
Comparative Whole-Genome Sequence Analyses of Fusarium Wilt Pathogen (Foc R1, STR4 and TR4) Infecting Cavendish (AAA) Bananas in India, with a Special Emphasis on Pathogenicity Mechanisms


 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Collection of Foc Races
2.2. DNA Extraction and Quantification
2.3. Genome Sequencing and Assembly
2.4. Mapping of Foc Genome
2.5. Identification and Analysis of Variants in the Foc Genome
2.6. Genome and Functional Annotation
2.7. Molecular Characterization of Indian VCGs for SIX genes
3. Results and Discussion
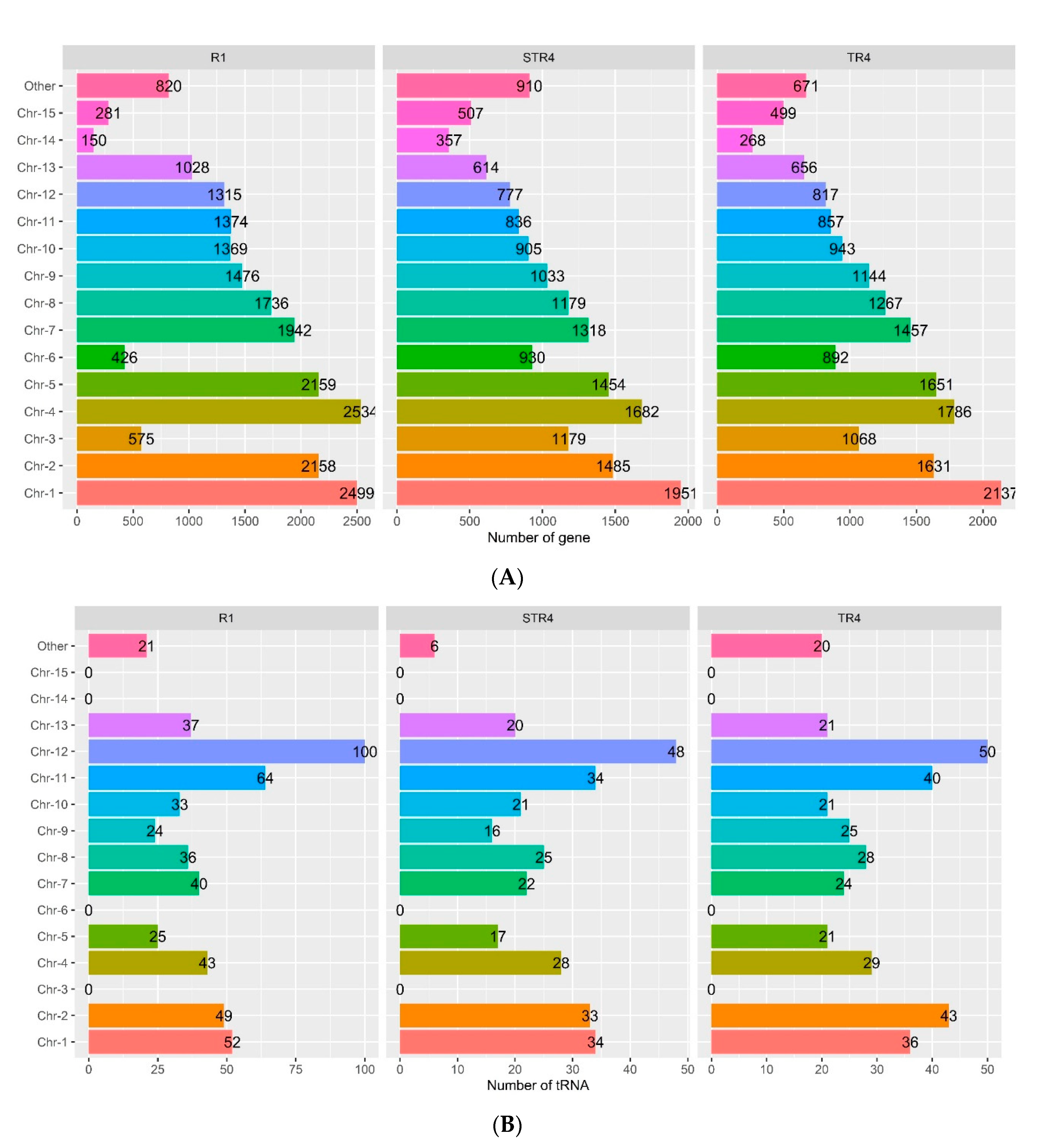
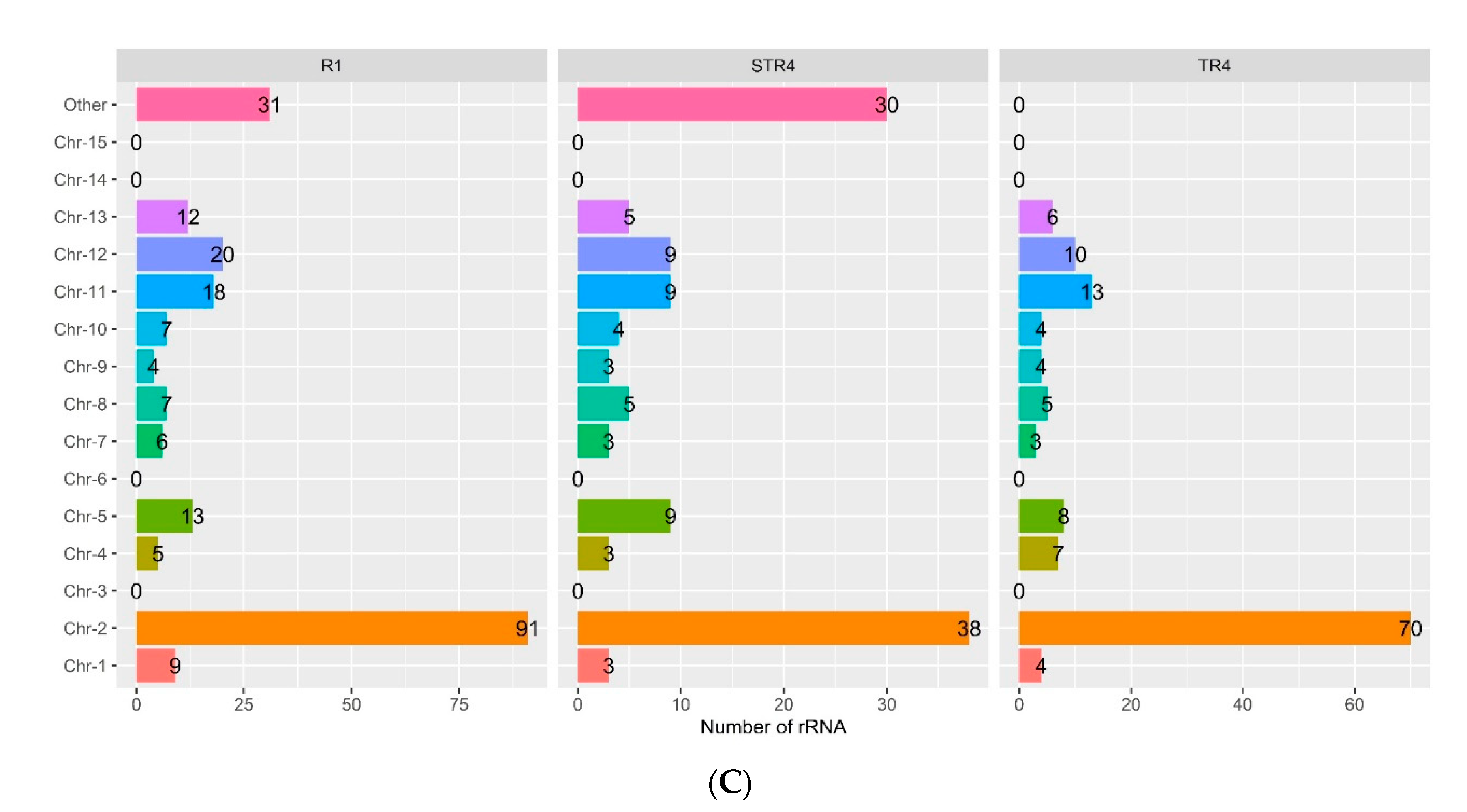
3.1. Genome Sequencing and General Features
3.2. Transposable Elements in the Foc Genomes
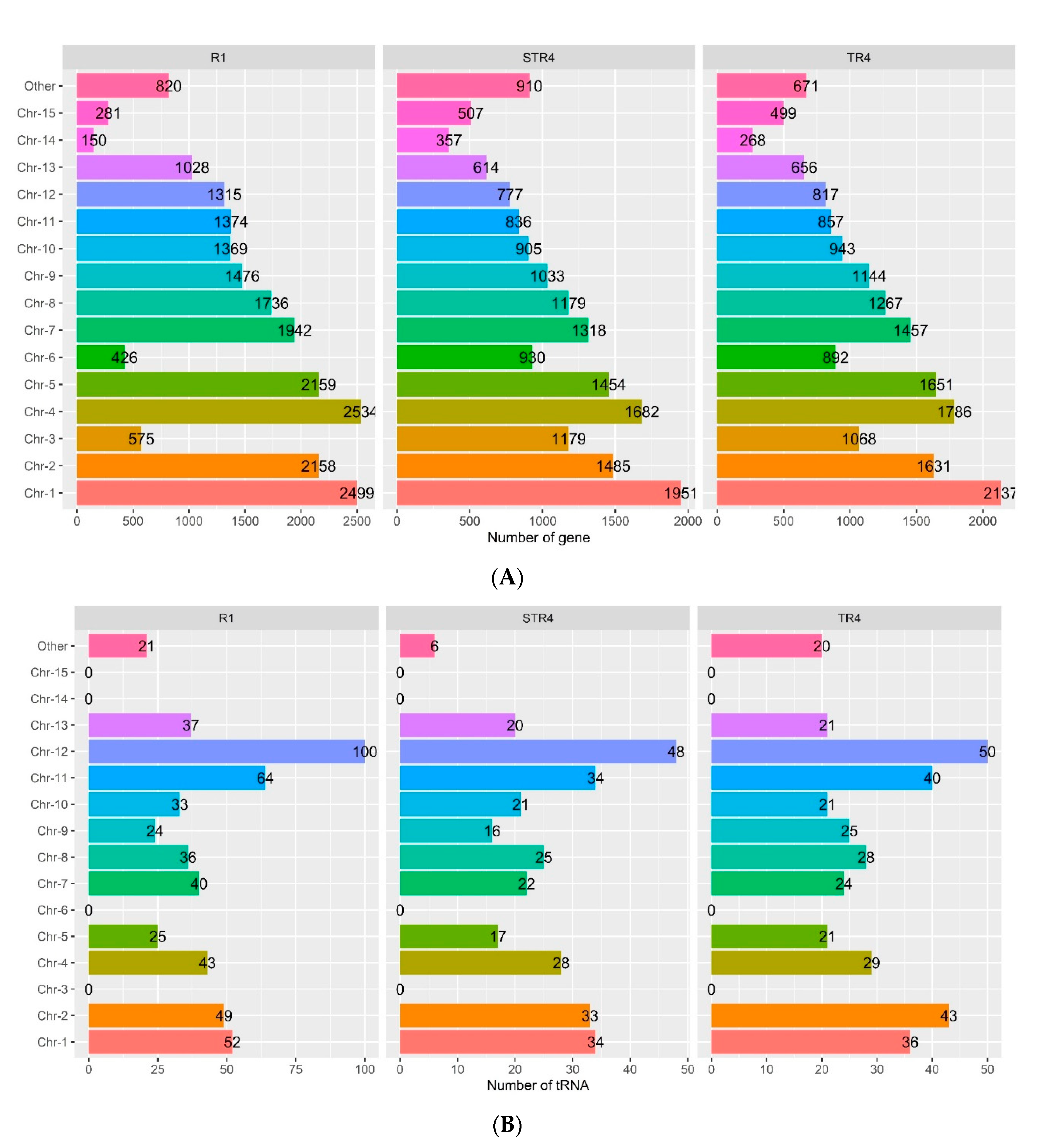
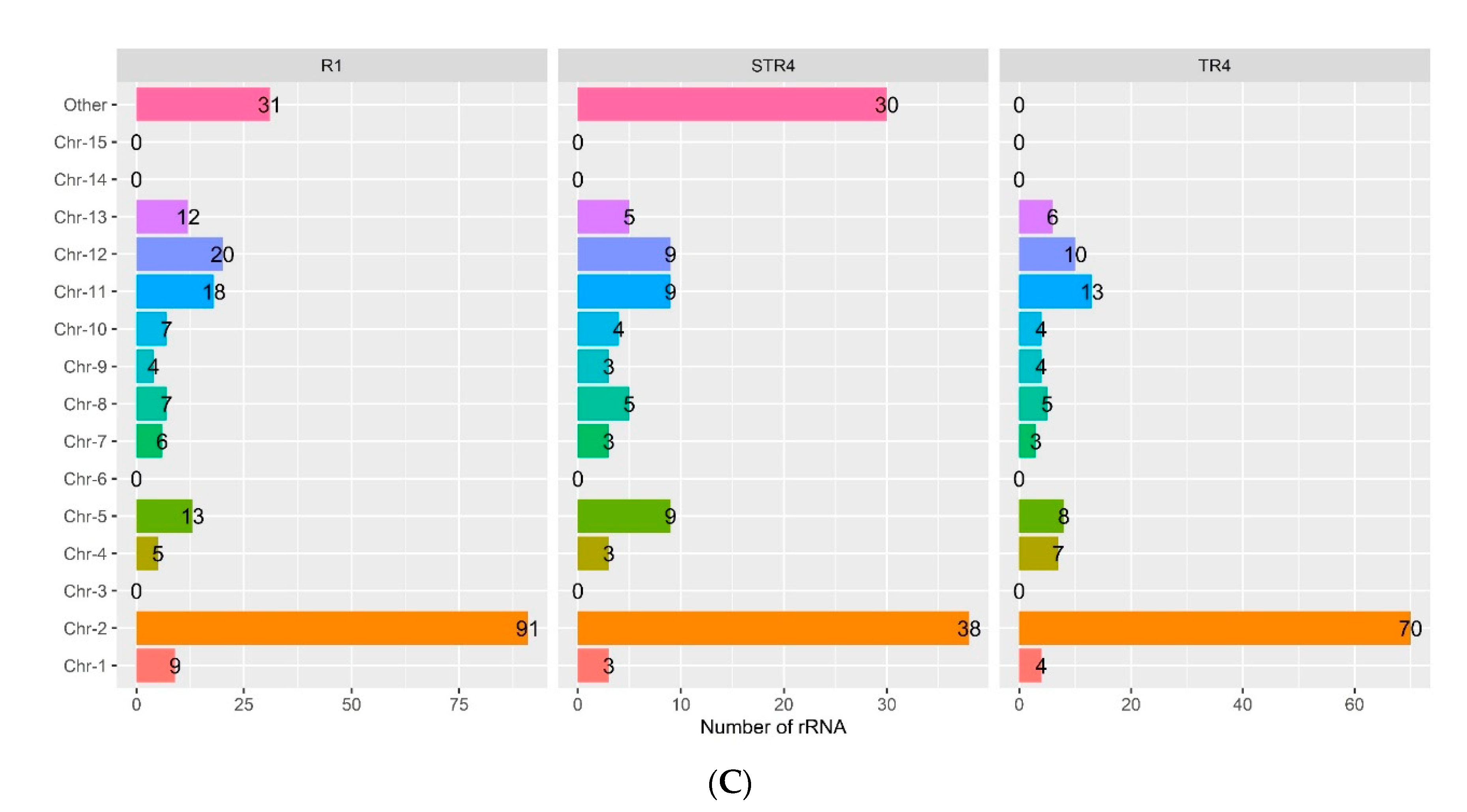
3.3. Gene Content in the Foc Genomes
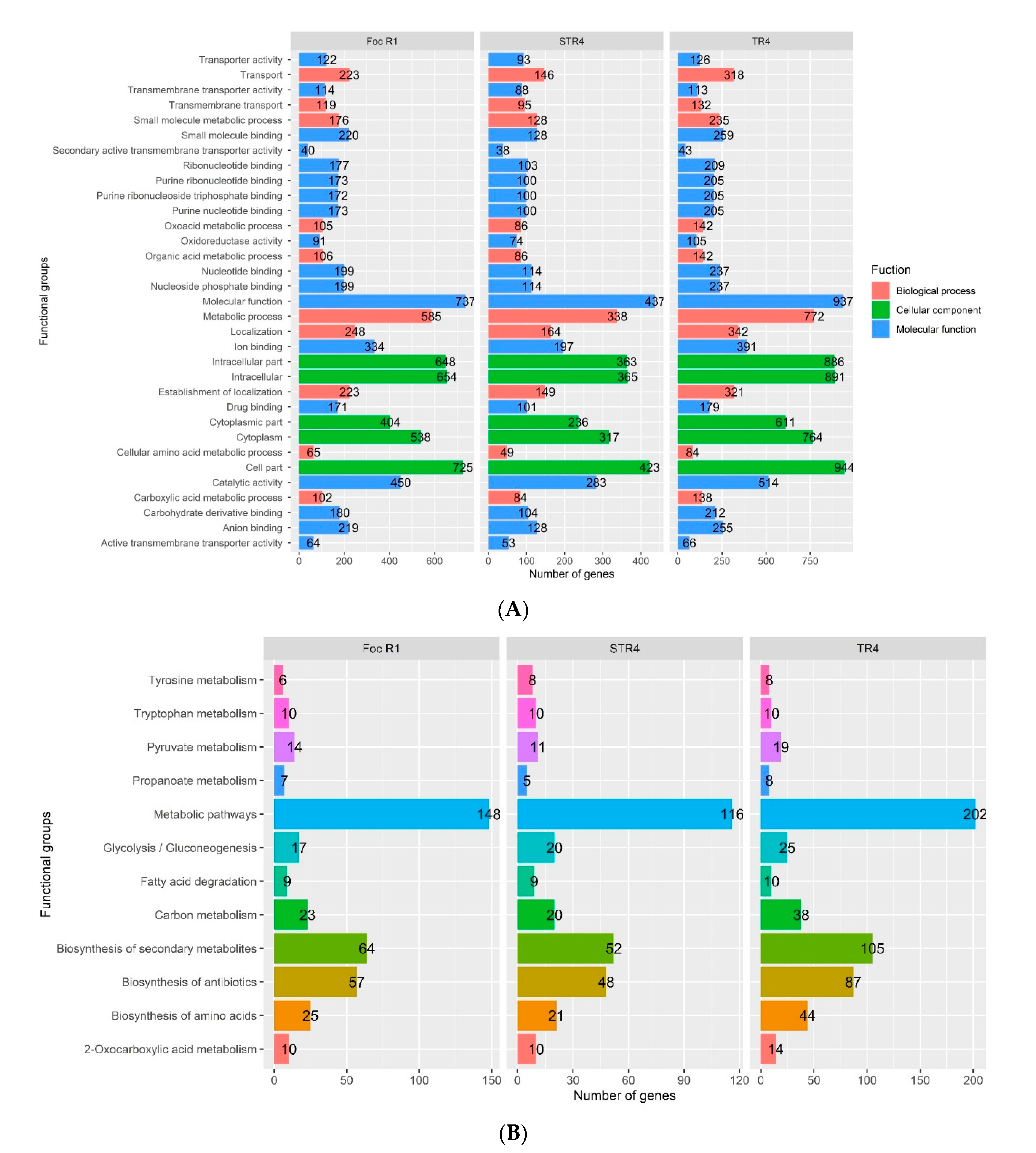
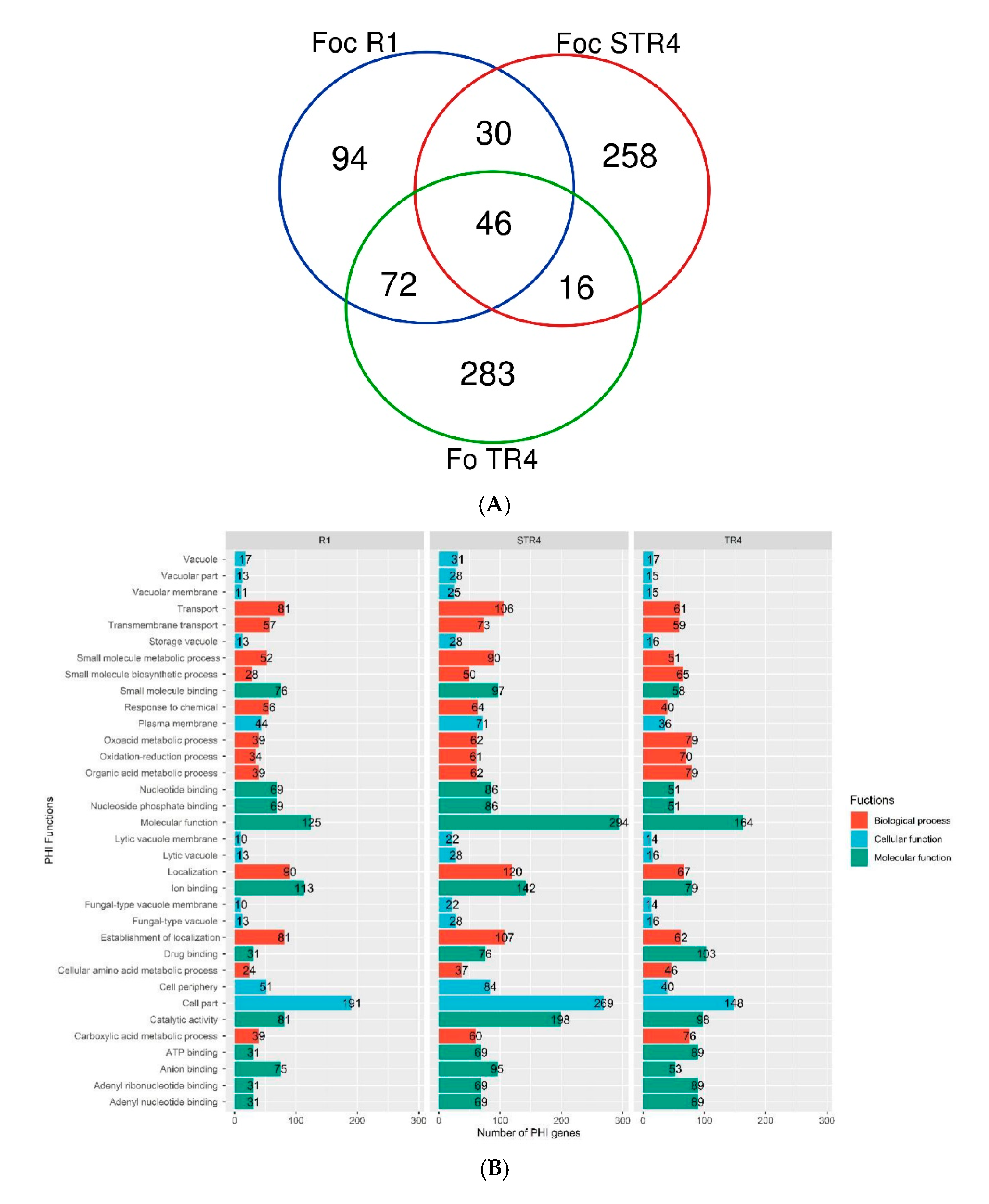
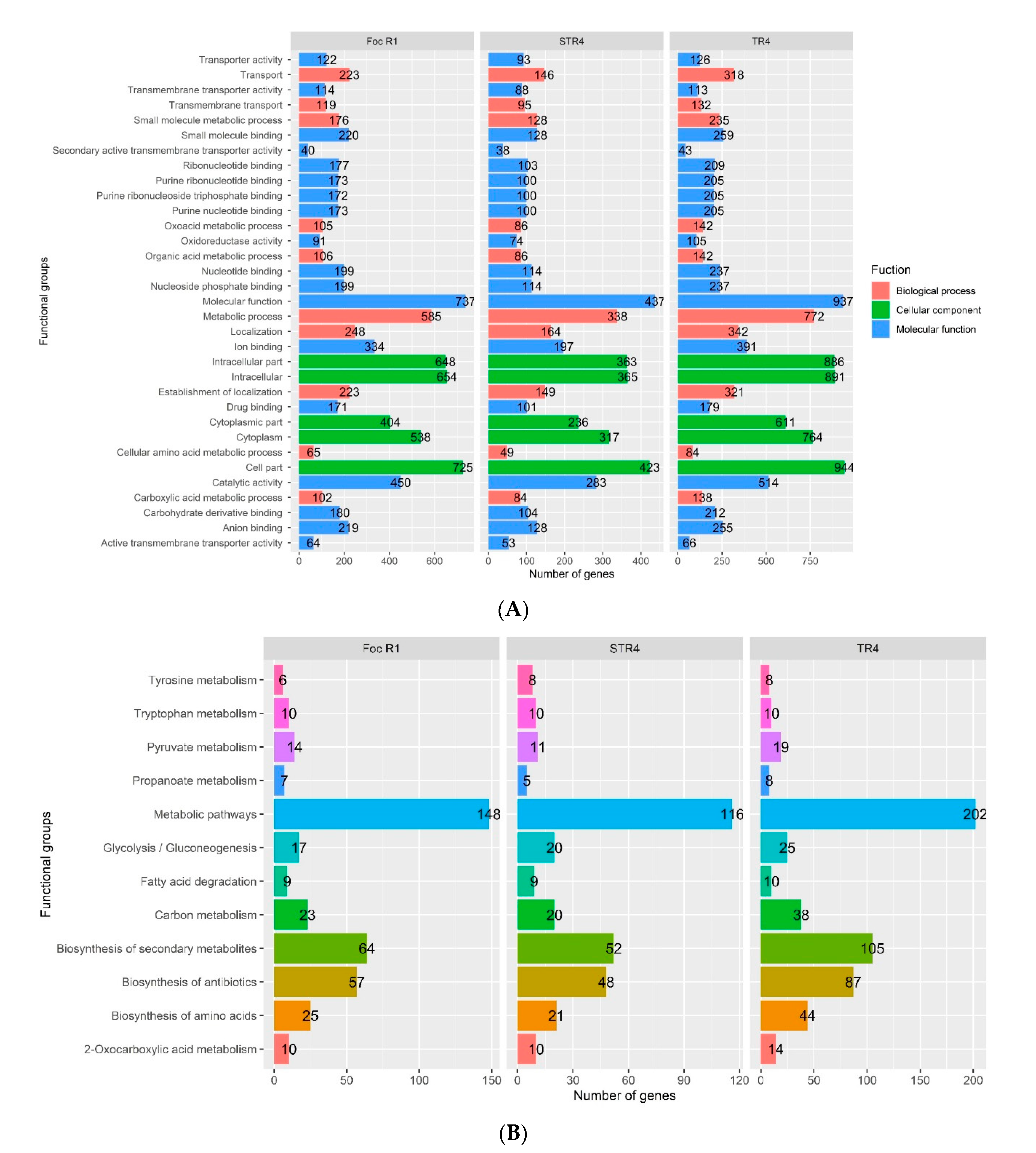
3.4. GO and KEGG in the Foc Genome
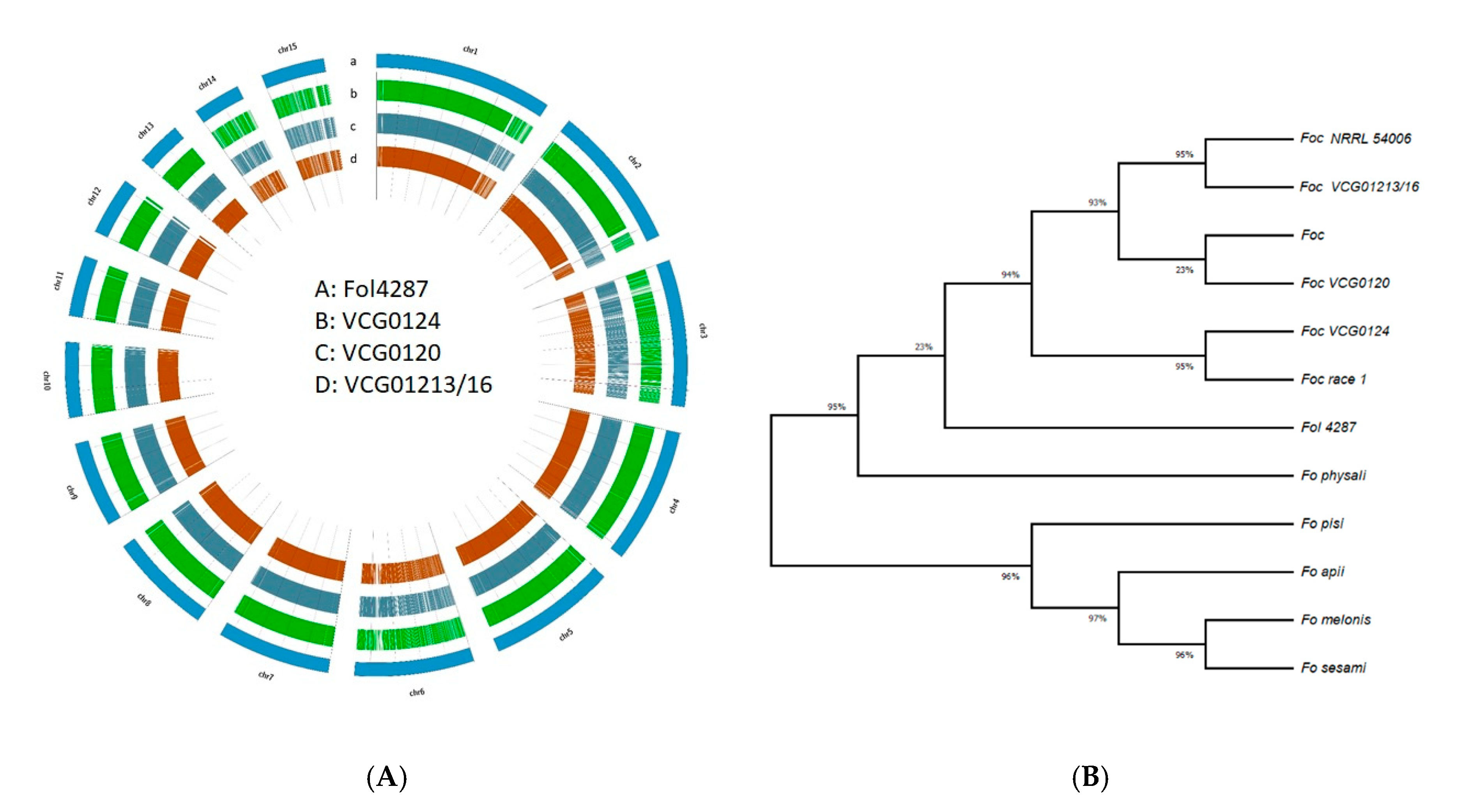
