Triplex Hybridization-Based Nanosystem for the Rapid Screening of Pneumocystis Pneumonia in Clinical Samples


 ,
,  , , and
, , and
Abstract
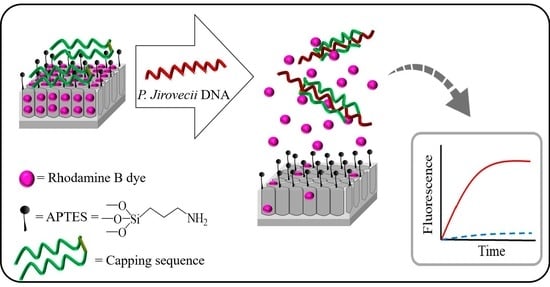
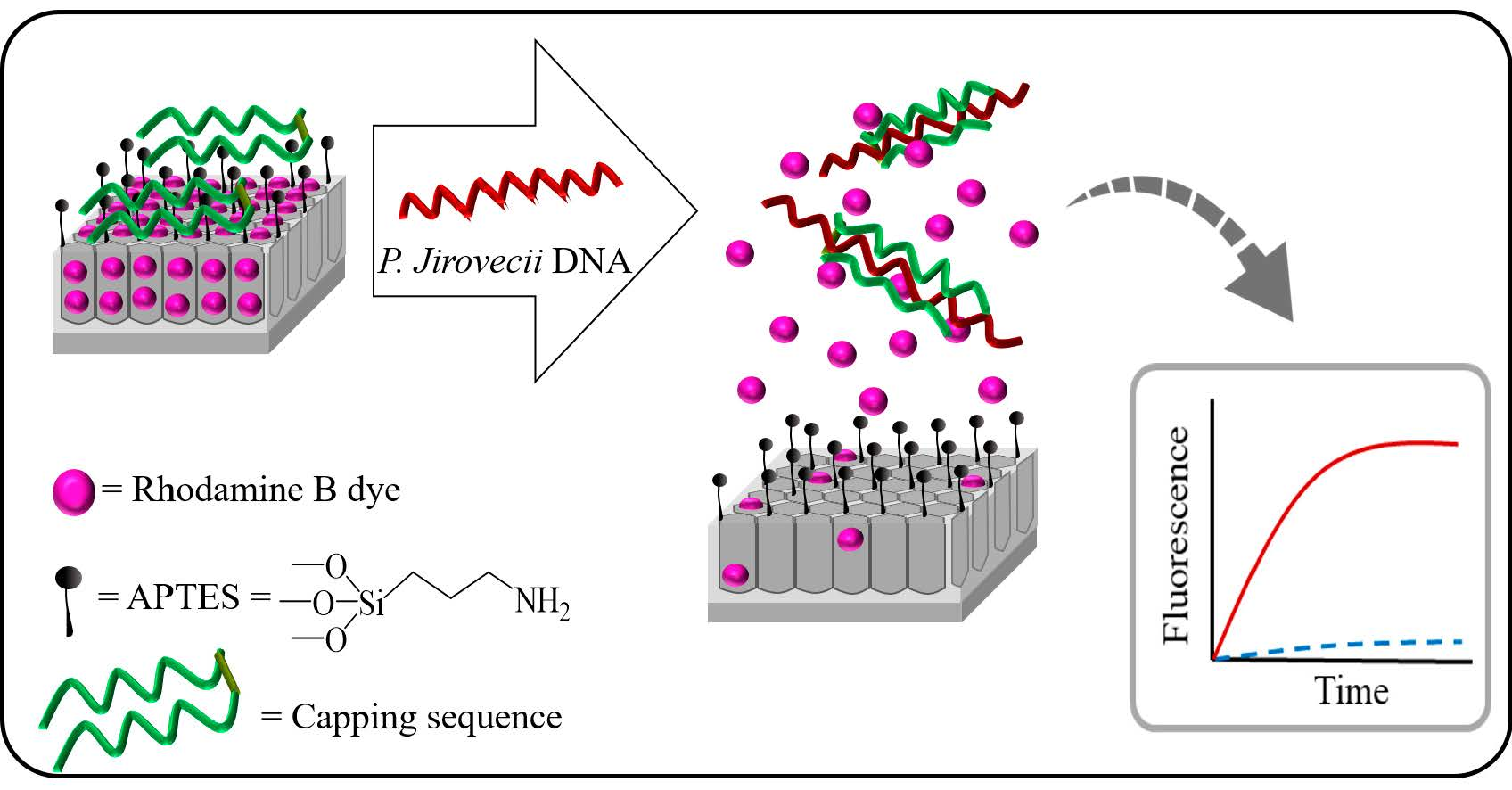
1. Introduction
2. Materials and Methods
2.1. General Techniques
2.2. Chemicals
2.3. Synthesis of Oligonucleotides
2.4. Synthesis of Nanomaterials S0, S1 (Duplex), S2 (Clamp), and S3 (Control)
2.5. Release Kinetics
2.6. Real Media Experiments
2.7. Response to Different Target Concentrations
2.8. Selectivity to Possible Interferents
2.9. Validation of the Method in Clinical Samples
3. Results and Discussion
3.1. Synthesis and Characterization of the Biosensors
3.2. Delivery Kinetics
3.3. Analytical Performance: Sensitivity and Specificity Studies
3.4. P. jirovecii Detection in Clinical Samples
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fauchier, T.; Hasseine, L.; Gari-Toussaint, M.; Casanova, V.; Marty, P.M.; Pomares, C. Detection of Pneumocystis jirovecii by Quantitative PCR To Differentiate Colonization and Pneumonia in Immunocompromised HIV-Positive and HIV-Negative Patients. J. Clin. Microbiol. 2016, 54, 1487–1495. [Google Scholar] [CrossRef] [PubMed]
- Van de Meer, G.; Brug, S.L. Infection Àpneumocystis Chez L’homme et Chez Les Animax. Annales de la Société Belge de Médecine Tropicale 1942, 22, 301–308. [Google Scholar]
- Skalski, J.H.; Kottom, T.J.; Limper, A.H. Pathobiology of Pneumocystis pneumonia: Life cycle, cell wall and cell signal transduction. FEMS Yeast Res. 2015, 15, fov046. [Google Scholar] [CrossRef] [PubMed]
- Dellière, S.; Gits-Muselli, M.; Bretagne, S.; Alanio, A. Outbreak-Causing Fungi: Pneumocystis jirovecii. Mycopathologia 2020, 185, 783–800. [Google Scholar] [CrossRef] [PubMed]
- Montes-Cano, M.A.; Chabe, M.; Fontillon-Alberdi, M.; De La Horra, C.; Respaldiza, N.; Medrano, F.J.; Varela, J.M.; Dei-Cas, E.; Calderón, E.J. Vertical transmission of Pneumocystis jirovecii in humans. Emergy Infect. Dis. 2009, 15, 125–127. [Google Scholar] [CrossRef] [PubMed]
- Jarboui, M.A.; Sellami, A.; Cheikhrouhou, F.; Makni, F.; Ben Arab, N.; Ben Jemaa, M.; Ayadi, A.; Sellami, H. Molecular diagnosis of Pneumocystis jirovecipneumonia in immunocompromised patients. Mycoses 2009, 53, 329–333. [Google Scholar]
- Durand-Joly, I.; Chabã, M.; Soula, F.; Delhaes, L.; Camus, D.; Dei-Cas, E.; Chabe, M. Molecular diagnosis ofPneumocystispneumonia. FEMS Immunol. Med. Microbiol. 2005, 45, 405–410. [Google Scholar] [CrossRef]
- Song, Y.; Ren, Y.; Wang, X.; Li, R. The biosynthesis pathway of Swainsonine, a new anticancer drug from three endophytic fungi. Med. Mycol. 2016, 57, 111–116. [Google Scholar] [CrossRef]
