1. Introduction
Histoplasmosis is a systemic fungal disease caused by the inhalation of conidia of the dimorphic fungus
Histoplasma capsulatum sensu lato, with cases reported worldwide. Histoplasmosis is one of the most frequent fungal infections affecting persons living with HIV/AIDS. Histoplasmosis causes significant morbidity and mortality in HIV-infected individuals, particularly in those countries with limited access to rapid diagnostics or antiretroviral therapies, with a reported mortality up to 40% [
1,
2,
3,
4]. The initial pulmonary manifestations of histoplasmosis are often misdiagnosed as a bacterial or viral pneumonia or classified as another disease, e.g., tuberculosis, with time and effort spent looking for nonfungal infectious etiologies. Diagnosis has traditionally relied largely on conventional blood cultures, which are positive in only approximately 50% of cases and may take up to 6 weeks, thus delaying diagnosis and initiation of therapy [
1]. A major limitation of the immunological test is that in the presence of an active infection, they are negative in up to 50% of immunosuppressed patients, especially those with AIDS [
4]. Other assays such as enzyme immunoassays (EIA) for antigen detection can be performed in urine and serum samples, and some of them have recently become commercially available. Antigens tend to be concentrated in the urine, making
Histoplasma antigen detection more reliable; however, false-positive results may appear due to cross-reaction with other mycoses such as blastomycosis (or those caused by other dimorphic Onygenales) as they overlap in endemicity [
5]. Given the public health need to provide a reliable, rapid, and affordable diagnosis of histoplasmosis, there is a strong motivation to develop new and rapid diagnostic methods with high sensitivity and specificity, for example, by employing molecular techniques [
6,
7,
8].
Fungal infections can be diagnosed on the basis of morphological, immunological, clinical, and histopathological information. Imaging can also be suggestive of fungal infections, such as X ray or CT scans. These images could show patchy pneumonitis that eventually calcifies and could form hilar lymphadenopathy. Advanced stages of histoplasmosis could develop a mass lesion that resembles a fibroma (histoplasmoma), although it would be difficult to differentiate histoplasmosis from other respiratory mycoses [
1,
4]. Of these procedures, histopathology can provide important diagnostic information in a relatively short period of time, but identification is typically only tentative unless complemented by specialized techniques such as immunofluorescence, or when the etiological agent has distinct unique structures. Gomori methenamine or Grocott staining is useful for
H. capsulatum yeast identification, although via these or other methods, it is easily confused with other yeasts such as
Candida spp.,
Pneumocystes jirovecii,
Cryptococcus neoformans, or other infectious agents such as
Leishmania spp. and
Toxoplasma gondii [
4,
9]. The observation in any clinical sample of the small intracellular yeast is suggestive of histoplasmosis. However, some small cells such as poorly encapsulated
Cryptococcus spp.,
Candida glabrata,
Penicillium marneffei, Pneumocystis jirovecii, Toxoplasma gondii, and
Leishmania donovani can be confused, leading to misdiagnosis, in particular, when histoplasmosis is caused by
H. duboisii, which produces larger blastoconidia with a thicker cell wall than
H. capsulatum [
1,
5].
DNA-based diagnosis has not yet been established as a routine clinical diagnostic tool for histoplasmosis, but is used in some reference laboratories [
1,
10]. One molecular assay used for the detection of
Histoplasma spp. is a nested PCR assay based on a gene coding for a 100 kDa protein that is considered specific to
H. capsulatum [
11,
12,
13]. A 100 kDa PCR assay has also been applied to environmental detection of the fungus in composted organic fertilizers, soil samples from caves, and bird excreta.
A new line of interest has arisen as a result of the increased reliability of available fungal genome sequences during the last decade. Reference sequences that were utilized when some of the currently available assays were designed and validated have since been updated, sometimes with dramatic quality improvements in sequence and annotation accuracy [
14]. With the recent availability of finished or draft genome assemblies, or even from unassembled Illumina raw sequence reads, new target regions can be identified for developing more accurate molecular diagnostic assays. The regions of interest for assay design should ideally fulfill two conditions: their sequences should be specific to the fungus of interest (i.e., not present as a highly similar sequence in other organisms), and should also be conserved in all strains of the fungus that might be present in clinical contexts.