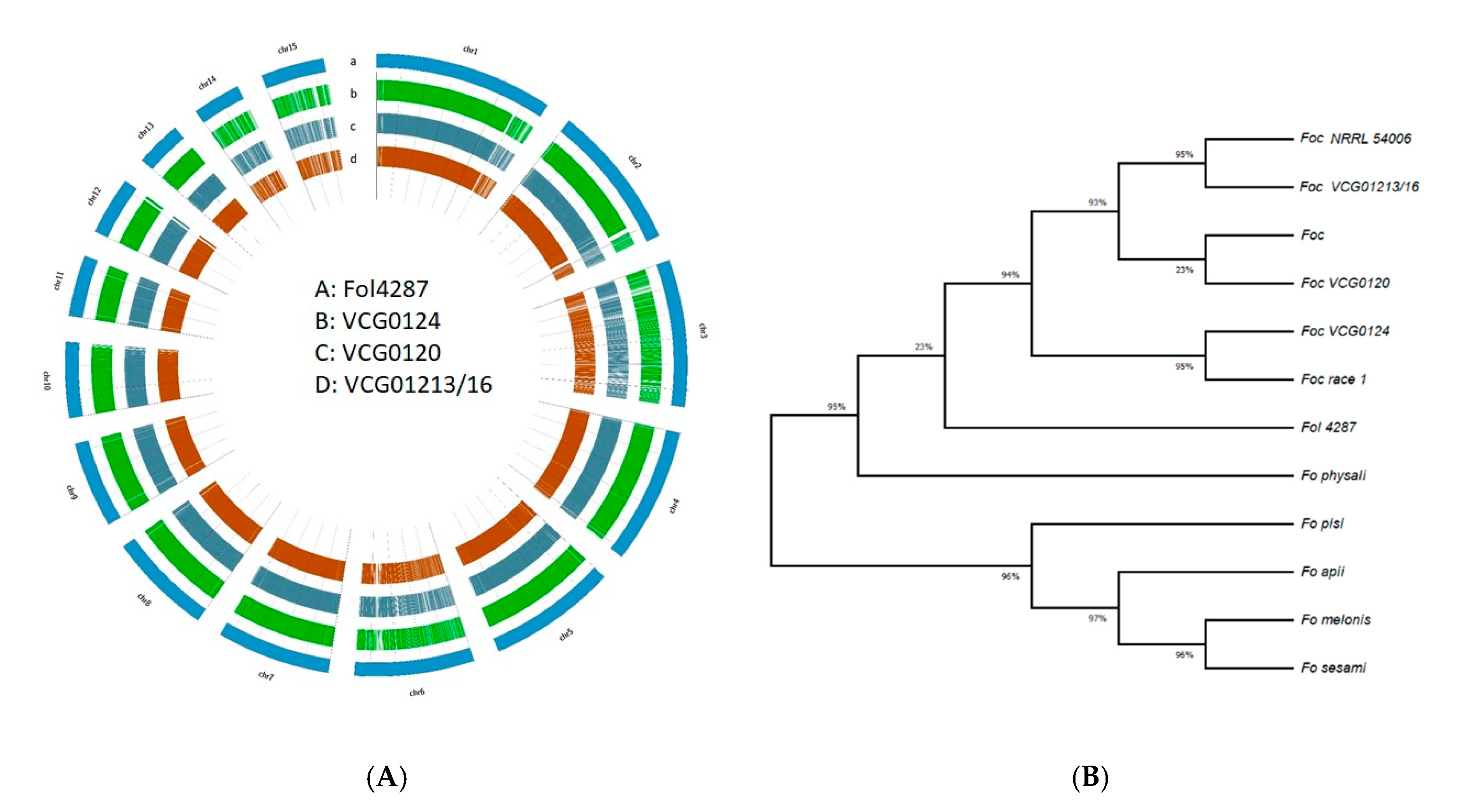
3.5. SNPs, InDels and Phylogenetic Relationship
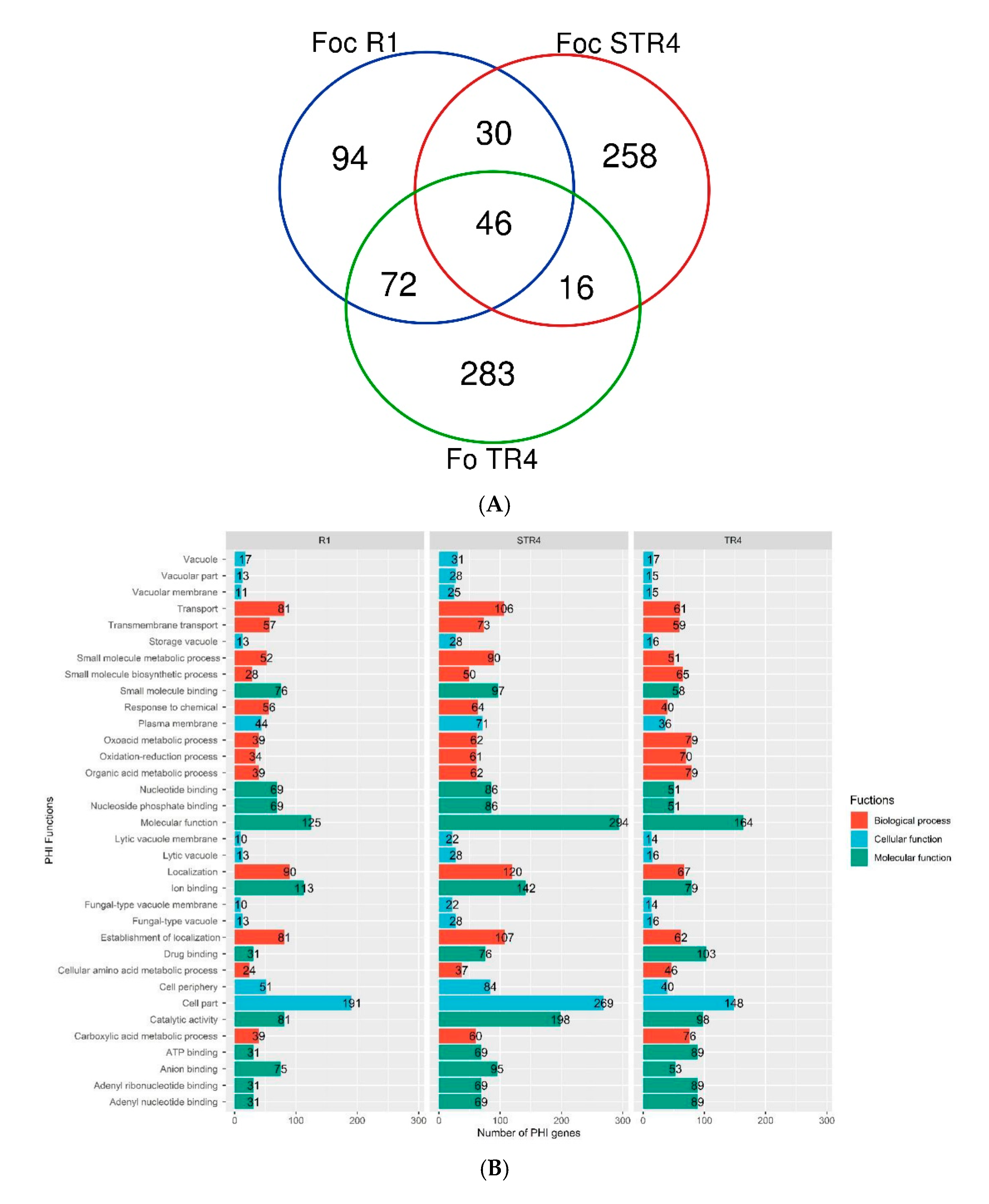
3.6. Virulent Genes and Their Functions in the Genome of Foc
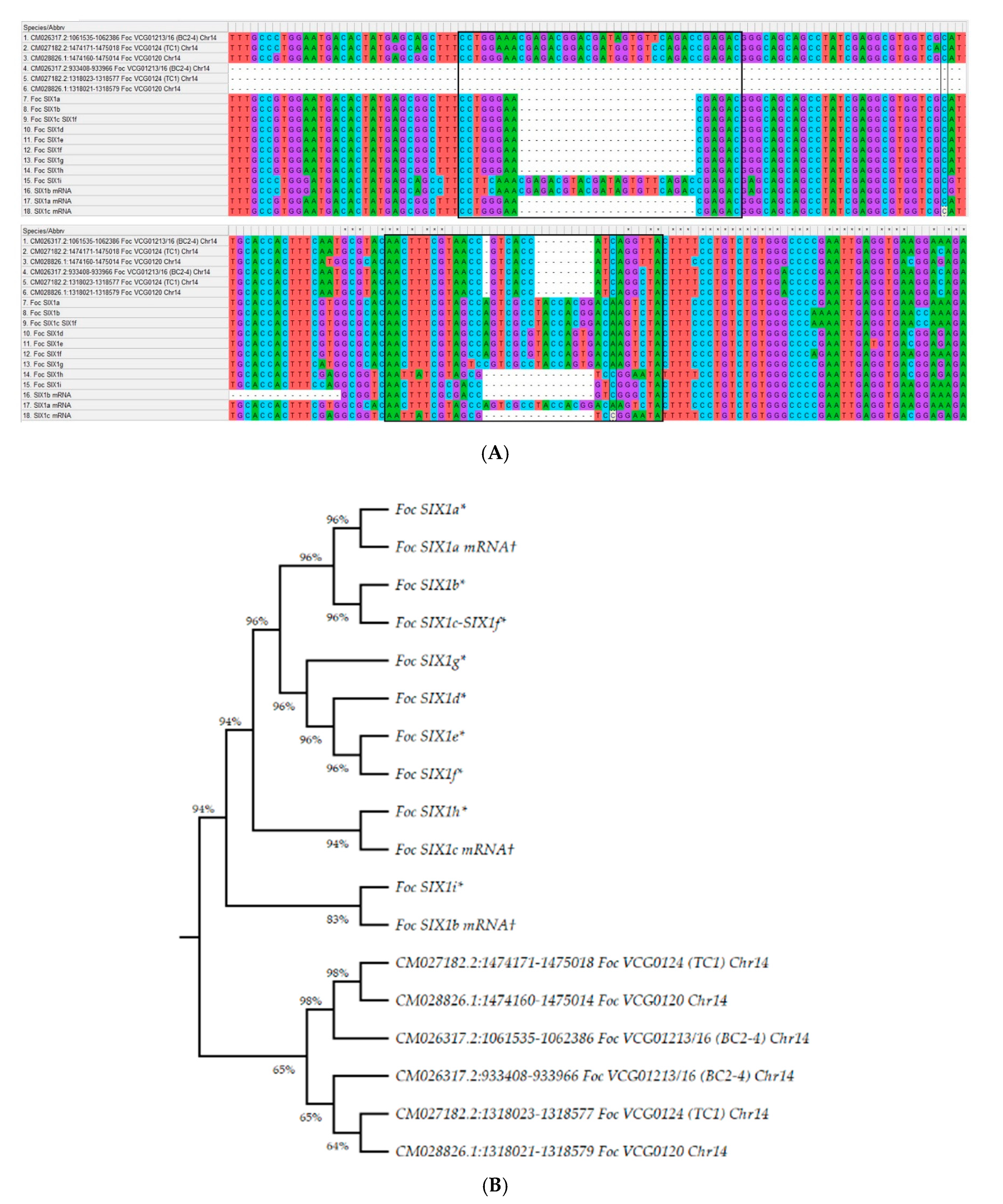
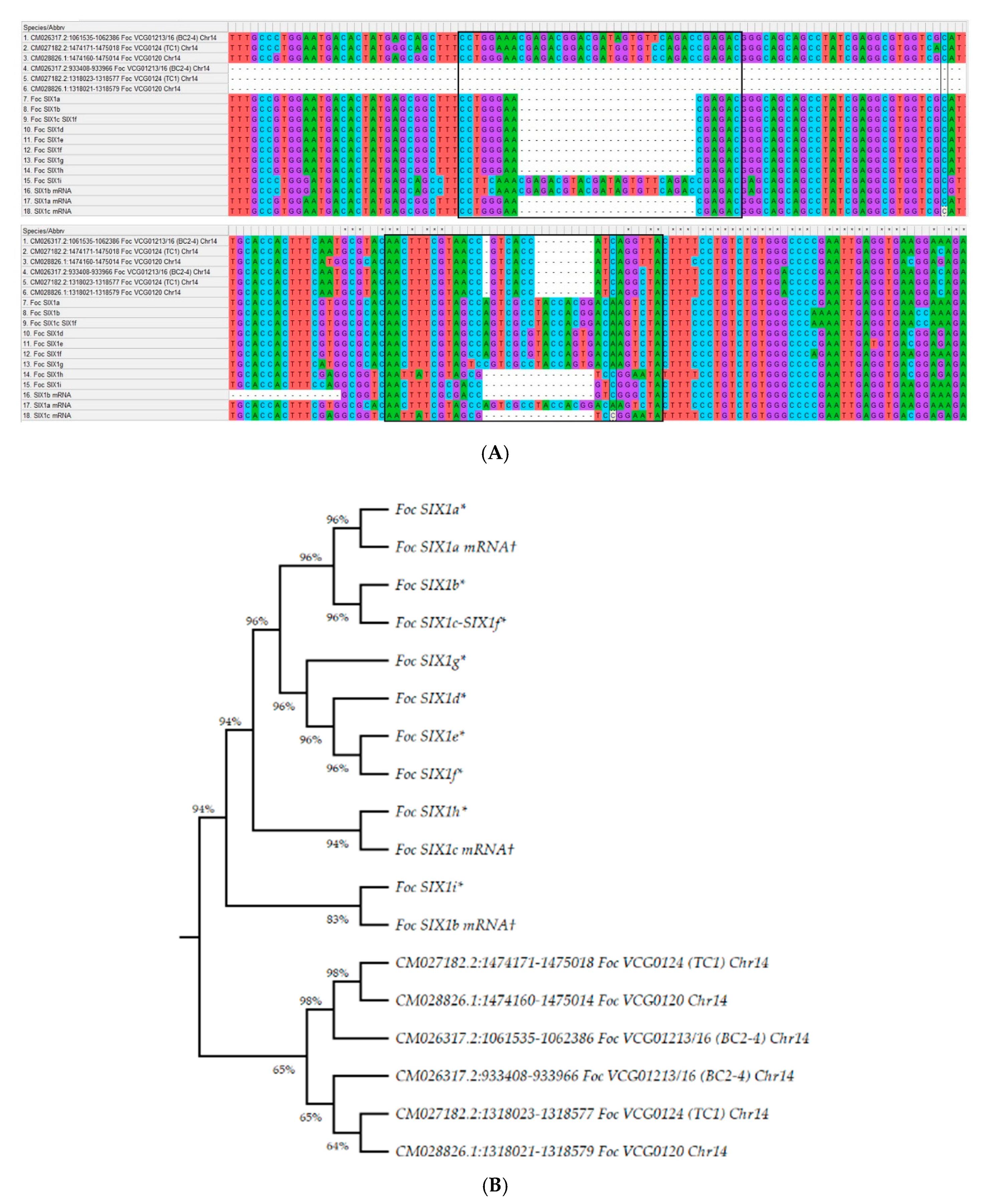
3.7. The Organisation of Secreted in Xylem Protein-Encoding Genes in Foc Genome
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sen, C.; Mishra, H.; Srivastav, P. Modified atmosphere packaging and active packaging of banana (Musa spp.): A review on control of ripening and