Identification of Antifungal Targets Based on Computer Modeling




Abstract
1. Introduction
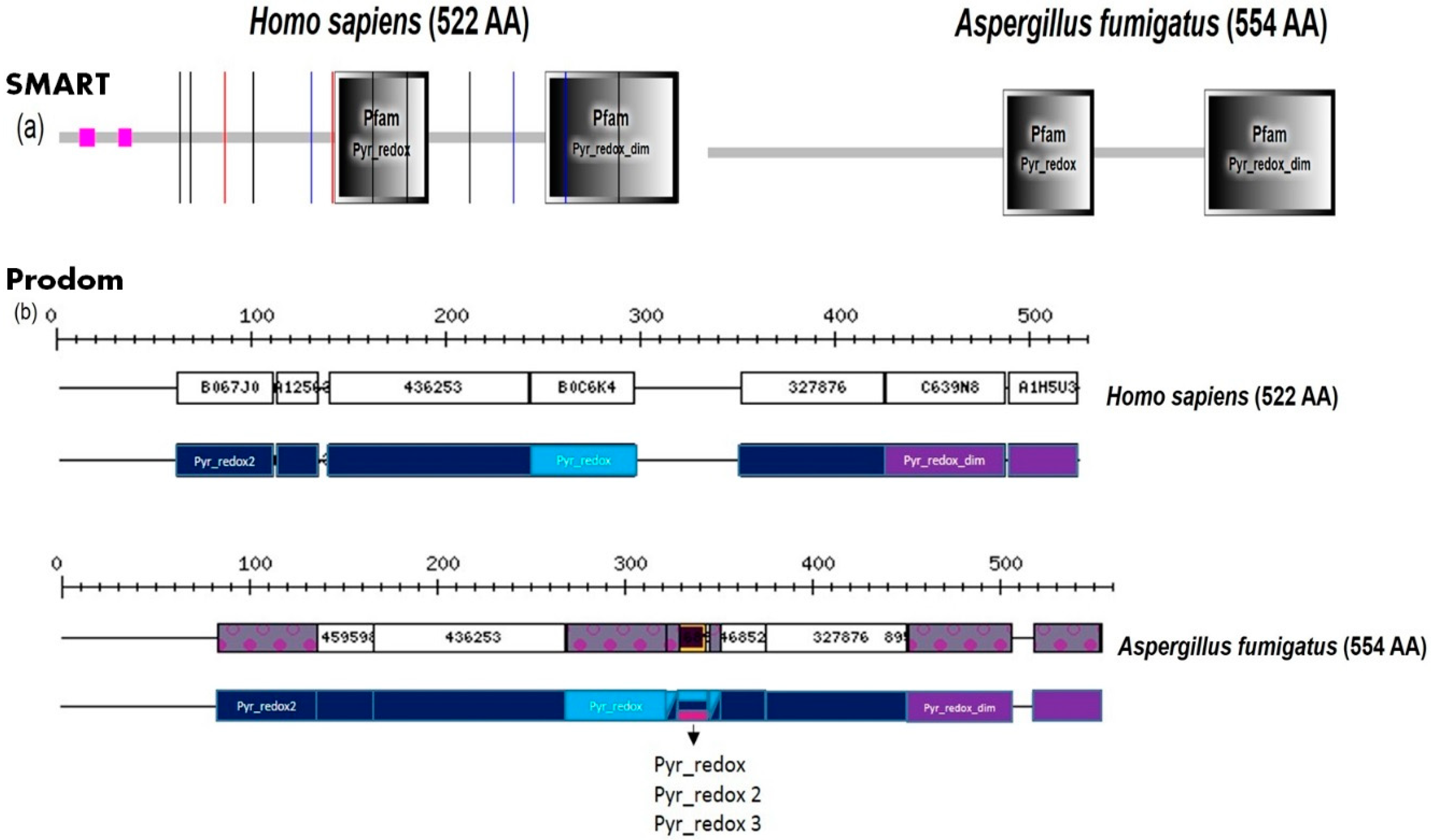
2. Fungal Diseases and Domain Analysis
3. Screens and Counter-Screens for Unique Fungal Protein Targets Using Protein–Protein Interaction Networks and Metabolic Networks
3.1. Interactome Based Approaches
3.2. Metabolic Modeling Approach
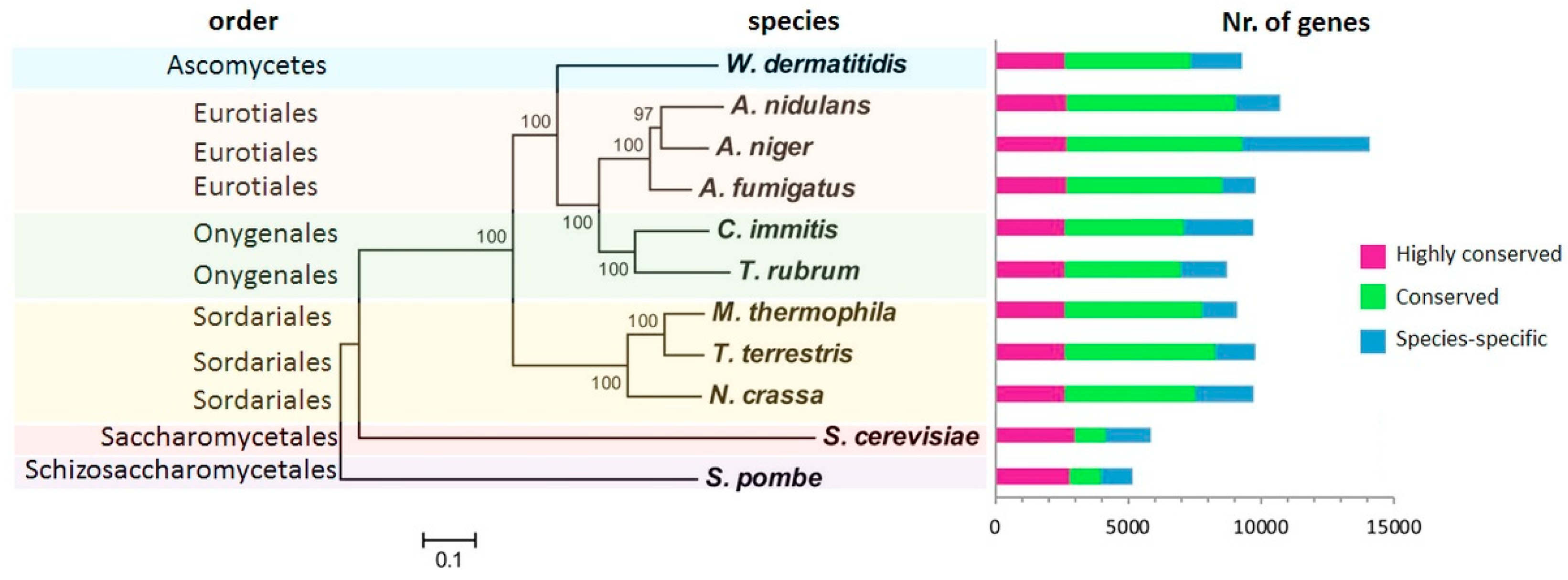
3.3. Homology-Based Approach
3.4. Ranking and Prioritization Criteria
4. Toxic Intermediates and Fungal Toxins
5. Validating and Verifying the Antifungal Target Quality and Antimycotic Drug Screening
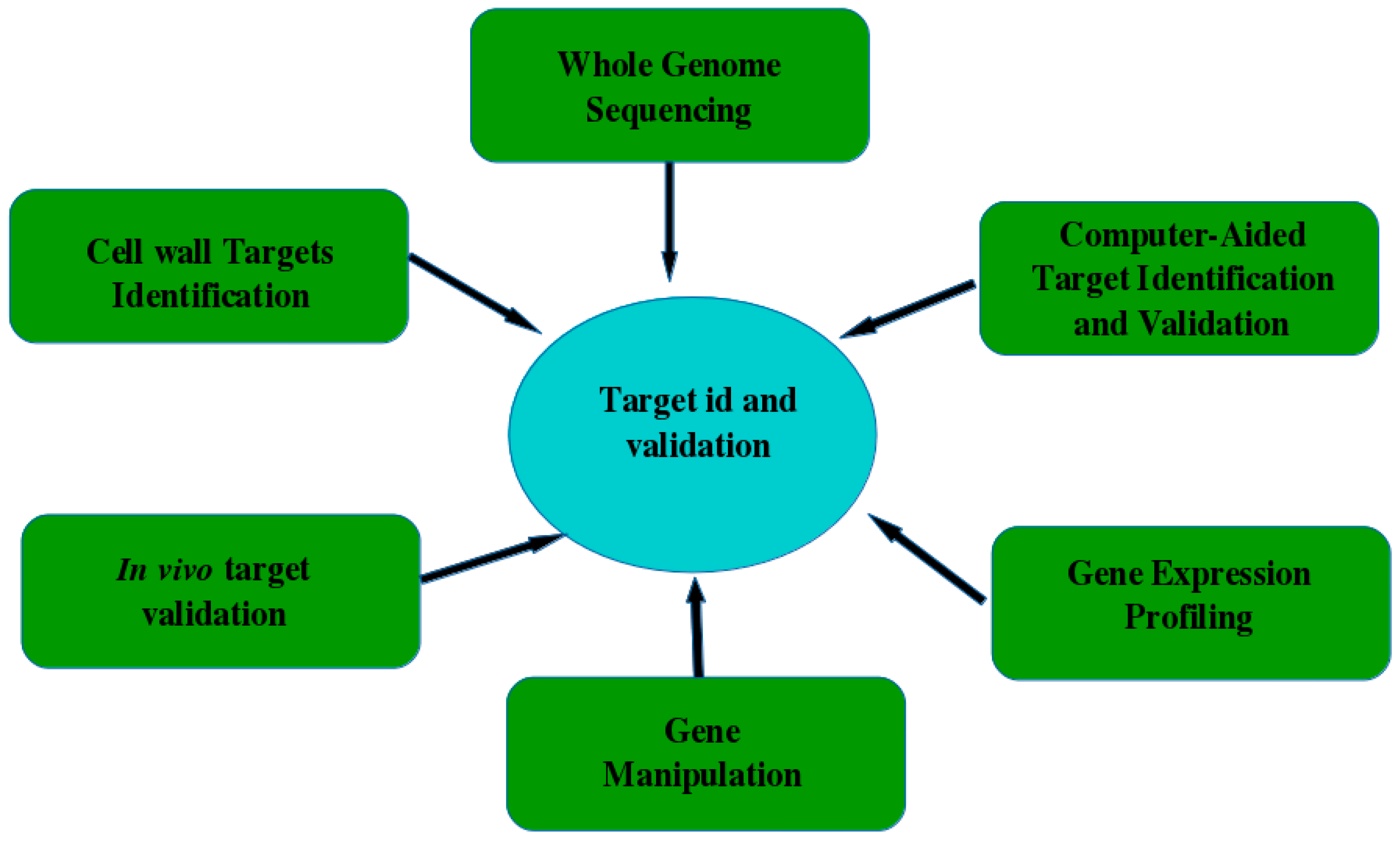
5.1. Target Identification
5.2. Target Validation
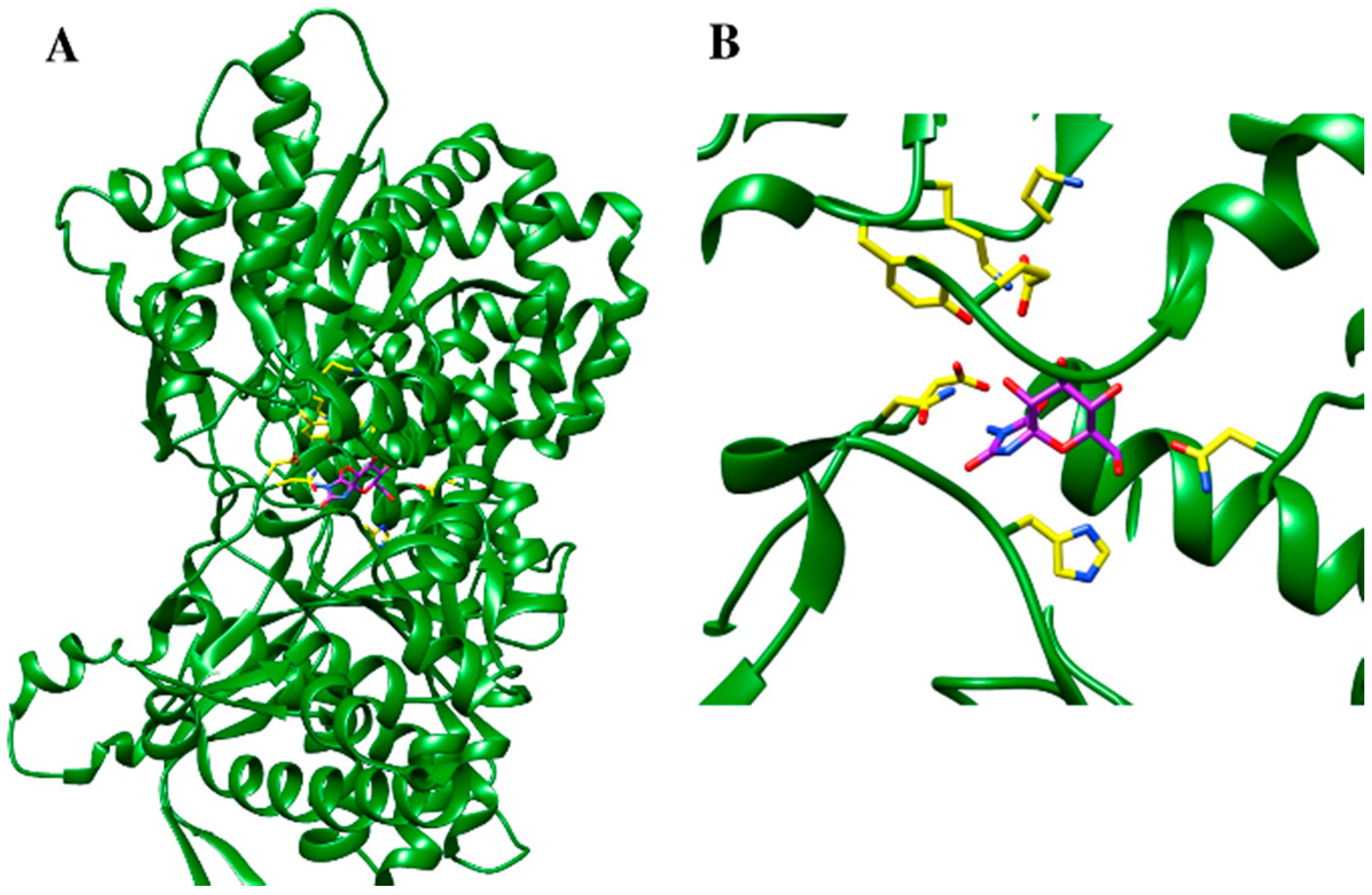
5.3. Protein Structure-Based Drug Design Strategies
6. Concluding Discussion
Methods and Their Perspective
7. Future and Outlook
Funding
Conflicts of Interest
References
- Richardson, J.P.; Naglik, J.R. Special Issue: Mucosal Fungal Infections. J. Fungi. 2018, 4, 43. [Google Scholar] [CrossRef] [PubMed]
- Denning, D.W. Invasive aspergillosis. Clin. Infect. Dis. 1998, 26, 781–803. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M.C.; Hawkins, N.J.; Sanglard, D.; Gurr, S.J. Worldwide emergence of resistance to antifungal drugs challenges human health and food security. Science 2018, 360, 739–742. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M.C.; Henk, D.A.; Briggs, C.J.; Brownstein, J.S.; Madoff, L.C.; McCraw, S.L.; Gurr, S.J. Emerging fungal threats to animal, plant and ecosystem health. Nature 2012, 484, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Spivak, E.S.; Hanson, K.E. Candida auris: An Emerging Fungal Pathogen. J. Clin. Microbiol. 2018, 56, e00080-18. [Google Scholar] [CrossRef] [PubMed]
- Havlickova, B.; Czaika, V.A.; Friedrich, M. Epidemiological trends in skin mycoses worldwide. Mycoses 2008, 51 (Suppl. 4), 2–15. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.D.; Denning, D.W.; Gow, N.A.; Levitz, S.M.; Netea, M.G.; White, T.C. Hidden killers: Human fungal infections. Sci. Transl. Med. 2012, 4, 165rv113. [Google Scholar] [CrossRef] [PubMed]
- Paulussen, C.; Hallsword, J.E.; Álvarez-Pérez, S.; Nierman, W.C.; Hamill, P.G.; Blain, D.; Rediers, H.; Lievens, B. Ecology of aspergillosis: Insights into the pathogenic potency of Aspergillus fumigatus and some other Aspergillus species. Microb. Biotechnol. 2017, 10, 296–322. [Google Scholar] [CrossRef] [PubMed]