Properly designed and clinically validated assays should provide the laboratory technician and clinician with a definitive diagnosis of the fungal pathogen via a PCR assay that is easy to implement. Low-income countries can be affected in the diagnosis of fungal pathogens as a result of inadequate infrastructure and high costs of importation, high costs of healthcare, and/or limited budgets of local healthcare facilities/research centers [
15], and the application of molecular assays in clinical settings should, if possible, not be limited to highly specialized reference laboratories. Although molecular biology equipment is costly, a local research center or small clinic may be able to easily acquire a thermal cycler for conventional PCR, or if the budget permits, the equipment needed for real-time PCR.
With possible economic or time constraints in mind, we aimed to design a diagnostic method using either conventional PCR or real-time PCR that does not require sequencing, and we reasoned that an easily deployable and affordable assay would be beneficial for the public health sector. Considering that the development of molecular assay methods for the diagnosis of fungal infections has sometimes been guided by anecdotal reports of promising loci rather than by rational principles, the present work explored a new route. We describe here a molecular diagnostic approach that should be capable of a high level of analytical specificity and sensitivity, and a high expected clinical specificity and sensitivity, in this case, for detecting the endemic genus Histoplasma in tissues where the fungus can be expected to be present. The novel strategy for rationally designing PCR assays should be particularly useful where the target fungus is closely related to distinct fungi, e.g., presenting different clinical manifestations or with different recommended therapies, and, in this sense, the choice of Histoplasma is an especially stringent test of the principle. Indeed, the Ajellomycetaceae family, which includes Histoplasma, also includes the causal agents of paracoccidioidomycosis, blastomycosis, adiaspiromycosis/emmonsiosis, and the emerging disease emergomycosis, fungi that could therefore cross-react if the genomic target region of the assay is not carefully chosen with this possibility in mind. The strategy can be applied where accurate whole genome sequences of multiple strains of the target species and its close relatives are available and can be aligned.
2. Materials and Methods
2.1. Finding of Regions Unique to H. capsulatum
The methodology utilized to find genome regions that are unique to
H. capsulatum was based on bioinformatic strategies explored by our group in the context of the closely related pathogenic fungal genera of the Ajellomycetaceae family (
Supplemental Table S1). We initially used whole genome alignment programs such as NUCmer and PROmer from the MUMMER package [
16] and BLAST [
17], in order to identify regions that are unique to
H. capsulatum, and absent or only poorly conserved in
Paracoccidioides spp. [
18],
Blastomyces dermatitidis,
Emmonsia crescens,
E. parva/B. parvum, as well as in outgroups within the Onygenales order, nonfungal pathogens, and humans. For
H. capsulatum, all publicly available genome sequences, including sequences from diverse strains within species, were included in the comparative genome analyses. The fungal species listed above must be considered for possible cross-reaction because of their phylogenetic proximity to
Histoplasma spp. and/or the likelihood that they might be present in similar clinical settings.
Three approaches were considered in order to search for Histoplasma-specific regions within current assemblies.
In the first approach, contigs were compared. All contigs/scaffolds of the reference assembly of
H. capsulatum were aligned to contigs/scaffolds of the other fungal species, particularly those of the closely related pathogen
Paracoccidioides spp., where we could vouch for high sequence quality [
14], followed by a second-pass verification for analytical sensitivity by aligning all available strain sequences of
H. capsulatum to ensure intraspecies inclusion, and then a final filtering step to ensure that they were not similar to known sequences of other pathogens or to human genomic DNA, via BLAST versus the nonredundant (nr) NCBI database.
The second approach identified genes that were unique to
Histoplasma. Protein-coding genes confirmed or predicted by gene annotations were used to search for genes that are present in
H. capsulatum but not in other fungal species; in these searches, we also used orthologous gene clusters obtained using OrthoMCL [
19], and a final step used a BLAST search versus nonredundant databases to verify that the gene or genes selected are not present in any known sequences of other organisms.
In the third approach, chromosomal segments of a specified length were sought that were unique to Histoplasma. We used a pairwise alignment algorithm based on a sliding window BLAST (with window sizes 500 and 250 bp) in order to search for sequences in H. capsulatum assemblies that are conserved within and only within H. capsulatum strains, which generates a cluster of genomic regions having a specified similarity range.
For all three strategies, H. capsulatum sequence segments that meet the criteria of no similarity in the non-Histoplasma assemblies, and that also meet the criteria of being conserved in the strains of H. capsulatum, are then considered to be potential candidates for primer design.
