
Genetic Analyses of Amphotericin B Susceptibility in Aspergillus fumigatus



Abstract
1. Introduction
2. Materials and Methods
2.1. Whole-Genome Sequences and Variant Calling
2.2. Genome-Wide Association Study and Linkage Disequilibrium
2.3. Mating and Ascospore Collection
2.4. AMB Susceptibility Testing
2.5. DNA Extraction of the Progeny Strains
2.6. Polymerase Chain Reaction and Restriction Fragment Length Polymorphism
3. Results
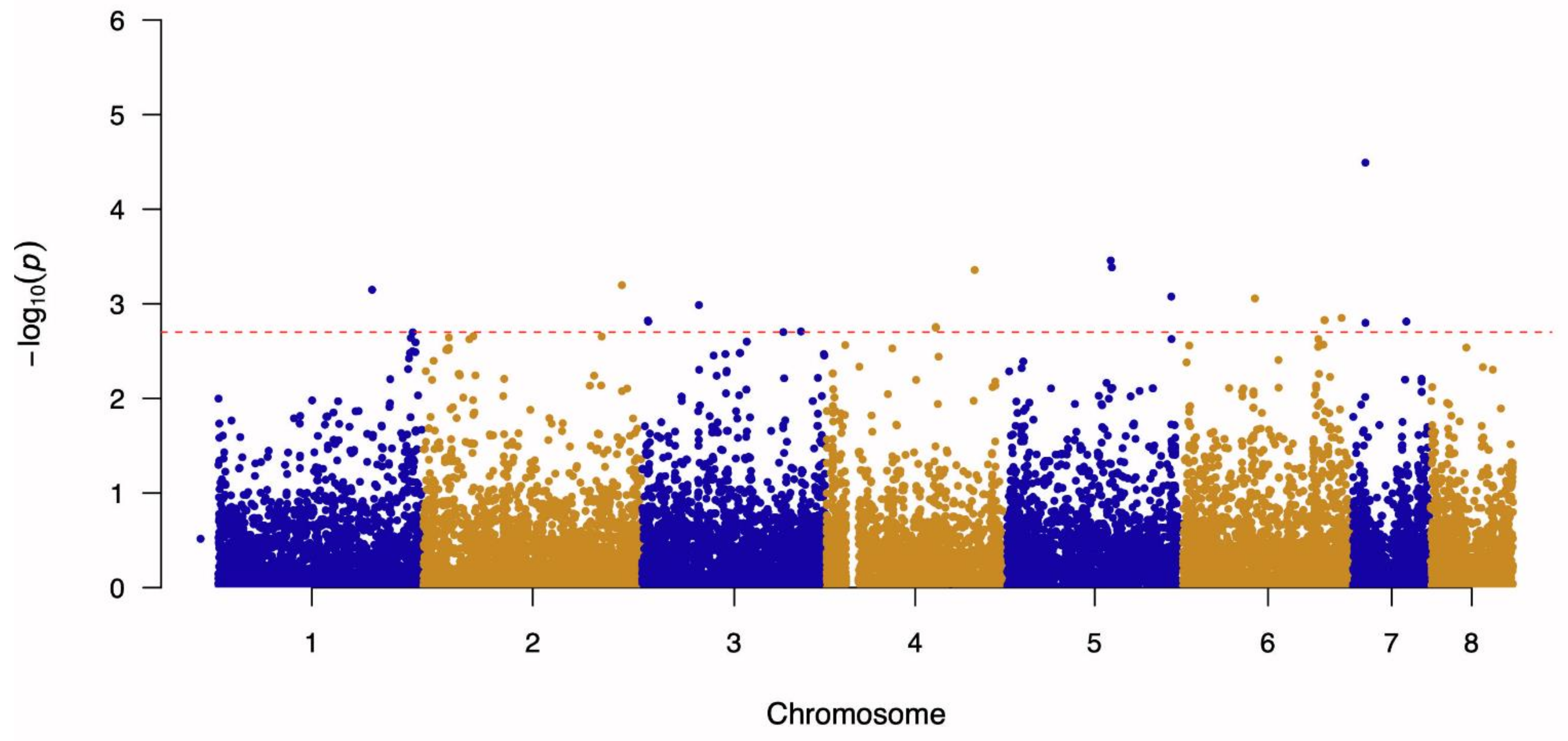
3.1. Genome-Wide Association Study and Linkage Disequilibrium Analysis
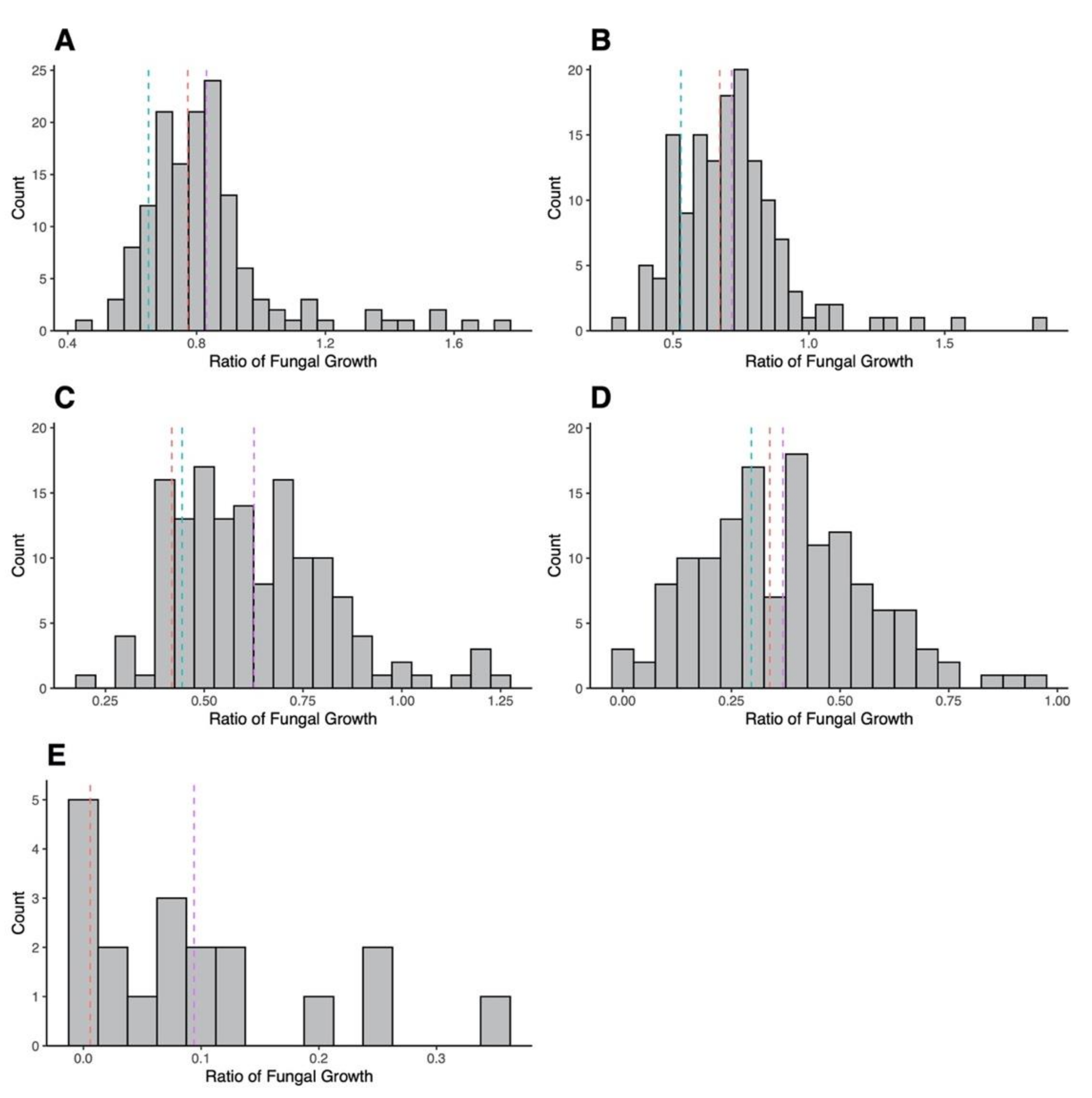
3.2. Mating Cross and AMB Susceptibility of Progeny
3.3. Progeny Genotyping
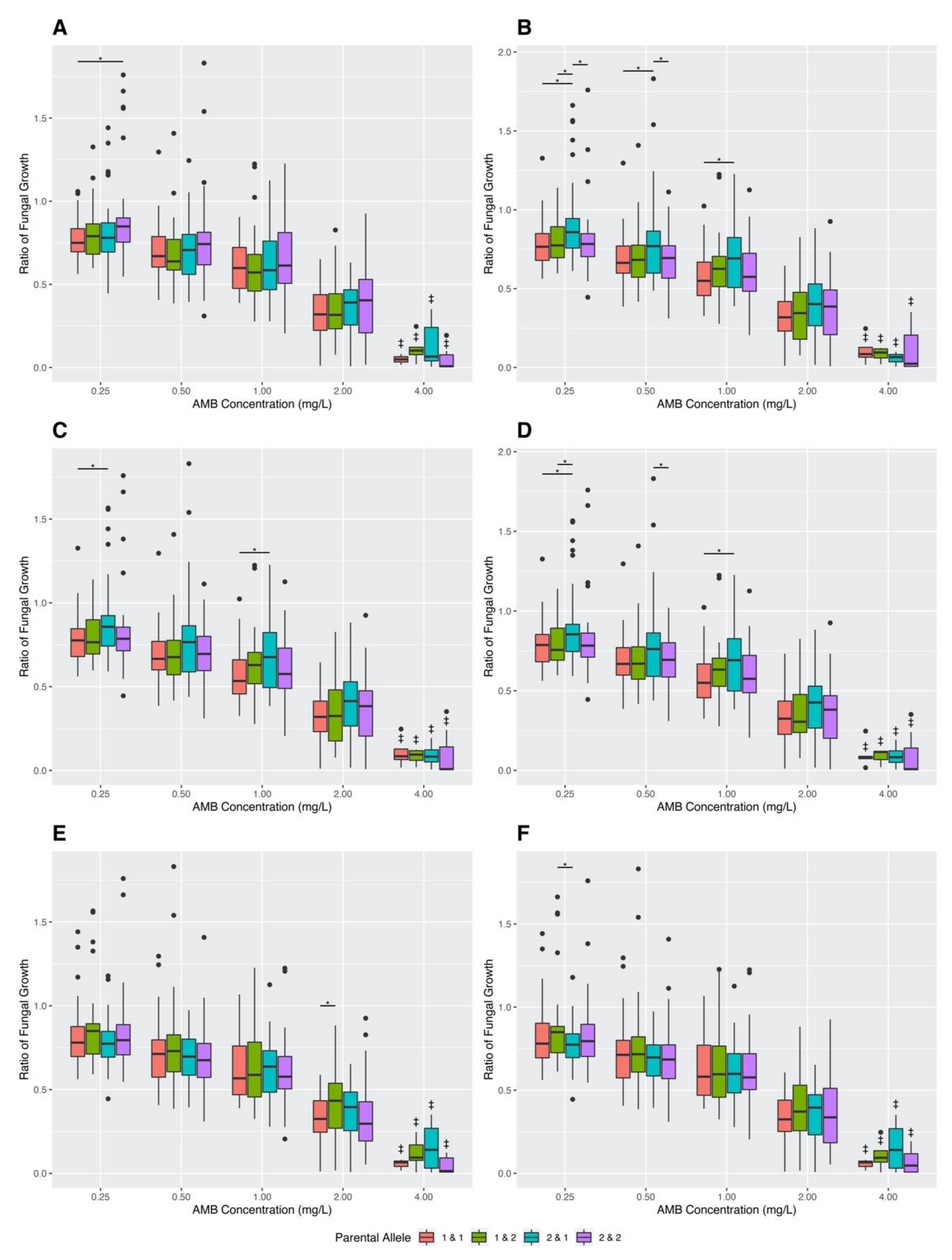
3.4. Association between Variant SNPs and AMB MIC and Growths at Different AMB Concentrations among Sexual Progeny Strains
3.4.1. Analyses Based on Individual SNPs
3.4.2. Analyses Based on Pairs of SNP Combinations
3.4.3. Analyses Based on Linked SNPs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
| AMB | Amphotericin B |
| GWAS | Genome-wide association study |
| MIC | Minimum inhibitory concentration |
| PCR | Polymerase chain reaction |
| RFLP | Restriction fragment length polymorphism |
| ROS | Reactive oxygen species |
| SNP | Single-nucleotide polymorphism |
References
- Amchentsev, A.; Kurugundla, N.; Saleh, A.G. Aspergillus-related lung disease. Respir. Med. CME 2008, 1, 205–215. [Google Scholar] [CrossRef][Green Version]
- Paulussen, C.; Hallsworth, J.E.; Álvarez-Pérez, S.; Nierman, W.C.; Hamill, P.G.; Blain, D.; Rediers, H.; Lievens, B. Ecology of aspergillosis: Insights into the pathogenic potency of Aspergillus fumigatus and some other Aspergillus species. Microb. Biotechnol. 2016, 10, 296–322. [Google Scholar] [CrossRef]
- Kwon-Chung, K.J.; Sugui, J.A. Aspergillus fumigatus—What makes the species a ubiquitous human fungal pathogen? PLoS Pathog. 2013, 9, e1003743. [Google Scholar] [CrossRef]
- Latgé, J.P. Aspergillus fumigatus and aspergillosis. Clin. Microbiol. Rev. 1999, 12, 310–350. [Google Scholar] [CrossRef] [PubMed]
- Bongomin, F.; Gago, S.; Oladele, R.O.; Denning, D.W. Global and multi-national prevalence of fungal diseases—estimate precision. J. Fungi 2017, 3, 57. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.D.; Denning, D.W.; Gow, N.A.R.; Levitz, S.M.; Netea, M.G.; White, T.C. Hidden killers: Human fungal infections. Sci. Transl. Med. 2012, 4, 165rv13. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, J.; Tovar-Torres, M.; Hingwe, A.; Cheema, F.; Welch, V.; Ford, K. The changing epidemiology of invasive aspergillosis in the non-traditional host: Risk factors and outcomes. Pulm. Crit. Care Med. 2016, 1, 67–71. [Google Scholar] [CrossRef]
- Arastehfar, A.; Carvalho, A.; Houbraken, J.; Lombardi, L.; Garcia-Rubio, R.; Jenks, J.D.; Rivero-Menendez, O.; Aljohani, R.; Jacobsen, I.D.; Berman, J.; et al. Aspergillus fumigatus and aspergillosis: From basics to clinics. Stud. Mycol. 2021, 100, 100115. [Google Scholar] [CrossRef]
- Garcia-Rubio, R.; Cuenca-Estrella, M.; Mellado, E. Triazole resistance in Aspergillus species: An emerging problem. Drugs 2017, 77, 599–613. [Google Scholar] [CrossRef]
- Nywening, A.V.; Rybak, J.M.; Rogers, P.D.; Fortwendel, J.R. Mechanisms of triazole resistance in Aspergillus fumigatus. Environ. Microbiol. 2020, 22, 4934–4952. [Google Scholar] [CrossRef]