- Zirkel, J.; Cecil, A.; Schafer, F.; Rahlfs, S.; Ouedraogo, A.; Xiao, K.; Sawadogo, S.; Coulibaly, B.; Becker, K.; Dandekar, T. Analyzing Thiol-Dependent Redox Networks in the Presence of Methylene Blue and Other Antimalarial Agents with RT-PCR-Supported in silico Modeling. Bioinform. Biol. Insights 2012, 6, 287–302. [Google Scholar] [CrossRef] [PubMed]
- Xiao, K.; Jehle, F.; Peters, C.; Reinheckel, T.; Schirmer, R.H.; Dandekar, T. CA/C1 peptidases of the malaria parasites Plasmodium falciparum and P. berghei and their mammalian hosts—A bioinformatical analysis. Biol. Chem. 2009, 390, 1185–1197. [Google Scholar] [CrossRef] [PubMed]
- Paulussen, C.; Boulet, G.A.; Cos, P.; Delputte, P.; Maes, L.J. Animal models of invasive aspergillosis for drug discovery. Drug Discov. Today 2014, 19, 1380–1386. [Google Scholar] [CrossRef] [PubMed]
- Wurster, S.; Thielen, V.; Weis, P.; Walther, P.; Elias, J.; Waaga-Gasser, A.M.; Dragan, M.; Dandekar, T.; Einsele, H.; Loffler, J.; et al. Mucorales spores induce a proinflammatory cytokine response in human mononuclear phagocytes and harbor no rodlet hydrophobins. Virulence 2017, 8, 1708–1718. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Martinez, D.A.; Gujja, S.; Sykes, S.M.; Zeng, Q.; Szaniszlo, P.J.; Wang, Z.; Cuomo, C.A. Comparative genomic and transcriptomic analysis of wangiella dermatitidis, a major cause of phaeohyphomycosis and a model black yeast human pathogen. G3: Genes Genomes Genet. 2014, 4, 561–578. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Healy, M.D.; Dougherty, B.A.; Esposito, K.M.; Maurice, T.C.; Mazzucco, C.E.; Bruccoleri, R.E.; Davison, D.B.; Frosco, M.; Barrett, J.F.; et al. Conserved Fungal Genes as Potential Targets for Broad-Spectrum Antifungal Drug Discovery. Eukaryot. Cell 2006, 4, 638–649. [Google Scholar] [CrossRef] [PubMed]
- Coronado, J.E.; Mneimneh, S.; Epstein, S.L.; Qiu, W.G.; Lipke, P.N. Conserved processes and lineage-specific proteins in fungal cell wall evolution. Eukaryot. Cell 2007, 6, 2269–2277. [Google Scholar] [CrossRef] [PubMed]
- Fedorova, N.D.; Khaldi, N.; Joardar, V.S.; Maiti, R.; Amedeo, P.; Anderson, M.J.; Crabtree, J.; Silva, J.C.; Badger, J.H.; Albarraq, A.; et al. Genomic islands in the pathogenic filamentous fungus Aspergillus fumigatus. PLoS Genet. 2008, 4, e1000046. [Google Scholar] [CrossRef] [PubMed]
- Bateman, A.; Birney, E.; Durbin, R.; Eddy, S.R.; Howe, K.L.; Sonnhammer, E.L. The Pfam protein families database. Nucleic Acids Res. 2000, 28, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Sammut, S.J.; Finn, R.D.; Bateman, A. Pfam 10 years on: 10,000 families and still growing. Brief. Bioinform. 2008, 9, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Bairoch, A.; Apweiler, R.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M.; et al. The Universal Protein Resource (UniProt). Nucleic Acids Res. 2005, 33, D154–D159. [Google Scholar] [CrossRef] [PubMed]