2.2. Strains
Genomic DNA from fungal strain cultures listed in
Table 1 were obtained from several fungal pathogen DNA collections maintained at the Corporación para Investigaciones Biológicas (CIB, Medellín, Colombia) or the Centers for Disease Control and Prevention (CDC, Atlanta, GA). Genomic DNA for microbial strains used in analytical specificity tests were also obtained from these collections and are listed in
Table 1. The relative concentrations of the genomic DNA were determined with a NanoDrop ND1000 apparatus (Thermo Scientific, Wilmington, NC, USA).
2.3. Obtaining DNA from Clinical Samples
Formalin-fixed paraffin-embedded (FFPE) skin tissues from two patients from Clínica CES, Medellin, Colombia and previously diagnosed with disseminated histoplasmosis were analyzed. Samples were identified as 173196 and 5177. Both samples were recovered from skin biopsies and both patients had been previously classified as being HIV-positive and with AIDS. DNA extraction and purification were performed with the QIAamp DNA FFPE tissue kit (Qiagen; Valencia, CA, USA), with the following minor modifications of the manufacturer’s protocol: (i) DNA was purified from ten sections per sample, (ii) the samples were incubated with 20 μL of Proteinase K (20 mg/mL, Qiagen) overnight (almost 17 h) at 37 °C instead of 1 h at 65 °C, (iii) the DNA purification was performed with phenol:chloroform:isoamyl alcohol [
20] and precipitation with sodium acetate and isopropanol, and (iv) the pellet was re-suspended in 50 μL of TE 1X.
2.4. Primer Design
The primers were designed using the Histoplasma-unique regions selected via the bioinformatic analysis and were subsequently analyzed using OligoAnalyzer 3.1, adhering to quality control guidelines provided by Integrated DNA Technologies, Inc. (IDT). Some quality control aspects that were checked include: primers must be between 20 and 23 nucleotides in length, the ideal GC content of primers should be between 40 and 60%, melting temperatures (Tm) of primers should be between 42 and 65 °C, primers in a pair should have Tm’s within 2 °C of each other, and secondary structures (i.e., hairpins) within primers and potential dimerization between the primers should be avoided.
The primers designed were subjected to a BLAST search against the GenBank sequence database, in order to avoid cross-homology with other microorganisms or the human genome. The primers that were selected are listed in