- Tomás, A.L.; Matos, O. Pneumocystis jirovecii Pneumonia: Current Advances in Laboratory Diagnosis. OBM Genet. 2018, 2, 1. [Google Scholar] [CrossRef]
- Urabe, N.; Sakamoto, S.; Sano, G.; Ito, A.; Sekiguchi, R.; Homma, S. Serial change in serum biomarkers during treatment of Non-HIV Pneumocystis pneumonia. J. Infect. Chemother. 2019, 25, 936–942. [Google Scholar] [CrossRef]
- Esteves, F.; Calé, S.; Badura, R.; De Boer, M.; Maltez, F.; Calderón, E.; Van Der Reijden, T.; Márquez-Martín, E.; Antunes, F.; Matos, O. Diagnosis of Pneumocystis pneumonia: Evaluation of four serologic biomarkers. Clin. Microbiol. Infect. 2015, 21, 379.e1–379.e10. [Google Scholar] [CrossRef] [PubMed]
- Tomás, A.L.; Cardoso, F.; Esteves, F.; Matos, O. Serological diagnosis of pneumocystosis: Production of a synthetic recombinant antigen for immunodetection of Pneumocystis jirovecii. Sci. Rep. 2016, 6, 36287–36295. [Google Scholar] [CrossRef] [PubMed]
- Arvanitis, M.; Anagnostou, T.; Fuchs, B.B.; Caliendo, A.M.; Mylonakis, E. Molecular and Nonmolecular Diagnostic Methods for Invasive Fungal Infections. Clin. Microbiol. Rev. 2014, 27, 490–526. [Google Scholar] [CrossRef] [PubMed]
- Tomás, A.L.; Cardoso, F.; Pinto, M.; de Almeida, M.P.; de Sousa, B.; Pereira, E. In Proceedings of the 28th European Congress of Clinical Microbiology and Infectious Diseases, Madrid, Spain, 21–24 April 2018. Available online: https://www.eurekalert.org/pub_releases/2018-04/esoc-2ec041018.php (accessed on 29 October 2020).
- Tomás, A.L.; De Almeida, M.P.; Cardoso, F.; Pinto, M.; Pereira, E.; Franco, R.; Matos, O. Development of a Gold Nanoparticle-Based Lateral-Flow Immunoassay for Pneumocystis Pneumonia Serological Diagnosis at Point-of-Care. Front. Microbiol. 2019, 10, 2917. [Google Scholar] [CrossRef] [PubMed]
- García-Fernández, A.; Aznar, E.; Martínez-Máñez, R.; Sancenón, F. New Advances in In Vivo Applications of Gated Mesoporous Silica as Drug Delivery Nanocarriers. Small 2020, 16, e1902242. [Google Scholar] [CrossRef] [PubMed]
- Aznar, E.; Coll, C.; Marcos, M.D.; Martínez-Máñez, R.; Sancenón, F.; Soto, J.; Amorós, P.; Cano, J.; Ruiz, E. Controlled delivery systems using antibody-capped mesoporous nanocontainers. Chem. Eur. J. 2009, 15, 6877–6888. [Google Scholar] [CrossRef]
- Sancenón, F.; Pascual, L.; Oroval, M.; Aznar, E.; Martínez-Máñez, R. Gated Silica Mesoporous Materials in Sensing Applications. ChemistryOpen 2015, 4, 418–437. [Google Scholar] [CrossRef] [PubMed]