extension of shelf life. J. Stored Prod. Postharvest Res. 2012, 3, 122–132. [Google Scholar] [CrossRef]
- Ploetz, R.C. Fusarium wilt of banana. Phytopathology 2015, 105, 1512–1521. [Google Scholar] [CrossRef] [Green Version]
- Thangavelu, R.; Loganathan, M.; Artheem, R.; Prabakaran, M.; Uma, S. Fusarium wilt: A threat to banana cultivation and its management. Cab Rev. 2020, 15, 24. [Google Scholar] [CrossRef]
- Thangavelu, R.; Muthukathan, G.; Pushpakanth, P.; Murugan, L.; Edwin, R.E.; Marimuthu, N.; Uma, S. First report of Fusarium oxysporum f.sp. cubense VCG 0125 and VCG 01220 of Race 1 infecting Cavendish bananas (Musa sp. AAA) in India. Plant Dis. 2020, 105. [Google Scholar] [CrossRef]
- Stover, R.H. Fusarial Wilt (Panama Disease) of Bananas and Other Musa Species; Commonwealth Mycological Institute: London, UK, 1962. [Google Scholar]
- ICAR National Research Centre for Banana. Anonymous. Annual Report 2018–‘19; ICAR-National Research Centre for Banana: Tiruchirappalli, India, 2019; Available online: http://nrcb.res.in (accessed on 11 May 2020).