Table 2. These primers were designed for use in the conventional and real-time PCR assays.
2.5. Conventional PCR Assay
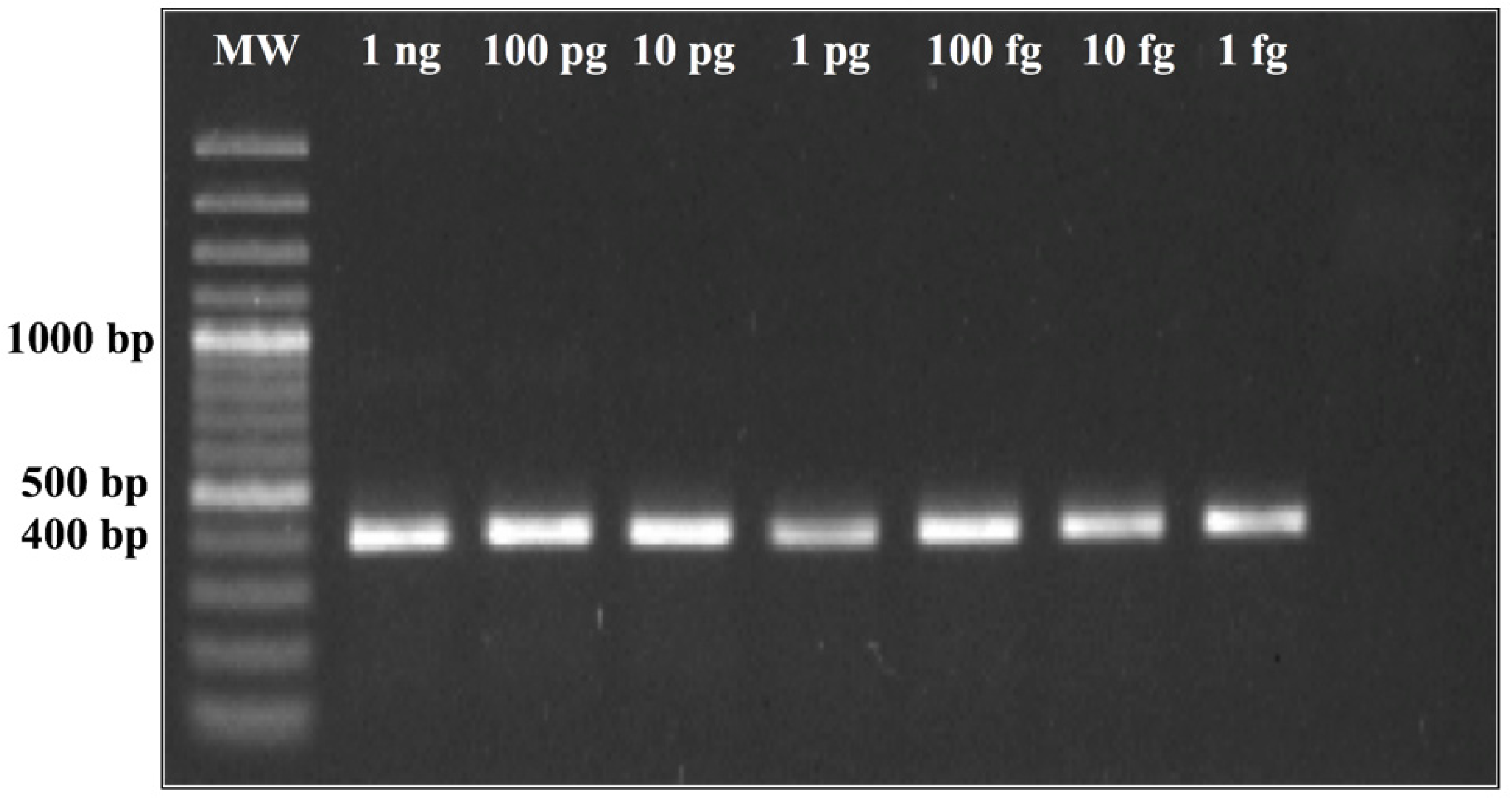
The genomic regions found to be unique for H. capsulatum by computational prediction were experimentally assessed by conventional PCR. Thermocycler conditions were standardized via a temperature gradient of 54 °C–60 °C using a T100 Bio-Rad Thermal Cycler. The amplification products were analyzed on agarose gel and visualized with ethidium bromide under UV light. The PCR conditions selected were as follows: an initial step of 95 °C for 10 min, followed by 40 cycles of 95 °C for 30 s, 60 °C for 30 s, and 72 °C for 1 min.
2.6. Real-Time PCR Assay
Real-time PCR (qPCR) was performed using SYBR Green Real-Time PCR Master Mix, according to the manufacturer’s instructions (Thermo Fisher Scientific Inc.: Waltham, MA, USA) and using conditions standardized via conventional PCR. The CFX96 Real-Time PCR Detection System (Bio-Rad, Headquarters Hercules, CA, USA) was used to carry out the amplification. PCR reactions were performed in a 20 μL final volume containing qPCR master mix 2x. Each experiment was carried out in triplicate. The real-time PCR conditions were as follows: an initial step of 95 °C for 10 min, followed by 45 cycles of 95 °C for 30 s, 60 °C for 30 s, and 70 °C for 1 min, with a melting curve at 60 °C to 95 °C in increments of 0.5 °C each 0.05 s.
2.7. Determining Analytical Specificity of Primers In Vitro
The analytical specificity of the primer sets was evaluated by conventional PCR and corresponding real-time PCR using purified DNA from different isolates of
H. capsulatum, as well as from collections of other related fungal pathogens and
Mycobacterium tuberculosis maintained by the Corporación para Investigaciones Biológicas (CIB) and the Centers for Disease Control and Prevention (CDC, Atlanta, GA, USA). The isolates were tested at a concentration of 1 ng/μL. For analytical sensitivity tests, all strains of
H. capsulatum were tested for amplification with our chosen primers. A total of 62
H. capsulatum isolates were used, including isolates from North America (
H. capsulatum CDC/Thon and
H. capsulatum G217B), Central and South America (
H. capsulatum CIB 1980,
H. capsulatum G184B, and
H. capsulatum CDC 3670/CDC2787), and Africa (
H. duboisii CDC5822/CDC5823). Isolates of other pathogens used for determining analytical specificity are listed in
Table 1.