- Mondragón, L.; Mas, N.; Ferragud, V.; De La Torre, C.; Agostini, A.; Martínez-Máñez, R.; Sancenón, F.; Amorós, P.; Pérez-Payá, E.; Orzáez, M. Enzyme-Responsive Intracellular-Controlled Release Using Silica Mesoporous Nanoparticles Capped with ε-Poly-l-lysine. Chem. A Eur. J. 2014, 20, 5271–5281. [Google Scholar] [CrossRef]
- Giménez, C.; De La Torre, C.; Gorbe, M.; Aznar, E.; Sancenón, F.; Murguía, J.R.; Martínez-Máñez, R.; Marcos, M.D.; Amorós, P. Gated Mesoporous Silica Nanoparticles for the Controlled Delivery of Drugs in Cancer Cells. Langmuir 2015, 31, 3753–3762. [Google Scholar] [CrossRef]
- Aznar, E.; Villalonga, R.; Giménez, C.; Sancenón, F.; Marcos, M.D.; Martínez-Máñez, R.; Díez, P.; Pingarrón, J.M.; Amorós, P. Glucose-triggered release using enzyme-gated mesoporous silica nanoparticles. Chem. Commun. 2013, 49, 6391–6393. [Google Scholar] [CrossRef]
- Ouyang, C.; Zhang, S.; Xue, C.; Yu, X.; Xu, H.; Wang, Z.; Lu, Y.; Wu, Z.-S. Precision-Guided Missile-Like DNA Nanostructure Containing Warhead and Guidance Control for Aptamer-Based Targeted Drug Delivery into Cancer Cells in Vitro and in Vivo. J. Am. Chem. Soc. 2020, 142, 1265–1277. [Google Scholar] [CrossRef] [PubMed]
- Argoubi, W.; Sánchez, A.; Parrado, C.; Raouafi, N.; Villalonga, R. Label-free electrochemical aptasensing platform based on mesoporous silica thin film for the detection of prostate specific antigen. Sens. Actuators B Chem. 2018, 255, 309–315. [Google Scholar] [CrossRef]
- Llopis-Lorente, A.; Lozano-Torres, B.; Bernardos, A.; Martínez-Máñez, R.; Sancenón, F. Mesoporous silica materials for controlled delivery based on enzymes. J. Mater. Chem. B 2017, 5, 3069–3083. [Google Scholar] [CrossRef] [PubMed]
- Pla, L.; Xifre-Perez, E.; Ribes, À.; Aznar, E.; Marcos, M.D.; Marsal, L.F.; Martínez-Máñez, R.; Sancenón, F. A Mycoplasma Genomic DNA Probe using Gated Nanoporous Anodic Alumina. ChemPlusChem 2017, 82, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Ribes, A.; Aznar, E.; Santiago-Felipe, S.; Xifre-Perez, E.; Tormo-Mas, M.Á.; Pemán, J.; Marsal, L.F.; Martínez-Máñez, R. Selective and Sensitive Probe Based in Oligonucleotide-Capped Nanoporous Alumina for the Rapid Screening of Infection Produced by Candida albicans. ACS Sens. 2019, 4, 1291–1298. [Google Scholar] [CrossRef] [PubMed]
- Goñi, J.R.; Vaquerizas, R.E.-G.A.S.J.M.; Dopazo, J.; Orozco, M. Exploring the reasons for the large density of triplex-forming oligonucleotide target sequences in the human regulatory regions. BMC Genomic 2006, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Frank-Kamenetskii, M.D.; Mirkin, S.M. Triplex DNA structures. Annu. Rev. Biochem. 1995, 64, 65–95. [Google Scholar] [CrossRef] [PubMed]