- Birney, E.; Ensembl, T. Ensembl: A genome infrastructure. Cold Spring Harb. Symp. Quant. Biol. 2003, 68, 213–215. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2014, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 2018, 46, D493–D496. [Google Scholar] [CrossRef] [PubMed]
- Bru, C.; Courcelle, E.; Carrere, S.; Beausse, Y.; Dalmar, S.; Kahn, D. The ProDom database of protein domain families: More emphasis on 3D. Nucleic Acids Res. 2005, 33, D212–D215. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L.; Koonin, E.V.; Lipman, D.J. A genomic perspective on protein families. Science 1997, 278, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Galperin, M.Y.; Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 2015, 43, D261–D269. [Google Scholar] [CrossRef] [PubMed]
- Galperin, M.Y.; Kristensen, D.M.; Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Microbial genome analysis: The COG approach. Brief. Bioinform. 2017. [Google Scholar] [CrossRef] [PubMed]
- Jensen, L.J.; Julien, P.; Kuhn, M.; von Mering, C.; Muller, J.; Doerks, T.; Bork, P. eggNOG: Automated construction and annotation of orthologous groups of genes. Nucleic Acids Res. 2008, 36, D250–D254. [Google Scholar] [CrossRef] [PubMed]
- Kaltdorf, M.; Srivastava, M.; Gupta, S.K.; Liang, C.; Binder, J.; Dietl, A.M.; Meir, Z.; Haas, H.; Osherov, N.; Krappmann, S.; et al. Systematic Identification of Anti-Fungal Drug Targets by a Metabolic Network Approach. Front. Mol. Biosci. 2016, 3, 22. [Google Scholar] [CrossRef] [PubMed]
- Menche, J.; Sharma, A.; Kitsak, M.; Ghiassian, S.D.; Vidal, M.; Loscalzo, J.; Barabasi, A.L. Disease networks. Uncovering disease-disease relationships through the incomplete interactome. Science 2015, 347, 1257601. [Google Scholar] [CrossRef] [PubMed]
- Boros, L.G.; Serkova, N.J.; Cascante, M.S.; Lee, W.-N.P. Use of metabolic pathway flux information in targeted cancer drug design. Drug Discov. Today: Ther. Strateg. 2004, 1, 435–443. [Google Scholar] [CrossRef]
- Gottlieb, A.; Stein, G.Y.; Ruppin, E.; Sharan, R. PREDICT: A method for inferring novel drug indications with application to personalized medicine. Mol. Syst. Biol. 2011, 7, 496. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Zhao, G.; Xu, J.R.; Kistler, H.C.; Gao, L.; Ma, L.J. Compartmentalized gene regulatory network of the pathogenic fungus Fusarium graminearum. New Phytol. 2016, 211, 527–541. [Google Scholar] [CrossRef] [PubMed]