2.8. Generation of Positive-Control Plasmids
Positive-control plasmids were constructed for
H. capsulatum using the primers designed from species-specific regions or genes, as described in
Table 2. The amplified targets were cloned into the pCR 2.1 vector using the pCR 2.1 TOPO TA cloning kit (Invitrogen Corporation: Carlsbad, CA, USA) according to the manufacturer’s instructions. The plasmid construct was then purified using the PureLink
® Quick Plasmid Miniprep Kit (Thermo Fisher Scientific Inc.: Waltham, MA, USA). The ligation reactions were transformed in TOP10 chemically competent cells. Colonies were selected in LB plates containing 50 μg/mL of kanamycin (two for each transformation [
21]).
2.9. Determining Diagnostic Sensitivity
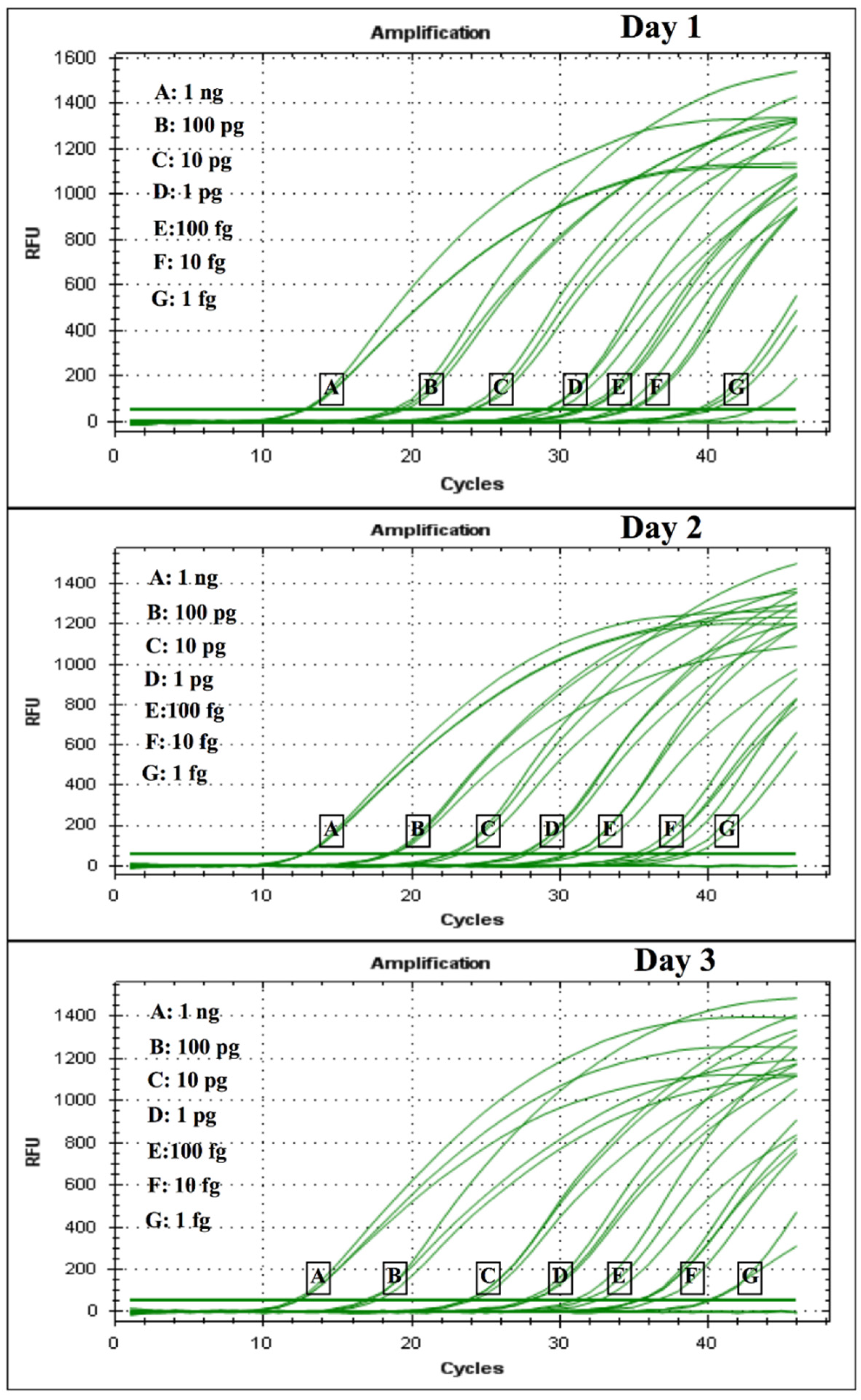