- Lestrade, P.P.A.; Meis, J.F.; Arends, J.P.; van der Beek, M.T.; de Brauwer, E.; van Dijk, K.; de Greeff, S.C.; Haas, P.J.; Hodiamont, C.J.; Kuijper, E.J.; et al. Diagnosis and management of aspergillosis in the Netherlands: A national survey. Mycoses 2016, 59, 101–107. [Google Scholar] [CrossRef]
- Vermeulen, E.; Maertens, J.; De Bel, A.; Nulens, E.; Boelens, J.; Surmont, I.; Mertens, A.; Boel, A.; Lagrou, K. Nationwide surveillance of azole resistance in Aspergillus diseases. Antimicrob. Agents Chemother. 2015, 59, 4569–4576. [Google Scholar] [CrossRef]
- Resendiz Sharpe, A.; Lagrou, K.; Meis, J.F.; Chowdhary, A.; Lockhart, S.R.; Verweij, P.E. Triazole resistance surveillance in Aspergillus fumigatus. Med. Mycol. 2018, 56, S83–S92. [Google Scholar] [CrossRef]
- Seufert, R.; Sedlacek, L.; Kahl, B.; Hogardt, M.; Hamprecht, A.; Haase, G.; Gunzer, F.; Haas, A.; Grauling-Halama, S.; MacKenzie, C.R.; et al. Prevalence and characterization of azole-resistant Aspergillus fumigatus in patients with cystic fibrosis: A prospective multicentre study in Germany. J. Antimicrob. Chemother. 2018, 73, 2047–2053. [Google Scholar] [CrossRef]
- Prigitano, A.; Esposto, M.C.; Biffi, A.; De Lorenzis, G.; Favuzzi, V.; Koncan, R.; Lo Cascio, G.; Barao Ocampo, M.; Colombo, C.; Pizzamiglio, G.; et al. Triazole resistance in Aspergillus fumigatus isolates from patients with cystic fibrosis in Italy. J. Cyst. Fibros. 2017, 16, 64–69. [Google Scholar] [CrossRef][Green Version]
- Choukri, F.; Botterel, F.; Sitterlé, E.; Bassinet, L.; Foulet, F.; Guillot, J.; Costa, J.M.; Fauchet, N.; Dannaoui, E. Prospective evaluation of azole resistance in Aspergillus fumigatus clinical isolates in France. Med. Mycol. 2015, 53, 593–596. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wu, C.J.; Wang, H.C.; Lee, J.C.; Lo, H.J.; Dai, C.T.; Chou, P.H.; Ko, W.C.; Chen, Y.C. Azole-resistant Aspergillus fumigatus isolates carrying TR34/L98H mutations in Taiwan. Mycoses 2015, 58, 544–549. [Google Scholar] [CrossRef]
- Chowdhary, A.; Sharma, C.; Meis, J.F. Azole-resistant aspergillosis: Epidemiology, molecular mechanisms, and treatment. J. Infect. Dis. 2017, 216, S436–S444. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.; Zhang, L.; Ji, Y.; Verweij, P.E.; Tsui, K.M.; Hagen, F.; Houbraken, J.; Meis, J.F.; Abliz, P.; Wang, X.; et al. Triazole phenotypes and genotypic characterization of clinical Aspergillus fumigatus isolates in China. Emerg. Microbes Infect. 2017, 6, 1–6. [Google Scholar] [CrossRef]
- Zhou, D.; Korfanty, G.A.; Mo, M.; Wang, R.; Li, X.; Li, H.; Li, S.; Wu, J.Y.; Zhang, K.Q.; Zhang, Y.; et al. Extensive genetic diversity and widespread azole resistance in greenhouse populations of Aspergillus fumigatus in Yunnan, China. mSphere 2021, 6, e00066-21. [Google Scholar] [CrossRef] [PubMed]
- Talbot, J.J.; Subedi, S.; Halliday, C.L.; Hibbs, D.E.; Lai, F.; Lopez-Ruiz, F.J.; Harper, L.; Park, R.F.; Cuddy, W.S.; Biswas, C.; et al. Surveillance for azole resistance in clinical and environmental isolates of Aspergillus fumigatus in Australia and cyp51A homology modelling of azole-resistant isolates. J. Antimicrob. Chemother. 2018, 73, 2347–2351. [Google Scholar] [CrossRef]
- Chowdhary, A.; Sharma, C.; van den Boom, M.; Yntema, J.B.; Hagen, F.; Verweij, P.E.; Meis, J.F. Multi-azole-resistant Aspergillus fumigatus in the environment in Tanzania. J. Antimicrob. Chemother. 2014, 69, 2979–2983. [Google Scholar] [CrossRef]
- Verweij, P.E.; Lestrade, P.P.A.; Melchers, W.J.G.; Meis, J.F. Azole resistance surveillance in Aspergillus fumigatus: Beneficial or biased? J. Antimicrob. Chemother. 2016, 71, 2079–2082. [Google Scholar] [CrossRef]