- Mostert, D.; Molina, A.B.; Daniells, J.; Fourie, G.; Hermanto, C.; Chao, C.P.; Fabregar, E.; Sinohin, V.G.; Masdek, N.; Thangavelu, R.; et al. The distribution and host range of the banana Fusarium wilt fungus, Fusarium oxysporum F. Sp. Cubense, in Asia. PLoS ONE 2017, 12, e0181630. [Google Scholar] [CrossRef] [Green Version]
- Thangavelu, R.; Mostert, D.; Gopi, M.; Devi, P.G.; Padmanaban, B.; Molina, A.B.; Viljoen, A. Cavendish banana in India First detection of Fusarium oxysporum f sp. cubense tropical race 4 (TR4) on Cavendish banana in India. Eur. J. Plant Pathol. 2019, 154, 777–786. [Google Scholar] [CrossRef]
- Ordoñez, N.; García-Bastidas, F.; Laghari, H.B.; Akkary, M.Y.; Harfouche, E.N.; al Awar, B.N.; Kema, G.H.J. First Report of Fusarium oxysporum f. sp. cubense Tropical Race 4 Causing Panama Disease in Cavendish Bananas in Pakistan and Lebanon. Plant Dis. 2016, 100, 209. [Google Scholar] [CrossRef]
- Maryani, N.; Lombard, L.; Poerba, Y.S.; Subandiyah, S.; Crous, P.W.; Kema, G.H.J. Phylogeny and genetic diversity of the banana Fusarium wilt pathogen Fusarium oxysporum f. sp. cubense in the Indonesian centre of origin. Stud. Mycol. 2019, 92, 155–194. [Google Scholar] [CrossRef] [PubMed]
- Dita, M.A.; Waalwijk, C.; Buddenhagen, I.W.; Souza Jr, M.T.; Kema, G.H.J. A molecular diagnostic for tropical race 4 of the banana fusarium wilt pathogen. Plant Pathol. 2010, 59, 348–357. [Google Scholar] [CrossRef]
- Moore, N.; Hargreaves, P.; Pegg, K.; Irwin, J. Characterisation of Strains of Fusarium oxysporum f.sp. cubense by Production of Volatiles. Aust. J. Bot. 1991, 39, 161. [Google Scholar] [CrossRef]
- Ploetz, R.C. Panama disease: Return of the first banana menace. Int. J. Pest Manag. 1994, 40, 326–336. [Google Scholar] [CrossRef]
- Bentley, S.; Pegg, K.G.; Dale, J.L. Genetic variation among a world-wide collection of isolates of Fusarium oxysporum f. sp. cubense analysed by RAPD-PCR fingerprinting. Mycol. Res. 1995, 99, 1378–1384. [Google Scholar] [CrossRef]
- Koenig, R.L.; Ploetz, R.C.; Kistler, H.C. Fusarium oxysporum f. sp. cubense consists of a small number of divergent and globally distributed clonal lineages. Phytopathology 1997, 87, 915–923. [Google Scholar] [CrossRef] [Green Version]
- Groenewald, S.; Van Den Berg, N.; Marasas, W.F.O.; Viljoen, A. The application of high-throughput AFLP’s in assessing genetic diversity in Fusarium oxysporum f. sp. cubense. Mycol. Res. 2006, 110, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Carvalhais, L.C.; Henderson, J.; Rincon-Florez, V.A.; O’Dwyer, C.; Czislowski, E.; Aitken, E.A.B.; Drenth, A. Molecular Diagnostics of Banana Fusarium Wilt Targeting Secreted-in-Xylem Genes. Front. Plant Sci. 2019, 10, 547. [Google Scholar] [CrossRef]
- O’Donnell, K.; Kistlerr, H.C.; Cigelnik, E.; Ploetz, R.C. Multiple evolutionary origins of the fungus causing panama disease of banana: Concordant evidence from nuclear and mitochondrial gene genealogies. Proc. Natl. Acad. Sci. USA 1998, 95, 2044–2049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, L.; Han, L.; Yang, L.; Zeng, H.; Fan, D.; Zhu, Y.; Feng, Y.; Wang, G.; Peng, C.; Jiang, X.; et al. Genome and Transcriptome Analysis of the Fungal Pathogen Fusarium oxysporum f. sp. cubense Causing Banana Vascular Wilt Disease. PLoS ONE 2014, 9, e95543. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Ji, C.; Li, Y.; Wang, Z. Comparative transcriptomic analysis of race 1 and race 4 of Fusarium oxysporum f. sp. cubense induced with different carbon sources. G3 Genes Genomes Genet. 2017, 7, 2125–2138. [Google Scholar] [CrossRef] [Green Version]
- Yun, Y.; Song, A.; Bao, J.D.; Chen, S.; Lu, S.; Cheng, C.; Zheng, W.; Wang, Z.; Zhang, L. Genome data of Fusarium oxysporum f. sp. cubense race 1 and tropical race 4 isolates using long-read sequencing. Mol. Plant-Microbe Interact. 2019, 32, 1270–1272. [Google Scholar] [CrossRef] [PubMed]
- Poon, N.K.; Teo, C.H.; Othman, R.Y. Differential gene expression analysis of Secreted in Xylem (SIX) genes from Fusarium oxysporum f. sp. cubense tropical race 4 in Musa acuminata cv. Berangan and potential application for early detection of infection. J. Gen. Plant Pathol. 2020, 86, 13–23. [Google Scholar] [CrossRef]
- Asai, S.; Ayukawa, Y.; Gan, P.; Masuda, S.; Komatsu, K.; Shirasu, K.; Arie, T. High-Quality Draft Genome Sequence of Fusarium oxysporum f. sp. cubense Strain 160527, a Causal Agent of Panama Disease. Microbiol. Resour. Announc. 2019, 8, e00654-19. [Google Scholar] [CrossRef] [Green Version]
- Czislowski, E.; Zeil-Rolfe, I.; Aitken, E.A.B. Effector Profiles of Endophytic Fusarium Associated with Asymptomatic Banana (Musa sp.) Hosts. Int. J. Mol. Sci. 2021, 22, 2508. [Google Scholar] [CrossRef] [PubMed]
- Warmington, R.J.; Kay, W.; Jeffries, A.; O’Neill, P.; Farbos, A.; Moore, K.; Bebber, D.P.; Studholme, D.J. High-Quality Draft Genome Sequence of the Causal Agent of the Current Panama Disease Epidemic. Microbiol. Resour. Announc. 2019, 8, e00904-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urbaniak, C.; Massa, G.; Hummerick, M.; Khodadad, C.; Schuerger, A.; Venkateswaran, K. Draft genome sequences of two Fusarium oxysporum isolates cultured from infected Zinnia hybrida plants grown on the International Space Station. Genome Announc. 2018, 6, e00326-18. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Shao, J.; Wang, Y.; Li, W.; Guo, D.; Yan, B.; Xia, Y.; Peng, M. Analysis of banana transcriptome and global gene expression profiles in banana roots in response to infection by race 1 and tropical race 4 of Fusarium oxysporum f. sp. cubense. BMC Genom. 2013, 14, 851. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, S.; Taylor, A.; Jackson, A.C.; Armitage, A.D.; Bates, H.J.; Mead, A.; Harrison, R.J.; Clarkson, J.P. Identification and Expression of Secreted in Xylem Pathogenicity Genes in Fusarium oxysporum f. sp. pisi. Front. Microbiol. 2021, 12, 788. [Google Scholar] [CrossRef]
- Thangavelu, R.; Mustaffa, M.M. First Report on the Occurrence of a Virulent Strain of Fusarium Wilt Pathogen (Race-1) Infecting Cavendish (AAA) Group of Bananas in India. Plant Dis. 2010, 94, 1379. [Google Scholar] [CrossRef]
- Niemeyer, J.L.; Andrade, M. de A basic procedure for single spore isolation of Fusarium verticillioides and Fusarium subglutinans. Glob. J. Food Agric. Sci. 2016, 3, 145–148. [Google Scholar]
- Möller, E.M.; Bahnweg, G.; Sandermann, H.; Geiger, H.H. A simple and efficient protocol for isolation of high molecular weight DNA from filamentous fungi, fruit bodies, and infected plant tissues. Nucleic Acids Res. 1992, 20, 6115–6116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimin, A.V.; Marçais, G.; Puiu, D.; Roberts, M.; Salzberg, S.L.; Yorke, J.A. The MaSuRCA genome assembler. Bioinformatics 2013, 29, 2669–2677. [Google Scholar] [CrossRef] [Green Version]