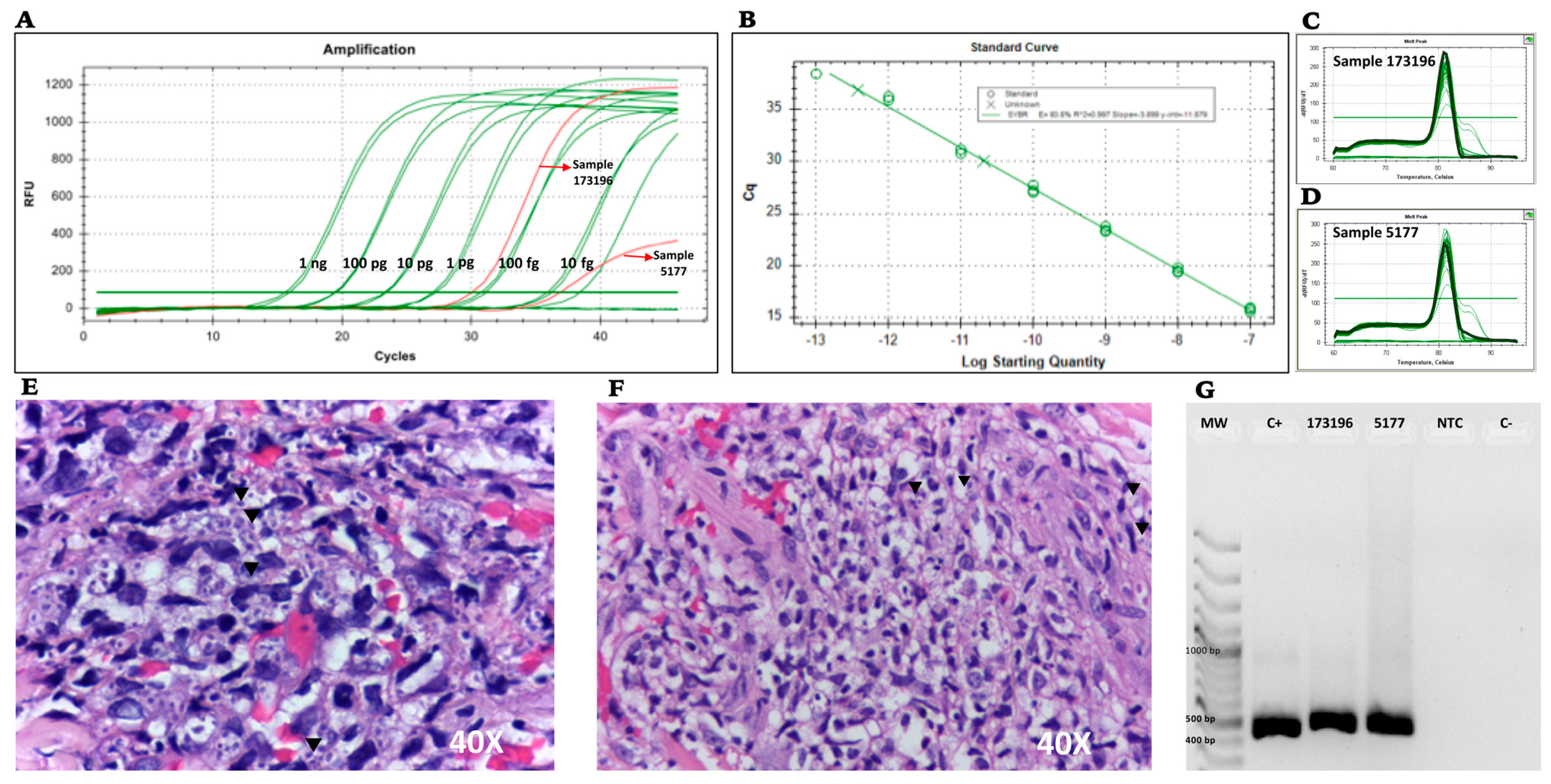
The diagnostic specificity was evaluated by conventional PCR and corresponding real-time PCR via two routes: (i) by testing a dilution series of H. capsulatum control plasmids, where a 10-fold serial dilution of the plasmid was performed in TE buffer (105 copies/μL serially diluted to 10 copies/μL) and was used to construct the standard curve for the limit of detection (LOD), and cycle threshold (CT) values for each dilution series were determined in triplicates in 3 different experiments consisting of 3 different tubes corresponding to the specific dilution of the curve on 3 different days; (ii) by testing DNA obtained from two FFPE skin tissues from two patients previously diagnosed with disseminated histoplasmosis and AIDS, provided by Clínica CES Medellín, Colombia.