- Nabili, M.; Shokohi, T.; Moazeni, M.; Khodavaisy, S.; Aliyali, M.; Badiee, P.; Zarrinfar, H.; Hagen, F.; Badali, H. High prevalence of clinical and environmental triazole-resistant Aspergillus fumigatus in Iran: Is it a challenging issue? J. Med. Microbiol. 2016, 65, 468–475. [Google Scholar] [CrossRef]
- Lestrade, P.; Buil, J.; Beek, M.; Kuijper, E.; Dijk, K.; Kampinga, G.; Rijnders, B.; Vonk, A.; Greeff, S.; Schoffelen, A.; et al. Paradoxal trends in azole-resistant Aspergillus fumigatus in a national multicenter surveillance program, The Netherlands, 2013–2018. Emerg. Infect. Dis. 2020, 26, 1447–1455. [Google Scholar] [CrossRef]
- Abdolrasouli, A.; Petrou, M.A.; Park, H.; Rhodes, J.L.; Rawson, T.M.; Moore, L.S.P.; Donaldson, H.; Holmes, A.H.; Fisher, M.C.; Armstrong-James, D. Surveillance for azole-resistant Aspergillus fumigatus in a centralized diagnostic mycology service, London, United Kingdom, 1998–2017. Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Seyedmousavi, S.; Melchers, W.J.G.; Mouton, J.W.; Verweij, P.E. Pharmacodynamics and dose-response relationships of liposomal amphotericin B against different azole-resistant Aspergillus fumigatus isolates in a murine model of disseminated aspergillosis. Antimicrob. Agents Chemother. 2013, 57, 1866–1871. [Google Scholar] [CrossRef]
- Verweij, P.E.; Ananda-Rajah, M.; Andes, D.; Arendrup, M.C.; Brüggemann, R.J.; Chowdhary, A.; Cornely, O.A.; Denning, D.W.; Groll, A.H.; Izumikawa, K.; et al. International expert opinion on the management of infection caused by azole-resistant Aspergillus fumigatus. Drug Resist. Updat. 2015, 21–22, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Cavassin, F.B.; Baú-Carneiro, J.L.; Vilas-Boas, R.R.; Queiroz-Telles, F. Sixty years of amphotericin B: An overview of the main antifungal agent used to treat invasive fungal infections. Infect. Dis. Ther. 2021, 10, 115–147. [Google Scholar] [CrossRef]
- Chang, Y.L.; Yu, S.J.; Heitman, J.; Wellington, M.; Chen, Y.L. New facets of antifungal therapy. Virulence 2017, 8, 222–236. [Google Scholar] [CrossRef] [PubMed]
- Alcazar-Fuoli, L.; Mellado, E. Ergosterol biosynthesis in Aspergillus fumigatus: Its relevance as an antifungal target and role in antifungal drug resistance. Front. Microbiol. 2013, 3, 439. [Google Scholar] [CrossRef]
- Kamiński, D.M. Recent progress in the study of the interactions of amphotericin B with cholesterol and ergosterol in lipid environments. Eur. Biophys. J. 2014, 43, 453–467. [Google Scholar] [CrossRef]
- Zavrel, M.; Esquivel, B.D.; White, T.C. The ins and outs of azole antifungal drug resistance: Molecular mechanisms of transport. In Handbook of Antimicrobial Resistance; Berghuis, A., Matlashewski, G., Wainberg, M.A., Sheppard, D., Eds.; Springer: New York, NY, USA, 2017; pp. 423–452. [Google Scholar]
- Reichert-Lima, F.; Lyra, L.; Pontes, L.; Moretti, M.L.; Pham, C.D.; Lockhart, S.R.; Schreiber, A.Z. Surveillance for azoles resistance in Aspergillus spp. highlights a high number of amphotericin B-resistant Isolates. Mycoses 2018, 61, 360–365. [Google Scholar] [CrossRef]
- Ashu, E.E.; Korfanty, G.A.; Samarasinghe, H.; Pum, N.; You, M.; Yamamura, D.; Xu, J. Widespread amphotericin B-resistant strains of Aspergillus fumigatus in Hamilton, Canada. Infect. Drug Resist. 2018, 11, 1549–1555. [Google Scholar] [CrossRef]