- Guthke, R.; Gerber, S.; Conrad, T.; Vlaic, S.; Durmus, S.; Cakir, T.; Sevilgen, F.E.; Shelest, E.; Linde, J. Data-based Reconstruction of Gene Regulatory Networks of Fungal Pathogens. Front. Microbiol. 2016, 7, 570. [Google Scholar] [CrossRef] [PubMed]
- Chordia, N.; Kumar, A. In Silico Approaches for Determination of Drug Targets. Front. Anti-Infect. Drug Discov. 2017, 4, 150. [Google Scholar]
- Rezola, A.; Pey, J.; Tobalina, L.; Rubio, Á.; Beasley, J.E.; Planes, F.J. Advances in network-based metabolic pathway analysis and gene expression data integration. Brief. Bioinform. 2014, 16, 265–279. [Google Scholar] [CrossRef] [PubMed]
- Csermely, P.; Korcsmaros, T.; Kiss, H.J.; London, G.; Nussinov, R. Structure and dynamics of molecular networks: A novel paradigm of drug discovery: A comprehensive review. Pharmacol. Ther. 2013, 138, 333–408. [Google Scholar] [CrossRef] [PubMed]
- Dicko, A.; Roh, M.E.; Diawara, H.; Mahamar, A.; Soumare, H.M.; Lanke, K.; Bradley, J.; Sanogo, K.; Kone, D.T.; Diarra, K.; et al. Efficacy and safety of primaquine and methylene blue for prevention of Plasmodium falciparum transmission in Mali: A. phase 2, single-blind, randomised controlled trial. Lancet Infect. Dis. 2018, 18, 627–639. [Google Scholar] [CrossRef]
- Ansari, M.A.; Fatima, Z.; Hameed, S. Antifungal Action of Methylene Blue Involves Mitochondrial Dysfunction and Disruption of Redox and Membrane Homeostasis in C. albicans. Open Microbiol. J. 2016, 10, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Dyer, M.D.; Murali, T.M.; Sobral, B.W. The landscape of human proteins interacting with viruses and other pathogens. PLoS Pathog. 2008, 4, e32. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Ke, Y.; Wang, J.; Tan, Y.; Myeni, S.K.; Li, D.; Shi, Q.; Yan, Y.; Chen, H.; Guo, Z.; et al. Insight into bacterial virulence mechanisms against host immune response via the Yersinia pestis-human protein–protein interaction network. Infect Immun. 2011, 79, 4413–4424. [Google Scholar] [CrossRef] [PubMed]
- Crua Asensio, N.; Munoz Giner, E.; de Groot, N.S.; Torrent Burgas, M. Centrality in the host–pathogen interactome is associated with pathogen fitness during infection. Nat. Commun. 2017, 8, 14092. [Google Scholar] [CrossRef] [PubMed]