4. Discussion
We systematically designed and tested two primer pairs for detecting the fungal pathogen
Histoplasma spp. that could be used for conventional or real-time PCR using similar conditions and the same primer sets. We focused on a general design to work for both PCR technologies, as specialized real-time PCR equipment may not always be accessible for molecular laboratories in resource-limited settings. The genomic regions in which the primer pairs were located correspond to two coding genes, culture filtrate protein 4 (
CFP4) and a putative protein kinase,
PPK. In view of its smaller PCR product size and more homogeneous amplification curves during LOD testing, we identified the PPK primer pair as the more promising choice for assay testing. The PPK primer pair was able to detect in vitro down to 1 fg of the control plasmid using both real-time and conventional PCR designs. The CFP4 primer pair gave good results, validating its analytic reliability and uniqueness to the
Histoplasma genus. Although CFP4 was designed and considered, the large amplicon size did present some difficulties in consistent amplification, in the efficiency of real-time PCR curves and the linearity of the standard curves. In the practice, shorter amplicons with SYBR
® Green can record lower CTs than longer ones [
23,
24]. We observed in the CFP4 qPCR, which has a larger amplicon size (800 bp), higher CT values, low fluorescence intensity, and a very low efficiency (<50%). However, in the conventional PCR, the CFP4 amplicon showed more efficient amplifications in all strains tested. After these initial experiments, we decided to continue with the PPK primer design that, at 400 base pairs, did not show these difficulties during the PCR runs.
We focused on the idea of creating primer sets that should work both for conventional and real-time PCR, and that do not involve a nested PCR design. Nested PCR requires two amplification steps, which can increase the complexity of the assay and the laboratory processing times and costs, as well as the probability of contamination. Assays using primer pairs obtained via our screening methods should not need a secondary amplification step, and in the designs we tested, the limits of detection were low enough to be comparable to a previous nested PCR assay of Bialek et al. in 2002 [
11] targeting a region of a gene for a 100 kDa protein.
More recently, Lopez et al. [
12] implemented real-time PCR assays for the detection of
H. capsulatum, targeting Hc 100 kDa, H, and M antigens [
11,
12,
13].
Our assay with PPK primers showed positive detections in 2 of 2 FFPE biopsies from patients with confirmed histoplasmosis. This finding suggests that the implementation of the PPK primer pair is likely to have good specificity for diagnosing histoplasmosis also in routine clinical settings.
In an environmental context, Gómez et al. [
13] applied a protocol based on the amplification of the Hc100 nested PCR to search for
H. capsulatum in composted organic fertilizers, soil samples from caves, and bird excreta, where only 10% of the samples were positive for the 100 kDa marker.
The recently emerging fungal pathogen genus
Emergomyces, which was named several years ago but has recently been more fully characterized, i.e., stably distinguished from its close relatives that are currently assigned to the genera
Emmonsia and
Blastomyces, is now represented by several genomic sequences, which also confirms its appreciable similarity to some other fungi from the Ajellomycetaceae family (see [
25] and refs. therein). During our analysis, the only sequences that were available for
Blastomyces were
B. dermatitidis. Species such as
B. percursus or
B. emzantsi were not considered, due to the lack of sequences available. The latter should be considered for future analysis. In particular, this similarity has apparently led to noted problems in identifying
Emergomyces in clinical settings where
Histoplasma may also be present or vice versa, due to cross-reactions [
25,
26]. Specifically, our bioinformatic analysis shows that the 100 kDa primer sets have a 92.5% similarity with
Emergomyces spp., where, e.g., in the genome sequence of
Emergomyces orientalis (strain 5z489, scaffold 7), 37 out of 40 nucleotides corresponding to the forward and reverse primers have an exact match.