- Fan, Y.; Wang, Y.; Xu, J. Comparative genome sequence analyses of geographic samples of Aspergillus fumigatus-relevance for amphotericin B resistance. Microorganisms 2020, 8, 1673. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Wang, Y.; Korfanty, G.A.; Archer, M.; Xu, J. Genome-wide association analysis for triazole resistance in Aspergillus fumigatus. Pathogens 2021, 10, 701. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Garrison, E.; Marth, G. Haplotype-based variant detection from short-read sequencing. arXiv 2012, arXiv:1207.3907. [Google Scholar]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef]
- Sugui, J.A.; Losada, L.; Wang, W.; Varga, J.; Ngamskulrungroj, P.; Abu-Asab, M.; Chang, Y.C.; O’Gorman, C.M.; Wickes, B.L.; Nierman, W.C.; et al. Identification and characterization of an Aspergillus fumigatus “supermater” pair. mBio 2011, 2, e00234-11. [Google Scholar] [CrossRef]
- Ashton, G.D.; Dyer, P.S. Culturing and mating of Aspergillus fumigatus. Curr. Protoc. Microbiol. 2019, 54. [Google Scholar] [CrossRef] [PubMed]
- Clinical and Laboratory Standards Institute (CLSI). Reference method for Broth Dilution Antifungal Susceptibility Testing of Filamentous fungi. Approved Standard—Second Edition. M38-A2; Clinical and Laboratory Standards Institute: Annapolis Junction, MD, USA, 2008; pp. 1–35. [Google Scholar]
- Xu, J.; Ramos, A.R.; Vilgalys, R.; Mitchell, T.G. Clonal and spontaneous origins of fluconazole resistance in Candida albicans. J Clin. Microbiol. 2000, 38, 1214–1220. [Google Scholar] [CrossRef] [PubMed]
- Arendrup, M.C.; Friberg, N.; Mares, M.; Kahlmeter, G.; Meletiadis, J.; Guinea, J.; Arendrup, M.C.; Meletiadis, J.; Guinea, J.; Friberg, N.; et al. How to interpret MICs of antifungal compounds according to the revised clinical breakpoints v. 10.0 European Committee on Antimicrobial Susceptibility Testing (EUCAST). Clin. Microbiol. Infect. 2020, 26, 1464–1472. [Google Scholar] [CrossRef]
- Loureiro, I.; Faria, J.; Clayton, C.; Ribeiro, S.M.; Roy, N.; Santarém, N.; Tavares, J.; Cordeiro-da-Silva, A. Knockdown of asparagine synthetase A renders Trypanosoma brucei auxotrophic to asparagine. PLoS Negl. Trop. Dis. 2013, 7, e2578. [Google Scholar] [CrossRef]
- Shekhova, E.; Kniemeyer, O.; Brakhage, A.A. Induction of mitochondrial reactive oxygen species production by itraconazole, terbinafine, and amphotericin B as a mode of action against Aspergillus fumigatus. Antimicrob. Agents Chemother. 2017, 61, e00978-17. [Google Scholar] [CrossRef]
- Ying, X.; Ma, K. Characterization of a zinc-containing alcohol dehydrogenase with stereoselectivity from the hyperthermophilic archaeon Thermococcus guaymasensis. J. Bacteriol. 2011, 193, 3009–3019. [Google Scholar] [CrossRef]
- Grahl, N.; Puttikamonkul, S.; Macdonald, J.M.; Gamcsik, M.P.; Ngo, L.Y.; Hohl, T.M.; Cramer, R.A. In vivo hypoxia and a fungal alcohol dehydrogenase influence the pathogenesis of invasive pulmonary aspergillosis. PLoS Pathog. 2011, 7, e1002145. [Google Scholar] [CrossRef]
- Shekhova, E.; Ivanova, L.; Krüger, T.; Stroe, M.C.; Macheleidt, J.; Kniemeyer, O.; Brakhage, A.A. Redox proteomic analysis reveals oxidative modifications of proteins by increased levels of intracellular reactive oxygen species during hypoxia adaptation of Aspergillus fumigatus. Proteomics 2019, 19, 1800339. [Google Scholar] [CrossRef] [PubMed]
- Costa, D.M.A.; Gómez, S.V.; de Araújo, S.S.; Pereira, M.S.; Alves, R.B.; Favaro, D.C.; Hengge, A.C.; Nagem, R.A.P.; Brandão, T.A.S. Catalytic mechanism for the conversion of salicylate into catechol by the flavin-dependent monooxygenase salicylate hydroxylase. Int. J. Biol. Macromol. 2019, 129, 588–600. [Google Scholar] [CrossRef]