- Garrison, E.; Marth, G. Haplotype-based variant detection from short-read sequencing. arXiv 2012, arXiv:1207.3907. [Google Scholar]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [Green Version]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Stanke, M.; Keller, O.; Gunduz, I.; Hayes, A.; Waack, S.; Morgenstern, B. Augustus: Ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006, 34, W435–W439. [Google Scholar] [CrossRef] [Green Version]
- Tatusov, R.L.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Kiryutin, B.; Koonin, E.V.; Krylov, D.M.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; et al. The COG database: An updated version includes eukaryotes. BMC Bioinform. 2003, 4, 41. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, 277–280. [Google Scholar] [CrossRef] [Green Version]
- Winnenburg, R. PHI-base: A new database for pathogen host interactions. Nucleic Acids Res. 2006, 34, D459–D464. [Google Scholar] [CrossRef]
- Winnenburg, R.; Urban, M.; Beacham, A.; Baldwin, T.K.; Holland, S.; Lindeberg, M.; Hansen, H.; Rawlings, C.; Hammond-Kosack, K.E.; Kohler, J. PHI-base update: Additions to the pathogen host interaction database. Nucleic Acids Res. 2007, 36, D572–D576. [Google Scholar] [CrossRef]
- Schmidt, S.M.; Houterman, P.M.; Schreiver, I.; Ma, L.; Amyotte, S.; Chellappan, B.; Boeren, S.; Takken, F.L.W.; Rep, M. MITEs in the promoters of effector genes allow prediction of novel virulence genes in Fusarium oxysporum. BMC Genom. 2013, 14, 119. [Google Scholar] [CrossRef] [Green Version]
- Taylor, A.; Vágány, V.; Jackson, A.C.; Harrison, R.J.; Rainoni, A.; Clarkson, J.P. Identification of pathogenicity-related genes in Fusarium oxysporum f. sp. cepae. Mol. Plant Pathol. 2016, 17, 1032–1047. [Google Scholar] [CrossRef] [Green Version]
- Czislowski, E.; Fraser-Smith, S.; Zander, M.; O’Neill, W.T.; Meldrum, R.A.; Tran-Nguyen, L.T.T.; Batley, J.; Aitken, E.A.B. Investigation of the diversity of effector genes in the banana pathogen, Fusarium oxysporum f. sp. cubense, reveals evidence of horizontal gene transfer. Mol. Plant Pathol. 2018, 19, 1155–1171. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.-J.; van der Does, H.C.; Borkovich, K.A.; Coleman, J.J.; Daboussi, M.-J.; Di Pietro, A.; Dufresne, M.; Freitag, M.; Grabherr, M.; Henrissat, B.; et al. Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature 2010, 464, 367–373. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. Busco: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [Green Version]
- Abrusan, G.; Grundmann, N.; DeMester, L.; Makalowski, W. TEclass—A tool for automated classification of unknown eukaryotic transposable elements. Bioinformatics 2009, 25, 1329–1330. [Google Scholar] [CrossRef] [Green Version]
- Thangavelu, R.; Edwinraj, E.; Gopi, M.; Loganathan, M.; Pushpakanth, P.; Marimuthu, N.; Prabaharan, M.; Sharmila, K.; Sasikala, R.; Uma, S. Comparative genome analyses and genome annotation of Foc VCGs infecting Cavendish banana in India. Mendeley Data 2021, V1. [Google Scholar] [CrossRef]
- Thangavelu, R.; Edwinraj, E.; Gopi, M.; Loganathan, M.; Pushpakanth, P.; Marimuthu, N.; Prabaharan, M.; Sharmila, K.; Sasikala, R.; Uma, S. Comparative data on transposable elements in the Fusarium wilt pathogen infecting Cavendish (AAA) bananas in India. Mendeley Data 2021, V1. [Google Scholar] [CrossRef]
- Mat Razali, N.; Cheah, B.H.; Nadarajah, K. Transposable Elements Adaptive Role in Genome Plasticity, Pathogenicity and Evolution in Fungal Phytopathogens. Int. J. Mol. Sci. 2019, 20, 3597. [Google Scholar] [CrossRef] [Green Version]
- Haas, B.J.; Kamoun, S.; Zody, M.C.; Jiang, R.H.Y.; Handsaker, R.E.; Cano, L.M.; Grabherr, M.; Kodira, C.D.; Raffaele, S.; Torto-Alalibo, T.; et al. Genome sequence and analysis of the Irish potato famine pathogen Phytophthora infestans. Nature 2009, 461, 393–398. [Google Scholar] [CrossRef]
- Zhou, E.; Jia, Y.; Singh, P.; Correll, J.C.; Lee, F.N. Instability of the Magnaporthe oryzae avirulence gene AVR-Pita alters virulence. Fungal Genet. Biol. 2007, 44, 1024–1034. [Google Scholar] [CrossRef]
- Yoshida, K.; Saunders, D.G.O.; Mitsuoka, C.; Natsume, S.; Kosugi, S.; Saitoh, H.; Inoue, Y.; Chuma, I.; Tosa, Y.; Cano, L.M.; et al. Host specialization of the blast fungus Magnaporthe oryzae is associated with dynamic gain and loss of genes linked to transposable elements. BMC Genom. 2016, 17, 370. [Google Scholar] [CrossRef] [Green Version]
- Grandaubert, J.; Balesdent, M.-H.; Rouxel, T. Evolutionary and Adaptive Role of Transposable Elements in Fungal Genomes. In Advances in Botanical Research; Elsevier: Oxford, UK, 2014; Volume 70, pp. 79–107. [Google Scholar]
- Liu, X.; Xing, M.; Kong, C.; Fang, Z.; Yang, L.; Zhang, Y.; Wang, Y.; Ling, J.; Yang, Y.; Lv, H. Genetic diversity, virulence, race profiling, and comparative genomic analysis of the fusarium oxysporum f. sp. Conglutinans Strains infecting cabbages in China. Front. Microbiol. 2019, 10, 1373. [Google Scholar] [CrossRef]
- Sharma, M.; Sengupta, A.; Ghosh, R.; Agarwal, G.; Tarafdar, A.; Nagavardhini, A.; Pande, S.; Varshney, R.K. Genome wide transcriptome profiling of Fusarium oxysporum f sp. ciceris conidial germination reveals new insights into infection-related genes. Sci. Rep. 2016, 6, 37353. [Google Scholar] [CrossRef] [PubMed]
- Dassa, E.; Bouige, P. The ABC of ABCs: A phylogenetic and functional classification of ABC systems in living organisms. Res. Microbiol. 2001, 152, 211–229. [Google Scholar] [CrossRef]
- Becher, R.; Weihmann, F.; Deising, H.B.; Wirsel, S.G. Development of a novel multiplex DNA microarray for Fusarium graminearum and analysis of azole fungicide responses. BMC Genom. 2011, 12, 52. [Google Scholar] [CrossRef] [Green Version]
- Glazebrook, J. Contrasting Mechanisms of Defense Against Biotrophic and Necrotrophic Pathogens. Annu. Rev. Phytopathol. 2005, 43, 205–227. [Google Scholar] [CrossRef] [PubMed]
- Hansen, L.H.; Johannesen, E.; Burmølle, M.; Sørensen, A.H.; Sørensen, S.J. Plasmid-encoded multidrug efflux pump conferring resistance to olaquindox in Escherichia coli. Antimicrob. Agents Chemother. 2004, 48, 3332–3337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sørensen, A.H.; Hansen, L.H.; Johannesen, E.; Sørensen, S.J. Conjugative plasmid conferring resistance to olaquindox. Antimicrob. Agents Chemother. 2003, 47, 798–799. [Google Scholar] [CrossRef] [Green Version]