As the PCR assay for the detection of
H. capsulatum amplifying the 100 kDa protein [
11] is widely used for molecular diagnostics, we searched for the sequence of this gene in our OrthoMCL results. The results did not include this gene as a unique gene for
H. capsulatum. For instance, BLAST results showed that the sequence for the 100 kDa gene, as well as the primers used in the assay, are similar to the closely related fungal species
B. dermatitidis and
P. lutzii. Success of the 100 kDa assay would therefore appear to be due to sufficient sequence dissimilarity in a gene that is, however, still present outside the
Histoplasma genus.
At the time that we performed our experiments, the Emergomyces species had not been described. Moreover, we did not have genomic sequences or DNA available for in silico or in vitro analysis. The lack of these data can be considered a limitation in our analysis. When genomic reads became available for Emergomyces spp., additional in silico analysis was performed. We found that the primer sequences of PPK presented here have similarity to sequences in a region of Emergomyces spp., but with four mismatches in the forward primer and four mismatches in the reverse primer compared with Histoplasma spp., and, even in the event of an amplification, the amplicon length would be very different; in contrast to the amplicon length of 400 bp for Histoplasma spp., Emergomyces spp. would instead have a much larger amplicon of 750 bp. This indicates that our design for PPK primers would probably differentiate Histoplasma spp. from Emergomyces spp. using a basic PCR approach.
The empirical results we present suggest that the putatively functional protein kinase gene used for the PPK assay may maintain evolutionary stability at the sequence level throughout strains of
H. capsulatum, thus allowing consistent sensitivity of assays targeting the gene. Indeed, we confirmed in vitro the analytical sensitivity of the primer pair via testing of the DNA from 62
H. capsulatum strains of different clades representing a large geographical diversity, as described by Kasuga et al. [
27], and we observed amplification in all 62 isolates tested. Further testing using two clinical samples positive for
H. capsulatum that were available confirmed amplification in both cases; moreover, in one of the two FFPE samples, the fungal burden was low, yet amplification was also achieved in this sample. Although the results are promising, further testing in clinical samples is needed for confirmation of the clinical validity of the assay.
5. Conclusions and Perspectives
The Histoplasma PCR assays presented and partly validated in this study (in silico, analytically in vitro, and with FFPE skin samples of patients from a clinic) compare favorably with existing assays in their expected sensitivity and specificity in future clinical settings. In Histoplasma, however, a fair clinical performance evaluation of these and other assays might require independent verification of the fungus’ presence via another method in the same sample taken for the PCR assay, rather than just via illness of the patient or Histoplasma presence in a sample taken from a different tissue or at a different time. Such verification might be challenging if the life history of the infecting Histoplasma does not guarantee the fungus’ presence at all times in blood or tissues that can be easily sampled.
The choice of Histoplasma is a particularly stringent or ‘worst-case’ proof of principle of the assay design methodology presented here, in the sense that this fungus has close evolutionary and sequence similarity to other clinically important pathogens in the same family (Ajellomycetaceae), thus increasing the risk of cross-reactions/false positives unless special care is taken in choosing the primers. Thus, we consider that the lack of cross-reaction we observed across 25 other fungi in vitro is a nontrivial and reassuring result.
Emerging fungal diseases of the past decade include emergomyces, in which new variants of previously existing strains from the
Blastomyces-Emmonsia group evolved and rapidly spread; as a result, some previously specific PCR or antigen assays for detecting
Histoplasma or other Ajellomycetaceae fungi may now have reduced discriminatory power in the clinic in countries where emergomyces has become endemic [
25,
26]. The approach we have presented here can frontally address the problem of a dynamically changing sequence landscape of pathogenic fungi, as it is based on empirical sequence comparison. Our design strategy would, in principle, allow the interleaving of assay updates with the arrival of new data streams from the molecular surveillance of endemic fungi (see, e.g., [
28]). In the event that an existing assay design becomes less reliable following the appearance of new variants of concern, a list of alternative assay designs could be maintained and dynamically re-tested against newly emerging isolates’ sequences. For the ranking of assay candidates in such a list, notions and algorithms from abstract sequence informatics may be helpful, for example, those developed in the context of searching for or evaluating the longest distinguishing substrings ([
29],
Section 2.4 and
Section 2.7). On a more general note, the need for agile assay designs that can be readily modified in response to emerging and clinically important species or strains (e.g.,
Emergomyces spp. or
Candida auris) is also a priority need in other areas of microbiology, as the recent evolution of coronaviruses has made clear.


 ,
, 



{kind=link}
{kind=link}
{kind=link}