- Graminha, M.A.S.; Rocha, E.M.F.; Prade, R.A.; Martinez-Rossi, N.M. Terbinafine resistance mediated by salicylate 1-monooxygenase in Aspergillus nidulans. Antimicrob. Agents Chemother. 2004, 48, 3530–3535. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Ge, W.; Watanabe, A.; Fortwendel, J.R.; Gibbons, J.G. Genome-wide association for itraconazole sensitivity in non-resistant clinical isolates of Aspergillus fumigatus. Front. Fungal Biol. 2021, 1, 6. [Google Scholar] [CrossRef]
- Archer, M.; Xu, J. Current practices for reference gene selection in RT-qPCR of Aspergillus: Outlook and recommendations for the future. Genes 2021, 12, 960. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Punekar, N.S. The metabolism of 4-aminobutyrate (GABA) in fungi. Mycol. Res. 1997, 101, 403–409. [Google Scholar] [CrossRef]
- Michelle, C.; Vourc’h, P.; Mignon, L.; Andres, C.R. What was the set of ubiquitin and ubiquitin-like conjugating enzymes in the eukaryote common ancestor? J. Mol. Evol. 2009, 68, 616–628. [Google Scholar] [CrossRef]
- Jogl, G.; Shen, Y.; Gebauer, D.; Li, J.; Wiegmann, K.; Kashkar, H.; Krönke, M.; Tong, L. Crystal structure of the BEACH domain reveals an unusual fold and extensive association with a novel PH domain. EMBO J. 2002, 21, 4785–4795. [Google Scholar] [CrossRef] [PubMed]
- Lev, S.; Li, C.; Desmarini, D.; Sorrell, T.C.; Saiardi, A.; Djordjevic, J.T. Fungal kinases with a sweet tooth: Pleiotropic roles of their phosphorylated inositol sugar products in the pathogenicity of Cryptococcus neoformans present novel drug targeting opportunities. Front. Cell. Infect. Microbiol. 2019, 9, 248. [Google Scholar] [CrossRef]
- Lyons, T.J.; Villa, N.Y.; Regalla, L.M.; Kupchak, B.R.; Vagstad, A.; Eide, D.J. Metalloregulation of yeast membrane steroid receptor homologs. Proc. Natl. Acad. Sci. USA 2004, 101, 5506–5511. [Google Scholar] [CrossRef] [PubMed]
- Serhan, G.; Stack, C.M.; Perrone, G.G.; Morton, C.O. The polyene antifungals, amphotericin B and nystatin, cause cell death in Saccharomyces cerevisiae by a distinct mechanism to amphibian-derived antimicrobial peptides. Ann. Clin. Microbiol. Antimicrob. 2014, 13, 18. [Google Scholar] [CrossRef] [PubMed]
- Zoll, J.; Verweij, P.E.; Melchers, W.J.G. Discovery and characterization of novel Aspergillus fumigatus mycoviruses. PLoS ONE 2018, 13, e0200511. [Google Scholar] [CrossRef]
- Takahashi-Nakaguchi, A.; Shishido, E.; Yahara, M.; Urayama, S.; Sakai, K.; Chibana, H.; Kamei, K.; Moriyama, H.; Gonoi, T. Analysis of an intrinsic mycovirus associated with reduced virulence of the human pathogenic fungus Aspergillus fumigatus. Front. Microbiol. 2020, 10, 3045. [Google Scholar] [CrossRef]
- Kotta-Loizou, I.; Coutts, R.H.A. Mycoviruses in Aspergilli: A comprehensive review. Front. Microbiol. 2017, 8, 1699. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SNP Site Number | Chromosome and Position (bp) | Primer Sequence (5′ to 3′) | Amplification Conditions | Restriction Enzyme |
|---|---|---|---|---|
| 1 | CHR 5—201,094 | F: ACAAACGCCCTTGATCGCTA R: TTTGAGCAGGCCGTAGAGTG | 95 °C for 10 min; 40 cycles: 95 °C for 30 s, 56 °C for 30 s, 72 °C for 1 min; 72 °C for 5 min. | FauI |
| 2 | CHR 5—2,362,267 (Represented by CHR 5—2,362,923) | F: CCCTAATGGGTCCGCCAAAA R: CCAGGTGGGGAGTATGGGTA | 95 °C for 10 min; 40 cycles: 95 °C for 30 s, 57 °C for 30 s, 72 °C for 1 min; 72 °C for 5 min. | HpyCH4IV |