- von Mering, C.; Krause, R.; Snel, B.; Cornell, M.; Oliver, S.G.; Fields, S.; Bork, P. Comparative assessment of large-scale data sets of protein–protein interactions. Nature 2002, 417, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Luscombe, N.M.; Lu, H.X.; Zhu, X.; Xia, Y.; Han, J.D.; Bertin, N.; Chung, S.; Vidal, M.; Gerstein, M. Annotation transfer between genomes: Protein–protein interologs and protein-DNA regulogs. Genome Res. 2004, 14, 1107–1118. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Chen, H.; Liu, K.; Sun, Z. Inferring protein function by domain context similarities in protein–protein interaction networks. BMC Bioinform. 2009, 10, 395. [Google Scholar] [CrossRef] [PubMed]
- Mahdavi, M.A.; Lin, Y.H. False positive reduction in protein–protein interaction predictions using gene ontology annotations. BMC Bioinform. 2007, 8, 262. [Google Scholar] [CrossRef] [PubMed]
- Saito, R.; Suzuki, H.; Hayashizaki, Y. Interaction generality, a measurement to assess the reliability of a protein–protein interaction. Nucleic Acids Res. 2002, 30, 1163–1168. [Google Scholar] [CrossRef] [PubMed]
- Sprinzak, E.; Sattath, S.; Margalit, H. How reliable are experimental protein–protein interaction data? J. Mol. Biol. 2003, 327, 919–923. [Google Scholar] [CrossRef]
- Remmele, C.W.; Luther, C.H.; Balkenhol, J.; Dandekar, T.; Muller, T.; Dittrich, M.T. Integrated inference and evaluation of host-fungi interaction networks. Front. Microbiol. 2015, 6, 764. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.C.; Lin, C.; Chuang, M.T.; Hsieh, W.P.; Lan, C.Y.; Chuang, Y.J.; Chen, B.S. Interspecies protein–protein interaction network construction for characterization of host–pathogen interactions: A Candida albicans-zebrafish interaction study. BMC Syst. Biol. 2013, 7, 79. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Gao, S.; Nguyen, N.N.; Fan, M.; Jin, J.; Liu, B.; Zhao, L.; Xiong, G.; Tan, M.; Li, S.; et al. Stringent homology-based prediction of H. sapiens-M. tuberculosis H37Rv protein–protein interactions. Biol. Direct. 2014, 9, 5. [Google Scholar] [CrossRef] [PubMed]
- Nierman, W.C.; Pain, A.; Anderson, M.J.; Wortman, J.R.; Kim, H.S.; Arroyo, J.; Berriman, M.; Abe, K.; Archer, D.B.; Bermejo, C.; et al. Genomic sequence of the pathogenic and allergenic filamentous fungus Aspergillus fumigatus. Nature 2005, 438, 1151–1156. [Google Scholar] [CrossRef] [PubMed]
- Itzhaki, Z.; Akiva, E.; Altuvia, Y.; Margalit, H. Evolutionary conservation of domain–domain interactions. Genome Biol. 2006, 7, R125. [Google Scholar] [CrossRef] [PubMed]
- Bateman, A.; Coin, L.; Durbin, R.; Finn, R.D.; Hollich, V.; Griffiths-Jones, S.; Khanna, A.; Marshall, M.; Moxon, S.; Sonnhammer, E.L.; et al. The Pfam protein families database. Nucleic Acids Res. 2004, 32, D138–D141. [Google Scholar] [CrossRef] [PubMed]
