Z.; Tanner, Y.; Le, H.; Sarah, E.; Peizhi, W.; Zhenglu, Y.; Busk, P.K.; Ying, X.; Yanbin, Y. dbCAN2: A meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2018, W95–W101. [Google Scholar] [CrossRef]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-Seq reads. Nat. Biotechnol. 2015, 33, 290. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Deng, Y.; Jianqi, L.I.; Songfeng, W.U.; Zhu, Y.; Chen, Y.; Fuchu, H.E. Integrated nr database in protein annotation system and its localization. Comput. Eng. 2006, 32, 71–72. [Google Scholar]
- Bateman, A.; Martin, M.J.; Orchard, S.; Magrane, M.; Agivetova, R.; Ahmad, S.; Alpi, E.; Bowler-Barnett, E.H.; Britto, R.; Bursteinas, B.; et al. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Consortium, T. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Kiryutin, B.; Koonin, E.V.; Krylov, D.M.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; et al. The COG database: An updated version includes eukaryotes. BMC Bioinform. 2003, 4, 41. [Google Scholar] [CrossRef] [PubMed]
- Minoru, K.; Susumu, G.; Shuichi, K.; Yasushi, O.; Masahiro, H. The KEGG resource for deciphering the genome. Nucleic Acids Res 2004, 32, D277–D280. [Google Scholar] [CrossRef]
- Kim, D.; Landmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Cingolani, P. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Chen, L.F.; Gong, Y.H.; Cai, Y.L.; Liu, W.; Zhou, Y.; Xiao, Y.; Xu, Z.Y.; Liu, Y.; Lei, X.Y.; Wang, G.Z.; et al. Genome sequence of the edible cultivated mushroom Lentinula edodes (Shiitake) reveals insights into lignocellulose degradation. PLoS ONE 2016, 11, e0160336. [Google Scholar] [CrossRef]
- Qu, J.B.; Huang, C.Y.; Zhang, J.X. Genome-wide functional analysis of SSR for an edible mushroom Pleurotus Ostreatus. Gene 2016, 575, 524–530. [Google Scholar] [CrossRef]
- Eastwood, D.C.; Floudas, D.; Binder, M.; Majcherczyk, A.; Schneider, P.; Aerts, A.; Asiegbu, F.O.; Baker, S.E.; Barry, K.; Bendiksby, M.; et al. The plant cell wall-decomposing machinery underlies the functional diversity of forest fungi. Science 2011, 333, 762–765. [Google Scholar] [CrossRef]
- Brown, A.J.; Casselton, L.A. Mating in mushrooms: Increasing the chances but prolonging the affair. Trends Genet. 2001, 17, 393–400. [Google Scholar] [CrossRef]
- Gilbertson, A. Cultural studies and genetics of sexuality of Ganoderma lucidum and G. tsugae in relation to the taxonomy of the G. lucidum complex. Mycologia 1986, 78, 694–705. [Google Scholar] [CrossRef]
- Casselton, L.A.; Olesnicky, N.S. Molecular genetics of mating recognition in basidiomycete fungi. Microbiol. Mol. Biol. Rev. 1998, 62, 55–70. [Google Scholar] [CrossRef] [PubMed]