| 3 | CHR 5—2,370,937 | F: GCCTACAGGGTCTTGCTTGT R: TGTCAGGACCGCCAATGAAA | 95 °C for 10 min; 40 cycles: 95 °C for 30 s, 56 °C for 30 s, 72 °C for 1 min; 72 °C for 5 min. | BbsI |
| 4 | CHR 5—2,399,121 | F: ATGAGGCAAGGGATCGTACC R: TGCCTACCTCAATCGCACTG | 95 °C for 10 min; 40 cycles: 95 °C for 30 s, 56 °C for 30 s, 72 °C for 1 min; 72 °C for 5 min. | HpyCH4III |
| 5 | CHR 6—1,608,813 (Represented by CHR 6—1,608,090) | F: AAGACAACTTCCGAGCCGTG R: GCCCCTCTTGGCCTCATTT | 95 °C for 10 min; 40 cycles: 95 °C for 30 s, 57 °C for 30 s, 72 °C for 1 min; 72 °C for 5 min. | BspDI |
| Chromosome | Position (bp) | Change | −log10 (p-Value) | Gene ID | Annotation | Predicted Effect |
|---|---|---|---|---|---|---|
| 7 | 278,099 | A to G | 4.49 | AFUA_7G01030-AFUA_7G01040 | Calcium-transporting ATPase—Cytidine deaminase, putative | Intergenic Region |
| 5 | 2,362,267 | G to A | 3.46 | AFUA_5G09190-AFUA_5G09200 | ABC bile acid transporter, putative—Ubiquitin conjugating enzyme (UbcC), putative | Intergenic Region |
| 5 | 2,386,509 | T to G | 3.38 | AFUA_5G09260-AFUA_5G09270 | Phosphatidylinositol transporter, putative—ER membrane protein complex subunit 1 | Intergenic Region |
| 4 | 3,275,045 | T to A | 3.36 | AFUA_4G12480 | Asparagine synthase-related protein | Missense Variant (Ser424Cys) |
| 2 | 4,385,926 | A to G | 3.20 | AFUA_2G16500-AFUA_2G16510 | Uncharacterized protein—Uncharacterized protein | Intergenic Region |
| 1 | 3,787,543 | A to G | 3.15 | AFUA_1G00400-AFUA_1G00420 | Uncharacterized protein—Carboxypeptidase | Intergenic Region |
| 5 | 3,698,701 | G to T | 3.08 | AFUA_5G14160-AFUA_5G14170 | Uncharacterized protein—Uncharacterized protein | Intergenic Region |
| 6 | 1,608,813 | C to T | 3.06 | AFUA_6G07160-AFUA_6G07170 | IZH family channel protein (Izh3), putative—Uncharacterized protein | Intergenic Region |
| 3 | 1,260,557 | T to C | 2.99 | AFUA_3G04310-AFUA_3G05320 | SnoRNA-binding protein, putative—C2H2 finger domain protein, putative | Intergenic Region |
| 6 | 3,521,360 | G to A | 2.85 | AFUA_6G13770-AFUA_6G13780 | C6 finger domain protein, putative—MFS multidrug transporter, putative | Intergenic Region |
| 6 | 3,141,751 | G to A | 2.83 | AFUA_6G12420 | SprT family metallopeptidase, putative | Missense Variant (Glu245Lys) |
| 6 | 3,149,653 | G to T | 2.83 | AFUA_6G12460 | Uncharacterized protein | Missense Variant (Asn213Lys) |
| 3 | 133,642 | T to C | 2.82 | AFUA_3G00600 | Uncharacterized protein | Missense Variant (Val519Ala) |
| 3 | 142,183 | A to C | 2.81 | AFUA_3G00620 | Zinc-containing alcohol dehydrogenase, putative | Missense Variant (His136Pro) |
| 7 | 1,182,007 | A to C | 2.81 | AFUA_7G05020-AFUA_7G05030 | Polysaccharide export protein (Cap59), putative—Pectin lyase B | Intergenic Region |
| 7 | 279,416 | T to C | 2.80 | AFUA_7G01050 | Salicylate hydroxylase, putative | Missense Variant (Gln396Arg) |
| 4 | 2,417,511 | A to G | 2.75 | AFUA_4G09240-AFUA_4G09250 | Uncharacterized protein—Uncharacterized protein | Intergenic Region |
| 4 | 2,417,525 | T to G | 2.75 | AFUA_4G09240-AFUA_4G09250 | Uncharacterized protein—Uncharacterized protein | Intergenic Region |
| 3 | 3,512,400 | T to C | 2.71 | AFUA_3G13230 | AT DNA-binding protein, putative | Synonymous Variant (Pro380Pro) |
| 3 | 3,122,663 | A to C | 2.70 | AFUA_3G11850-AFUA_3G11860 | Uncharacterized protein—Microtubule associated protein EB1, putative | Intergenic Region |
| Chromosome | Position | Gene ID | Predicted Effect (Amino Acid Substitution) | Description | Fisher’s Exact Tests (p-Value) |
|---|---|---|---|---|---|