- Goñi, J.R.; De La Cruz, X.; Orozco, M. Triplex-forming oligonucleotide target sequences in the human genome. Nucleic Acids Res. 2004, 32, 354–360. [Google Scholar] [CrossRef]
- Aviñó, A.; Frieden, M.; Morales, J.C.; García de la Torre, B.; Güimil García, R.; Azorín, F.; Gelpí, J.L.; Orozco, M.; González, C.; Eritja, R. Properties of triple helices formed by parallel-stranded hairpins containing 8-aminopurines. Nucleic Acids Res. 2002, 30, 2609–2619. [Google Scholar] [CrossRef]
- Nadal, A.; Eritja, R.; Esteve, T.; Pla, M. “Parallel” and “Antiparallel Tail-Clamps” Increase the Efficiency of Triplex Formation with Structured DNA and RNA Targets. ChemBioChem 2005, 6, 1034–1042. [Google Scholar] [CrossRef]
- Carrascosa, L.G.; Gómez-Montes, S.; Aviñó, A.; Nadal, A.; Pla, M.; Eritja, R.; Lechuga, L.M. Sensitive and label-free biosensing of RNA with predicted secondary structures by a triplex affinity capture method. Nucleic Acids Res. 2012, 40, e56. [Google Scholar] [CrossRef] [PubMed]
- Aviñó, A.; Huertas, C.S.; Lechuga, L.M.; Eritja, R. Sensitive and label-free detection of miRNA-145 by triplex formation. Anal. Bioanal. Chem. 2016, 408, 885–893. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Chen, G.; Jia, X.; Mao, X.; Chen, T.; Mao, D.; Zhang, W.; Xiong, W. Exponential amplification reaction and triplex DNA mediated aggregation of gold nanoparticles for sensitive colorimetric detection of microRNA. Anal. Chim. Acta 2020, 1095, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Ribes, À.; Santiago-Felipe, S.; Aviñó, A.; Candela-Noguera, V.; Eritja, R.; Sancenón, F.; Martínez-Máñez, R.; Aznar, E. Design of oligonucleotide-capped mesoporous silica nanoparticles for the detection of miRNA-145 by duplex and triplex formation. Sens. Actuators B Chem. 2018, 277, 598–603. [Google Scholar] [CrossRef]
- Pascual, L.; Baroja, I.; Aznar, E.; Sancenón, F.; Marcos, M.D.; Murguia, J.R.; Amoros, P.; Rurack, K.; Martínez-Máñez, R. Oligonucleotide-capped mesoporous silica nanoparticles as DNA-responsive dye delivery systems for genomic DNA detection. Chem. Commun. 2015, 51, 1414–1416. [Google Scholar] [CrossRef]
- Oroval, M.; Coll, C.; Bernardos, A.; Marcos, M.D.; Martínez-Máñez, R.; Shchukin, D.G.; Sancenón, F. Selective Fluorogenic Sensing of As(III) Using Aptamer-Capped Nanomaterials. ACS Appl. Mater. Interfaces 2017, 9, 11332–11336. [Google Scholar] [CrossRef]
- Vojkuvka, L.; Marsal, L.F.; Ferré-Borrull, J.; Formentin, P.; Pallares, J. Self-ordered porous alumina membranes with large lattice constant fabricated by hard anodization. Superlattices Microstruct. 2008, 44, 577–582. [Google Scholar] [CrossRef]