- Hunter, S.; Apweiler, R.; Attwood, T.K.; Bairoch, A.; Bateman, A.; Binns, D.; Bork, P.; Das, U.; Daugherty, L.; Duquenne, L.; et al. InterPro: The integrative protein signature database. Nucleic Acids Res. 2009, 37, D211–D215. [Google Scholar] [CrossRef] [PubMed]
- Yellaboina, S.; Tasneem, A.; Zaykin, D.V.; Raghavachari, B.; Jothi, R. DOMINE: A comprehensive collection of known and predicted domain–domain interactions. Nucleic Acids Res. 2011, 39, D730–D735. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.; Pagel, P.; Vilne, B.; Frishman, D. DIMA 3.0: Domain Interaction Map. Nucleic Acids Res. 2011, 39, D724–D729. [Google Scholar] [CrossRef] [PubMed]
- Remm, M.; Storm, C.E.; Sonnhammer, E.L. Automatic clustering of orthologs and in-paralogs from pairwise species comparisons. J. Mol. Biol. 2001, 314, 1041–1052. [Google Scholar] [CrossRef] [PubMed]
- Sonnhammer, E.L.; Ostlund, G. InParanoid 8: Orthology analysis between 273 proteomes, mostly eukaryotic. Nucleic Acids Res. 2015, 43, D234–D239. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Stoeckert, C.J., Jr.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [PubMed]
- Salwinski, L.; Miller, C.S.; Smith, A.J.; Pettit, F.K.; Bowie, J.U.; Eisenberg, D. The Database of Interacting Proteins: 2004 update. Nucleic Acids Res. 2004, 32, D449–D451. [Google Scholar] [CrossRef] [PubMed]
- Chatr-Aryamontri, A.; Oughtred, R.; Boucher, L.; Rust, J.; Chang, C.; Kolas, N.K.; O’Donnell, L.; Oster, S.; Theesfeld, C.; Sellam, A.; et al. The BioGRID interaction database: 2017 update. Nucleic Acids Res. 2017, 45, D369–D379. [Google Scholar] [CrossRef] [PubMed]
- Aranda, B.; Blankenburg, H.; Kerrien, S.; Brinkman, F.S.; Ceol, A.; Chautard, E.; Dana, J.M.; De Las Rivas, J.; Dumousseau, M.; et al. PSICQUIC and PSISCORE: Accessing and scoring molecular interactions. Nat. Methods 2011, 8, 528–529. [Google Scholar] [CrossRef] [PubMed]
- Horton, P.; Park, K.J.; Obayashi, T.; Fujita, N.; Harada, H.; Adams-Collier, C.J.; Nakai, K. WoLF PSORT: Protein localization predictor. Nucleic Acids Res. 2007, 35, W585–W587. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.N.; Chen, C.T.; Sung, T.Y.; Ho, S.Y.; Hsu, W.L. Protein subcellular localization prediction of eukaryotes using a knowledge-based approach. BMC Bioinform. 2009, 10 (Suppl. 15), S8. [Google Scholar] [CrossRef] [PubMed]