| 1 | 3,782,532 | AFUA_1G14160 | Missense Variant (Ser65Phe) | Uncharacterized protein | 1.96 × 10−1 |
| 1 | 3,787,813 | AFUA_1G00400-AFUA_1G00420 | Intergenic Region | Uncharacterized protein—Carboxypeptidase | 3.42 × 10−1 |
| 1 | 3,796,235 | AFUA_1G00400-AFUA_1G00420 | Intergenic Region | Uncharacterized protein—Carboxypeptidase | 3.43 × 10−1 |
| 1 | 3,800,222 | AFUA_1G00400-AFUA_1G00420 | Intergenic Region | Uncharacterized protein—Carboxypeptidase | 1.90 × 10−1 |
| 1 | 3,801,124 | AFUA_1G00400-AFUA_1G00420 | Intergenic Region | Uncharacterized protein—Carboxypeptidase | 1.96 × 10−1 |
| 1 | 3,801,488 | AFUA_1G00400-AFUA_1G00420 | Intergenic Region | Uncharacterized protein—Carboxypeptidase | 1.96 × 10−1 |
| 1 | 3,801,524 | AFUA_1G00400-AFUA_1G00420 | Intergenic Region | Uncharacterized protein—Carboxypeptidase | 1.96 × 10−1 |
| 1 | 3,801,974 | AFUA_1G00400-AFUA_1G00420 | Intergenic Region | Uncharacterized protein—Carboxypeptidase | 1.96 × 10−1 |
| 1 | 3,802,717 | AFUA_1G00400-AFUA_1G00420 | Intergenic Region | Uncharacterized protein—Carboxypeptidase | 1.88 × 10−1 |
| 1 | 3,803,746 | AFUA_1G14240 | Missense Variant (Glu467Asp) | Uncharacterized protein | 1.99 × 10−1 |
| 3 | 142,511 | AFUA_3G00620 | Synonymous Variant (Val245Val) | Zinc-containing alcohol dehydrogenase, putative | 6.67 × 10−1 |
| 3 | 3,129,756 | AFUA_3G11890 | Non-coding Transcript Variant | Thermolabile L-asparaginase, putative | 1.06 × 10−1 |
| 4 | 2,416,428 | AFUA_4G09240-AFUA_4G09250 | Intergenic Region | Uncharacterized protein—Uncharacterized protein | 3.39 × 10−7 * |
| 4 | 2,417,416 | AFUA_4G09240-AFUA_4G09250 | Intergenic Region | Uncharacterized protein—Uncharacterized protein | 1.28 × 10−6 * |
| 4 | 2,417,517 | AFUA_4G09240-AFUA_4G09250 | Intergenic Region | Uncharacterized protein—Uncharacterized protein | 2.96 × 10−4 * |
| 4 | 2,417,806 | AFUA_4G09240-AFUA_4G09250 | Intergenic Region | Uncharacterized protein—Uncharacterized protein | 2.58 × 10−4 * |
| 5 | 201,094 | AFUA_5G00700-AFUA_5G00710 | Intergenic Region | Uncharacterized protein—GABA permease, putative | 7.12 × 10−4 * |
| 5 | 201,751 | AFUA_5G00710 | Missense Variant (Arg37Lys) | GABA permease, putative | 7.12 × 10−4 * |
| 5 | 2,370,937 | AFUA_5G09220 | Missense Variant (Leu872Val) | BEACH domain protein | 5.15 × 10−4 * |
| 5 | 2,399,121 | AFUA_5G09320 | Non-coding Transcript Variant | Signal transduction protein (Syg1), putative | 7.64 × 10−4 * |
| 6 | 3,132,855 | AFUA_6G12400-AFUA_6G12410 | Intergenic Region | 1,3-beta-D-glucan-UDP glucosyltransferase—1,3-beta-glucanosyltransferase | 7.28 × 10−1 |
| 6 | 3,136,524 | AFUA_6G12400-AFUA_6G12410 | Intergenic Region | 1,3-beta-D-glucan-UDP glucosyltransferase—1,3-beta-glucanosyltransferase | 7.27 × 10−1 |
| 6 | 3,148,083 | AFUA_6G12440-AFUA_6G12450 | Intergenic Region | Uncharacterized protein—Chaperone/heat shock protein (Hsp12), putative | 7.40 × 10−1 |
| 7 | 1,184,553 | AFUA_7G05030-AFUA_7G05040 | Intergenic Region | Pectin lyase B—Rhamnosidase B, putative | 3.18 × 10−1 |
| Chromosome | Position (bp) | Gene | Amino Acid Substitution | Fisher’s Exact Test (p-Value) |
|---|---|---|---|---|
| 2 | 61,543 | AFUA_2G00320 (erg3) | Thr154Ile | 3.75 × 10−2 |
| 2 | 62,002 | AFUA_2G00320 (erg3) | Tyr286Phe | 3.75 × 10−2 |
| 2 | 145,934 | AFUA_2G00660 (tcsB) | Asp759Gly | 6.10 × 10−4 * |
| 2 | 146,469 | AFUA_2G00660 (tcsB) | Gly581Ser | 4.27 × 10−3 |
| 2 | 147,363 | AFUA_2G00660 (tcsB) | Arg283Gly | 1.32 × 10−3 * |
| 2 | 147,396 | AFUA_2G00660 (tcsB) | Ala272Pro | 4.39 × 10−4 * |