- James, T.Y.; Kues, U.; Rehner, S.A.; Vilgalys, R. Evolution of the gene encoding mitochondrial intermediate peptidase and its cosegregation with the A mating-type locus of mushroom fungi. Fungal Genet. Biol. 2004, 41, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Liu, J.K. Highly oxygenated lanostane triterpenoids from the fungus Ganoderma applanatum. Chem. Pharm. Bull. 2008, 56, 1035–1037. [Google Scholar] [CrossRef]
- Liu, J.Q.; Wang, C.F.; Li, Y.; Luo, H.R.; Qiu, M.H. Isolation and bioactivity evaluation of terpenoids from the medicinal fungus Ganoderma sinense. Planta Med. 2012, 78, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.R.; Wang, X.; Zhou, L.; Hou, B.; Zuo, Z.L.; Qiu, M.H. Ganocochlearic acid A, a rearranged hexanorlanostane triterpenoid, and cytotoxic triterpenoids from the fruiting bodies of Ganoderma cochlear. RSC Adv. 2015, 5, 95212–95222. [Google Scholar] [CrossRef]
- Zhao, X.R.; Zhang, B.J.; Deng, S.; Zhang, H.L.; Huang, S.S.; Huo, X.K.; Wang, C.; Liu, F.; Ma, X.C. Isolation and identification of oxygenated lanostane-type triterpenoids from the fungus Ganoderma lucidum. Phytochem. Lett. 2016, 16, 87–91. [Google Scholar] [CrossRef]
- Peng, X.R.; Liu, J.Q.; Han, Z.H.; Yuan, X.X.; Luo, H.R.; Qiu, M.H. Protective effects of triterpenoids from Ganoderma resinaceum on H2O2-induced toxicity in HepG2 cells. Food Chem. 2013, 141, 920–926. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Repeat Sequences | |||
|---|---|---|---|
| Item | Number | Length (bp) | Coverage |
| SINE | 6 | 488 | 0.00% |
| LINE | 121 | 22,417 | 0.05% |
| LTR | 1849 | 2,806,397 | 5.71% |
| DNA transposon | 538 | 1,013,038 | 2.06% |
| Satellite DNA | 26 | 2435 | 0.00% |
| Simple repeat | 7431 | 313,771 | 0.64% |
| Low complexity | 1008 | 51,127 | 0.10% |
| Other | 42 | 10,370 | 0.02% |
| Unknown | 6091 | 3,055,773 | 6.22% |
| Total | 17,112 | 7,275,816 | 14.80% |
| Non-Coding RNA | |||
| Class | Number | Total Length (bp) | Mean Length (bp) |
| rRNA | 13 | 17,274 | 1328 |
| sRNA | 2 | 709 | 354 |
| snRNA | 22 | 2869 | 130 |
| tRNA | 141 | 11,683 | 82 |
| Total | 178 | 32,535 | 182 |
| NO. | Compounds | Molecular Wght (Da) | Q1, m/z (Da) | Fragmentation, m/z (Da) |
|---|---|---|---|---|
| 1 | Ganoderic Acid SZ (C30H44O3) | 452.3 | 453.3 ([M+H]+) | 435.3, 185.1, 187.1, 239.2, 173.1, 201.2, 225.2 |
| 2 | Ganoderic Acid F (C30H46O3) | 454.3 | 455.3 ([M+H]+) | 437.3, 229.2, 299.3, 123.1, 201.2, 135.1 |
| 3 | Ganoderic Acid Jb (C30H46O4) | 470.3 | 471.3 ([M+H]+) | 435.3, 453.3, 201.2, 187.1, 471.3, 159.1, 175.1 |
| 4 | Ganodermanontriol (C30H48O4) | 472.4 | 473.4 ([M+H]+) | 329.2, 455.4, 415.3, 243.2, 261.2, 437.3, 189.1 |
| 5 | Ganoderic Acid T-Q (C32H46O5) | 510.3 | 511.3 ([M+H]+) | 433.3, 493.3, 311.2, 451.3, 293.2, 399.3, 337.2 |