- Briesemeister, S.; Rahnenfuhrer, J.; Kohlbacher, O. YLoc—An interpretable web server for predicting subcellular localization. Nucleic Acids Res. 2010, 38, W497–W502. [Google Scholar] [CrossRef] [PubMed]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Shlomi, T.; Cabili, M.N.; Herrgard, M.J.; Palsson, B.O.; Ruppin, E. Network-based prediction of human tissue-specific metabolism. Nat. Biotechnol. 2008, 26, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Oberhardt, M.A.; Palsson, B.Ø.; Papin, J.A. Applications of genome-scale metabolic reconstructions. Mol. Syst. Biol. 2009, 5, 320. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.S.; Burd, H.; Liu, J.; Almaas, E.; Wiest, O.; Barabasi, A.L.; Oltvai, Z.N.; Kapatral, V. Comparative genome-scale metabolic reconstruction and flux balance analysis of multiple Staphylococcus aureus genomes identify novel antimicrobial drug targets. J. Bacteriol. 2009, 191, 4015–4024. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.; Fell, D.A.; Dandekar, T. A general definition of metabolic pathways useful for systematic organization and analysis of complex metabolic networks. Nat Biotechnol. 2000, 18, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.C.; Hou, S.Y.; Chao, Y.P. Pathway analysis, engineering, and physiological considerations for redirecting central metabolism. Biotechnol. Bioeng. 1996, 52, 129–140. [Google Scholar] [CrossRef]
- Schuster, S.; Dandekar, T.; Fell, D.A. Detection of elementary flux modes in biochemical networks: A promising tool for pathway analysis and metabolic engineering. Trends Biotechnol. 1999, 17, 53–60. [Google Scholar] [CrossRef]
- Dandekar, T.; Moldenhauer, F.; Bulik, S.; Bertram, H.; Schuster, S. A method for classifying metabolites in topological pathway analyses based on minimization of pathway number. Biosystems 2003, 70, 255–270. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, R.; Liang, C.; Kaleta, C.; Kuhnel, M.; Hoffmann, E.; Kuznetsov, S.; Hecker, M.; Griffiths, G.; Schuster, S.; Dandekar, T. Integrated network reconstruction, visualization and analysis using YANAsquare. BMC Bioinform. 2007, 8, 313. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, R.; Musch, P.; von Kamp, A.; Engels, B.; Schirmer, H.; Schuster, S.; Dandekar, T. YANA—A software tool for analyzing flux modes, gene-expression and enzyme activities. BMC Bioinform. 2005, 6, 135. [Google Scholar] [CrossRef] [PubMed]
- Cecil, A.; Ohlsen, K.; Menzel, T.; François, P.; Schrenzel, J.; Fischer, A.; Dörries, K.; Selle, M.; Lalk, M.; Hantzschmann, J.; et al. Modelling antibiotic and cytotoxic isoquinoline effects in Staphylococcus aureus, Staphylococcus epidermidis and mammalian cells. Int. J. Med. Microbiol. 2015, 305, 96–109. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Liebeke, M.; Schwarz, R.; Zuhlke, D.; Fuchs, S.; Menschner, L.; Engelmann, S.; Wolz, C.; Jaglitz, S.; Bernhardt, J.; et al. Staphylococcus aureus physiological growth limitations: Insights from flux calculations built on proteomics and external metabolite data. Proteomics 2011, 11, 1915–1935. [Google Scholar] [CrossRef] [PubMed]