| 5 | 2,342,264 | AFUA_5G09100 (mpkC) | Trp330Ser | 4.43 × 10−5 * |
| 5 | 2,342,466 | AFUA_5G09100 (mpkC) | Ile378Thr | 4.43 × 10−5 * |
| 6 | 857,963 | AFUA_6G03890 (catA) | Asp328Asn | 5.28 × 10−2 |
| 6 | 858,366 | AFUA_6G03890 (catA) | Ser462Asn | 1.48 × 10−4 * |
| 6 | 2,533,399 | AFUA_6G10240 (fos1) | Ala532Asp | 8.17 × 10−2 |
| 6 | 3,232,955 | AFUA_6G12820 (mpkB) | Lys272Arg | 3.23 × 10−2 |
| SNP ID | Chromosome | Position (bp) | Gene ID | Annotation | Predicted Effect |
|---|---|---|---|---|---|
| 1 | 5 | 201,094 | AFUA_5G00700-AFUA_5G00710 | Uncharacterized protein—GABA permease, putative | Intergenic Region |
| 2 | 5 | 2,362,267 | AFUA_5G09190-AFUA_5G09200 | ABC bile acid transporter, putative—Ubiquitin conjugating enzyme (UbcC), putative | Intergenic Region |
| 3 | 5 | 2,370,937 | AFUA_5G09220 | BEACH domain protein | Missense Variant (Leu872Val) |
| 4 | 5 | 2,399,121 | AFUA_5G09320 | Signal transduction protein (Syg1), putative | Non-coding Transcript Variant |
| 5 | 6 | 1,608,813 | AFUA_6G07160-AFUA_6G07170 | IZH family channel protein (Izh3), putative—Uncharacterized protein | Intergenic Region |
| MIC = 2 mg/L | MIC = 4 mg/L | MIC = 8 mg/L | Fisher’s Exact Test (p-Value) | ||||
|---|---|---|---|---|---|---|---|
| Allele 1 | Allele 2 | Allele 1 | Allele 2 | Allele 1 | Allele 2 | ||
| SNP 1 | 0 | 4 | 64 | 56 | 7 | 12 | 4.89 × 10−2 |
| SNP 2 | 1 | 3 | 59 | 61 | 7 | 12 | 4.00 × 10−1 |
| SNP 3 | 1 | 3 | 60 | 60 | 8 | 11 | 6.00 × 10−1 |
| SNP 4 | 1 | 3 | 63 | 57 | 9 | 10 | 6.00 × 10−1 |
| SNP 5 | 3 | 1 | 53 | 67 | 8 | 11 | 5.42 × 10−1 |
| MIC = 2 mg/L | MIC = 4 mg/L | MIC = 8 mg/L | Fisher’s Exact Test (p-Values) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Alleles 1 and 1 | Alleles 1 and 2 | Alleles 2 and 1 | Alleles 2 and 2 | Alleles 1 and 1 | Alleles 1 and 2 | Alleles 2 and 1 | Alleles 2 and 2 | Alleles 1 and 1 | Alleles 1 and 2 | Alleles 2 and 1 | Alleles 2 and 2 | ||
| SNP 5 and 4 | 1 | 2 | 0 | 1 | 31 | 22 | 32 | 35 | 5 | 3 | 4 | 7 | 7.99 × 10−1 |
| SNP 5 and 3 | 1 | 2 | 0 | 1 | 29 | 24 | 31 | 36 | 4 | 4 | 4 | 7 | 8.64 × 10−1 |
| SNP 5 and 2 | 1 | 2 | 0 | 1 | 28 | 25 | 31 | 36 | 4 | 4 | 3 | 8 | 7.27 × 10−1 |
| SNP 5 and 1 | 0 | 3 | 0 | 1 | 29 | 24 | 35 | 32 | 2 | 6 | 5 | 6 | 2.15 × 10−1 |
| SNP 4 and 3 | 1 | 0 | 0 | 3 | 58 | 5 | 2 | 55 | 8 | 1 | 0 | 10 | 7.76 × 10−1 |
| SNP 4 and 2 | 1 | 0 | 0 | 3 | 56 | 7 | 3 | 54 | 7 | 2 | 0 | 10 | 7.57 × 10−1 |
| SNP 4 and 1 | 0 | 1 | 0 | 3 | 33 | 30 | 31 | 26 | 3 | 6 | 4 | 6 | 3.03 × 10−1 |
| SNP 3 and 2 | 1 | 0 | 0 | 3 | 58 | 2 | 1 | 59 | 7 | 1 | 0 | 11 | 5.07 × 10−1 |
| SNP 3 and 1 | 0 | 1 | 0 | 3 | 34 | 26 | 30 | 30 | 3 | 5 | 4 | 7 | 3.56 × 10−1 |
| SNP 2 and 1 | 0 | 1 | 0 | 3 | 33 | 26 | 31 | 30 | 3 | 4 | 4 | 8 | 3.07 × 10−1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, Y.; Korfanty, G.A.; Xu, J. Genetic Analyses of Amphotericin B Susceptibility in Aspergillus fumigatus. J. Fungi 2021, 7, 860. https://doi.org/10.3390/jof7100860
Fan Y, Korfanty GA, Xu J. Genetic Analyses of Amphotericin B Susceptibility in Aspergillus fumigatus. Journal of Fungi. 2021; 7(10):860. https://doi.org/10.3390/jof7100860
Chicago/Turabian StyleFan, Yuying, Gregory A. Korfanty, and Jianping Xu. 2021. "Genetic Analyses of Amphotericin B Susceptibility in Aspergillus fumigatus" Journal of Fungi 7, no. 10: 860. https://doi.org/10.3390/jof7100860
APA StyleFan, Y., Korfanty, G. A., & Xu, J. (2021). Genetic Analyses of Amphotericin B Susceptibility in Aspergillus fumigatus. Journal of Fungi, 7(10), 860. https://doi.org/10.3390/jof7100860