- Matsumura, Y.; Tsuchido, Y.; Yamamoto, M.; Nakano, S.; Nagao, M. Development of a fully automated PCR assay for the detection of Pneumocystis jirovecii using the GENECUBE system. Med. Mycol. J. 2019, 57, 841–847. [Google Scholar] [CrossRef]
- Yang, S.-L.; Wen, Y.-H.; Wu, Y.-S.; Wang, M.-C.; Chang, P.-Y.; Yang, S.; Lu, J.-J. Diagnosis of Pneumocystis pneumonia by real-time PCR in patients with various underlying diseases. J. Microbiol. Immunol. Infect. 2019, 5, 785–790. [Google Scholar] [CrossRef]
- Moodley, B.; Tempia, S.; Frean, J.A. Comparison of quantitative real-time PCR and direct immunofluorescence for the detection of Pneumocystis jirovecii. PLoS ONE 2017, 12, e0180589. [Google Scholar] [CrossRef]
- Alanio, A.; Desoubeaux, G.; Sarfati, C.; Hamane, S.; Bergeron, A.; Azoulay, É.; Molina, J.; DeRouin, F.; Menotti, J. Real-time PCR assay-based strategy for differentiation between active Pneumocystis jirovecii pneumonia and colonization in immunocompromised patients. Clin. Microbiol. Infect. 2011, 17, 1531–1537. [Google Scholar] [CrossRef] [PubMed]
- Marimuthu, S. Development of a Real-time PCR assay for Pneumocystis jirovecii on the Luminex ARIES® Platform. Univ. Louisev. J. Respir. Infect. 2019, 3, 5. [Google Scholar] [CrossRef]
- Cissé, O.H.; Almeida, J.M.G.C.F.; Fonseca, Á.; Kumar, A.A.; Salojärvi, J.; Overmyer, K.; Hauser, P.M.; Pagni, M. Genome Sequencing of the Plant Pathogen Taphrina deformans, the Causal Agent of Peach Leaf Curl. MBio 2013, 4, e00055-13. [Google Scholar] [CrossRef]
- Rojas, P.; Friaza, V.; García, E.; De La Horra, C.; Vargas, S.L.; Calderón, E.J.; Pavón, A. Early Acquisition of Pneumocystis jirovecii Colonization and Potential Association with Respiratory Distress Syndrome in Preterm Newborn Infants. Clin. Infect. Dis. 2017, 65, 976–981. [Google Scholar] [CrossRef] [PubMed]
- Kidd, S.E.; Chen, S.C.-A.; Meyer, W.; Halliday, C.L. A New Age in Molecular Diagnostics for Invasive Fungal Disease: Are We Ready? Front. Microbiol. 2020, 10, 2903. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oligonucleotide | Sequence (5′-3′) | |
|---|---|---|
| O1 | Duplex antiparallel | 5′-GAAAGGGAAACAGCCCAG-3′ |
| O2 | Clamp antiparallel | 5′-GACAAAGGGAAAG-TTTT-GAAAGGGAAACAGCCCAG-3′ |
| O3 | Control clamp antiparallel | 5′-AGAGCAGAAAGGA-TTTT-GAAAGGGAAACAGCCCAG-3′ |
| O4 | Target complementary | 5′-CTGGGCTGTTTCCCTTTC-3′ |
| C/Al | N/Al | P/Al | |
|---|---|---|---|
| S0 | 1.52 | 0.38 | - |
| S1 | 0.76 | 0.40 | 0.01 |
| S2 | 0.58 | 0.53 | 0.02 |
| S3 | 0.74 | 0.42 | 0.02 |
| Sample a | Biological Fluid | Reference Method (PCR) b | S2 c | |