| 6 | Ganoderic Acid Mf (C32H48O5) | 512.4 | 513.4 ([M+H]+) | 435.3, 495.3, 201.2, 295.2, 203.2, 453.3, 133.1 |
| 7 | Ganoderenic Acid C (C30H44O7) | 516.3 | 517.3 ([M+H]+) | 371.3, 499.3, 399.3, 463.3, 481.3, 353.2, 381.2 |
| 8 | Ganoderic Acid C2 (C30H46O7) | 518.3 | 519.3 ([M+H]+) | 355.3, 483.3, 501.3, 465.3, 447.3, 373.3 |
| 9 | Ganoderic Acid V (C32H48O6) | 528.3 | 529.3 ([M+H]+) | 469.3, 243.2, 423.3, 329.2, 451.3, 369.3, 355.3 |
| 10 | Ganoderic Acid Mj (C33H52O6) | 544.4 | 545.2 ([M+H]+) | 527.3, 449.3, 467.3, 431.3, 353.2, 421.3, 327.2 |
| 11 | Ganoderic Acid R (C34H50O6) | 554.4 | 555.4 ([M+H]+) | 435.3, 495.3, 201.2, 187.1, 145.1, 239.2, 341.2 |
| 12 | Ganoderic Acid Me (C34H50O6) | 554.4 | 555.4 ([M+H]+) | 435.3, 495.3, 295.2, 201.2, 203.2, 187.1, 189.2 |
| 13 | Ganoderic Acid Mk (C34H50O7) | 570.4 | 571.4 ([M+H]+) | 433.3, 451.3, 493.3, 201.2, 511.3, 293.2, 339.2 |
| 14 | Ganoderic Acid AP2 (C34H50O8) | 586.4 | 587.4 ([M+H]+) | 587.4, 569.4, 491.3, 509.3, 431.3, 395.3, 409.3, 463.3 |
| 15 | Ganoderic Acid T (C36H52O8) | 612.4 | 613.4 ([M+H]+) | 433.3, 493.3, 553.4, 201.2, 293.2, 451.3, 227.2 |
| 16 | Deacetyl Ganoderic acid F (C30H40O8) | 528.3 | 527.3 ([M-H]−) | 509.6, 479.4, 465.5, 435.6, 365.8, 315.2, 299.4 |
| 17 | Ganoderic acid A (C30H44O7) | 516.3 | 515.3 ([M-H]−) | 497.5, 453.6, 435.5, 355.3, 337.3, 299.5, 285.5 |
| 18 | Ganoderic acid GS-3 (C32H46O8) | 558.3 | 557.3 ([M-H]−) | 539.6, 497.4, 453.5, 435.6, 303.4, 287.4 |
| 19 | Ganoderic acid W (C34H52O7) | 572.4 | 571.4 ([M-H]−) | 553.6, 523.8, 509.3, 479.3, 465.4, 419.6, 345.5, 303.5, 285.4 |
| 20 | Ganochlearic acid A (C24H34O5) | 402.5 | 401.2 ([M-H]−) | 357.4, 329.4, 313.3, 287.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Q.; Liu, H.; Shi, Y.; Li, W.; Huang, J.; Xue, F.; Liu, Y.; Liu, G. Characteristics of the Genome, Transcriptome and Ganoderic Acid of the Medicinal Fungus Ganoderma lingzhi. J. Fungi 2022, 8, 1257. https://doi.org/10.3390/jof8121257
Wu Q, Liu H, Shi Y, Li W, Huang J, Xue F, Liu Y, Liu G. Characteristics of the Genome, Transcriptome and Ganoderic Acid of the Medicinal Fungus Ganoderma lingzhi. Journal of Fungi. 2022; 8(12):1257. https://doi.org/10.3390/jof8121257
Chicago/Turabian StyleWu, Qiang, Huan Liu, Yixin Shi, Wanting Li, Jia Huang, Feifei Xue, Yongnan Liu, and Gaoqiang Liu. 2022. "Characteristics of the Genome, Transcriptome and Ganoderic Acid of the Medicinal Fungus Ganoderma lingzhi" Journal of Fungi 8, no. 12: 1257. https://doi.org/10.3390/jof8121257
APA StyleWu, Q., Liu, H., Shi, Y., Li, W., Huang, J., Xue, F., Liu, Y., & Liu, G. (2022). Characteristics of the Genome, Transcriptome and Ganoderic Acid of the Medicinal Fungus Ganoderma lingzhi. Journal of Fungi, 8(12), 1257. https://doi.org/10.3390/jof8121257