- Dani, P.; Ujaoney, A.K.; Apte, S.K.; Basu, B. Regulation of potassium dependent ATPase (kdp) operon of Deinococcus radiodurans. PLoS ONE 2017, 12, e0188998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bialek, S.; Lavigne, J.; Chevalier, J. Membrane efflux and influx modulate both multidrug resistance and virulence of Klebsiella pneumoniae in a Caenorhabditis elegans model. Antimicrob. Agents Chemother. 2010, 54, 4373–4378. [Google Scholar] [CrossRef] [Green Version]
- Bialek-Davenet, S.; Lavigne, J.P.; Guyot, K.; Mayer, N.; Tournebize, R.; Brisse, S.; Leflon-Guibout, V.; Nicolas-Chanoine, M.H. Differential contribution of AcrAB and OqxAB efflux pumps to multidrug resistance and virulence in Klebsiella pneumoniae. J. Antimicrob. Chemother. 2015, 70, 81–88. [Google Scholar] [CrossRef] [Green Version]
- Peirs, P.; Lefèvre, P.; Boarbi, S.; Wang, X.-M.; Denis, O.; Braibant, M.; Pethe, K.; Locht, C.; Huygen, K.; Content, J. Mycobacterium tuberculosis with Disruption in Genes Encoding the Phosphate Binding Proteins PstS1 and PstS2 Is Deficient in Phosphate Uptake and Demonstrates Reduced In Vivo Virulence. Infect. Immun. 2005, 73, 1898–1902. [Google Scholar] [CrossRef] [Green Version]
- Soni, D.K.; Dubey, S.K.; Bhatnagar, R. ATP-binding cassette (ABC) import systems of Mycobacterium tuberculosis: Target for drug and vaccine development. Emerg. Microbes Infect. 2020, 9, 207–220. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, M.-P.; Park, J.; Cho, M.-H.; Lee, S.-W. Role of DetR in defence is critical for virulence of X anthomonas oryzae pv. oryzae. Mol. Plant. Pathol. 2016, 17, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Widinugraheni, S.; Niño-Sánchez, J.; Van Der Does, H.C.; Van Dam, P.; García-Bastidas, F.A.; Subandiyah, S.; Meijer, H.J.G.; Kistler, H.C.; Kema, G.H.J.; Rep, M. A SIX1 homolog in Fusarium oxysporum f.sp. Cubense tropical race 4 contributes to virulence towards Cavendish banana. PLoS ONE 2018, 13, e0205896. [Google Scholar] [CrossRef]
- Li, E.; Wang, G.; Xiao, J.; Ling, J.; Yang, Y.; Xie, B. A SIX1 homolog in Fusarium oxysporum f. sp. conglutinans is required for full virulence on cabbage. PLoS ONE 2016, 11, e0152273. [Google Scholar] [CrossRef]
- Gawehns, F.; Houterman, P.M.; Ichou, F.A.; Michielse, C.B.; Hijdra, M.; Cornelissen, B.J.C.; Rep, M.; Takken, F.L.W. The fusarium oxysporum effector six6 contributes to virulence and suppresses i-2-mediated cell death. Mol. Plant Microbe Interact. 2014, 27, 336–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraser-Smith, S.; Czislowski, E.; Meldrum, R.; Zander, M.; O’Neill, W.; Balali, G.; Aitken, E. Sequence variation in the putative effector gene SIX8 facilitates molecular differentiation of Fusarium oxysporum f. sp. cubense. Plant. Pathol. 2014, 63, 1044–1052. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Fol 4287 | Foc R1 (VCG0124) | Foc STR4 (VCG0120) | Foc TR4 (VCG01213/16) |
|---|---|---|---|---|
| Genome size (bp) | 61,386,934 | 61,471,473 | 61,380,681 | 63,220,715 |
| No. of scaffolds (Count) | 114 | 88 | 85 | 88 |
| No. of contig (Count) | 1362 | 1371 | 1362 | 6560 |
| Scaffold N50 (Mb) | 1,976,106 | 4,781,098 | 4,589,962 | 4,589,937 |
| Maximum contig length | 6,854,980 | 8,732,082 | 6,853,100 | 7,450,211 |
| Minimum contig length | 900 | 898 | 888 | 886 |
| Average contig length | 698,542 | 884,718 | 722,130 | 718,421 |
| Median contig length | 15,960 | 17,299 | 14,354 | 15,960 |
| GC content | 48.4 | 48.5 | 48.4 | 48.5 |
| Protein count | 27,347 | 22,151 | 18,946 | 19,651 |
| Total genes (Augustus) | 20,925 | 21,842 | 17,118 | 17,745 |
| BUSCO (%) | 96.4 | 98.4 | 96.9 | 98.9 |
| Secretome Genes | 1847 | 1949 | 1870 | 2330 |
| tRNA | 320 | 524 | 304 | 358 |
| rRNA | 125 | 223 | 121 | 134 |
| ORF | 892,698 | 1,149,990 | 891,736 | 926,420 |
| Repeats (bp) | 10,770,810 ‡ | 10,755,517 | 10,901,072 | 8,844,756 |
| Total transposable elements (bp) | 2,446,574 ‡ | 346,254 | 342,987 | 273,767 |
| Retroelements | 433,505 ‡ | 87,052 | 85,124 | 56,365 |
| DNA transposons | 506,130 ‡ | 2527 | 2591 | 2391 |
| Total interspersed repeats | 342,368 ‡ | 89,579 | 87,787 | 58,756 |
| Simple repeats | 115,058 | 110,011 | 99,987 | |
| Low complexity region | 141,331 | 144,912 | 114,926 |
| Accession | SIX1 | SIX2 | SIX3 | SIX4 | SIX5 | SIX6 | SIX7 | SIX8 | SIX9 | SIX10 | SIX11 | SIX12 | SIX13 | SIX14 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Fol 4287 | + | + | + | − | + | + | − | + | + | + | − | − | + | − |
| Foc VCG0124 (TC1 ‡) | + | − | − | + | − | + | − | − | − | − | − | − | + | − |
| Foc VCG0124 (TC1 *) | + | ++ | + | ++ | ++ | ++ | + | − | − | + | − | + | + | − |
| Foc VCG0120 STR4 ‡ | + | + | − | − | − | − | + | + | + | − | − | − | + | − |
| Foc VCG0120 STR4 * | + | ++ | + | ++ | ++ | ++ | + | + | ++ | + | − | − | + | − |
| Foc VCG01213/16 (BC2-4 ‡) | + | + | − | − | − | − | − | + | + | − | − | − | + | − |
| Foc VCG01213/16 (BC2-4 *) | + | ++ | + | ++ | ++ | + | + | + | ++ | + | − | ++ | + | − |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raman, T.; Edwin Raj, E.; Muthukathan, G.; Loganathan, M.; Periyasamy, P.; Natesh, M.; Manivasakan, P.; Kotteeswaran, S.; Rajendran, S.; Subbaraya, U. Comparative Whole-Genome Sequence Analyses of Fusarium Wilt Pathogen (Foc R1, STR4 and TR4) Infecting Cavendish (AAA) Bananas in India, with a Special Emphasis on Pathogenicity Mechanisms. J. Fungi 2021, 7, 717. https://doi.org/10.3390/jof7090717
Raman T, Edwin Raj E, Muthukathan G, Loganathan M, Periyasamy P, Natesh M, Manivasakan P, Kotteeswaran S, Rajendran S, Subbaraya U. Comparative Whole-Genome Sequence Analyses of Fusarium Wilt Pathogen (Foc R1, STR4 and TR4) Infecting Cavendish (AAA) Bananas in India, with a Special Emphasis on Pathogenicity Mechanisms. Journal of Fungi. 2021; 7(9):717. https://doi.org/10.3390/jof7090717
Chicago/Turabian StyleRaman, Thangavelu, Esack Edwin Raj, Gopi Muthukathan, Murugan Loganathan, Pushpakanth Periyasamy, Marimuthu Natesh, Prabaharan Manivasakan, Sharmila Kotteeswaran, Sasikala Rajendran, and Uma Subbaraya. 2021. "Comparative Whole-Genome Sequence Analyses of Fusarium Wilt Pathogen (Foc R1, STR4 and TR4) Infecting Cavendish (AAA) Bananas in India, with a Special Emphasis on Pathogenicity Mechanisms" Journal of Fungi 7, no. 9: 717. https://doi.org/10.3390/jof7090717
APA StyleRaman, T., Edwin Raj, E., Muthukathan, G., Loganathan, M., Periyasamy, P., Natesh, M., Manivasakan, P., Kotteeswaran, S., Rajendran, S., & Subbaraya, U. (2021). Comparative Whole-Genome Sequence Analyses of Fusarium Wilt Pathogen (Foc R1, STR4 and TR4) Infecting Cavendish (AAA) Bananas in India, with a Special Emphasis on Pathogenicity Mechanisms. Journal of Fungi, 7(9), 717. https://doi.org/10.3390/jof7090717