|---|---|---|---|---|
| Ct | Result | Result | ||
| 1 | Sputum | 28 | Infected | + |
| 2 | Sputum | 27.5 | Infected | + |
| 3 | BAL | 36.6 | Infected | + |
| 4 | Sputum | 28.5 | Infected | + |
| 5 | BAL | 38.8 | Infected | + |
| 6 | BAL | 19 | Infected | + |
| 7 | Sputum | 26.8 | Infected | + |
| 8 | Sputum | 30.28 | Infected | + |
| 9 | Sputum | 35.3 | Infected | + |
| 10 | Sputum | 34.9 | Infected | + |
| 11 | BAL | > 40 | Non-Infected | - |
| 12 | Sputum | > 40 | Non-Infected | - |
| 13 | NPA | - | Non- Infected | - |
| 14 | NPA | - | Non- Infected | - |
| 15 | NPA | - | Non- Infected | - |
| 16 | NPA | 33.6 | Infected | + |
| 17 | NPA | 37.2 | Infected | + |
| 18 | NPA | 34.9 | Infected | + |
| 19 | NPA | 28.8 | Infected | + |
| 20 | NPA | - | Non- Infected | - |
| 21 | NPA | 33.4 | Infected | + |
| 22 | NPA | - | Non- Infected | - |
| 23 | NPA | - | Non- Infected | - |
| 24 | NPA | - | Non- Infected | - |
| 25 | NPA | - | Non- Infected | + |
| 26 | NPA | - | Non- Infected | + |
| 27 | NPA | - | Non- Infected | - |
| 28 | NPA | 34.9 | Infected | - |
| 29 | NPA | 39.4 | Infected | - |
| 30 | NPA | 34.4 | Infected | + |
| 31 | NPA | 34.6 | Infected | + |
| 32 | NPA | 33.4 | Infected | + |
| 33 | NPA | 35.5 | Infected | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pla, L.; Aviñó, A.; Eritja, R.; Ruiz-Gaitán, A.; Pemán, J.; Friaza, V.; Calderón, E.J.; Aznar, E.; Martínez-Máñez, R.; Santiago-Felipe, S. Triplex Hybridization-Based Nanosystem for the Rapid Screening of Pneumocystis Pneumonia in Clinical Samples. J. Fungi 2020, 6, 292. https://doi.org/10.3390/jof6040292
Pla L, Aviñó A, Eritja R, Ruiz-Gaitán A, Pemán J, Friaza V, Calderón EJ, Aznar E, Martínez-Máñez R, Santiago-Felipe S. Triplex Hybridization-Based Nanosystem for the Rapid Screening of Pneumocystis Pneumonia in Clinical Samples. Journal of Fungi. 2020; 6(4):292. https://doi.org/10.3390/jof6040292
Chicago/Turabian StylePla, Luis, Anna Aviñó, Ramón Eritja, Alba Ruiz-Gaitán, Javier Pemán, Vicente Friaza, Enrique J. Calderón, Elena Aznar, Ramón Martínez-Máñez, and Sara Santiago-Felipe. 2020. "Triplex Hybridization-Based Nanosystem for the Rapid Screening of Pneumocystis Pneumonia in Clinical Samples" Journal of Fungi 6, no. 4: 292. https://doi.org/10.3390/jof6040292
APA StylePla, L., Aviñó, A., Eritja, R., Ruiz-Gaitán, A., Pemán, J., Friaza, V., Calderón, E. J., Aznar, E., Martínez-Máñez, R., & Santiago-Felipe, S. (2020). Triplex Hybridization-Based Nanosystem for the Rapid Screening of Pneumocystis Pneumonia in Clinical Samples. Journal of Fungi, 6(4), 292. https://doi.org/10.3390/jof6040292