- Yeh, I.; Hanekamp, T.; Tsoka, S.; Karp, P.D.; Altman, R.B. Computational analysis of Plasmodium falciparum metabolism: Organizing genomic information to facilitate drug discovery. Genome Res. 2004, 14, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Burn, P. Lipid metabolic enzymes: Emerging drug targets for the treatment of obesity. Nat. Rev. Drug Discov. 2004, 3, 695–710. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, A.; Fedorova, N.D.; Crabtree, J.; Yu, Y.; Kim, S.; Chen, D.; Loss, O.; Cairns, T.; Goldman, G.; Armstrong-James, D.; et al. Sub-telomere directed gene expression during initiation of invasive aspergillosis. PLoS Pathog. 2008, 4, e1000154. [Google Scholar] [CrossRef] [PubMed]
- Schrettl, M.; Beckmann, N.; Varga, J.; Heinekamp, T.; Jacobsen, I.D.; Jochl, C.; Moussa, T.A.; Wang, S.; Gsaller, F.; Blatzer, M.; et al. HapX-mediated adaption to iron starvation is crucial for virulence of Aspergillus fumigatus. PLoS Pathog. 2010, 6, e1001124. [Google Scholar] [CrossRef] [PubMed]
- Willger, S.D.; Puttikamonkul, S.; Kim, K.H.; Burritt, J.B.; Grahl, N.; Metzler, L.J.; Barbuch, R.; Bard, M.; Lawrence, C.B.; Cramer, R.A., Jr. A sterol-regulatory element binding protein is required for cell polarity, hypoxia adaptation, azole drug resistance, and virulence in Aspergillus fumigatus. PLoS Pathog. 2008, 4, e1000200. [Google Scholar] [CrossRef] [PubMed]
- Arnaud, M.B.; Cerqueira, G.C.; Inglis, D.O.; Skrzypek, M.S.; Binkley, J.; Chibucos, M.C.; Crabtree, J.; Howarth, C.; Orvis, J.; Shah, P.; et al. The Aspergillus Genome Database (AspGD): Recent developments in comprehensive multispecies curation, comparative genomics and community resources. Nucleic Acids Res. 2012, 40, D653–D659. [Google Scholar] [CrossRef] [PubMed]
- Schomburg, I.; Jeske, L.; Ulbrich, M.; Placzek, S.; Chang, A.; Schomburg, D. The BRENDA enzyme information system-From a database to an expert system. J. Biotechnol. 2017, 261, 194–206. [Google Scholar] [CrossRef] [PubMed]
- Joshi-Tope, G.; Gillespie, M.; Vastrik, I.; D’Eustachio, P.; Schmidt, E.; de Bono, B.; Jassal, B.; Gopinath, G.R.; Wu, G.R.; Matthews, L.; et al. Reactome: A knowledgebase of biological pathways. Nucleic Acids Res. 2005, 33, D428–D432. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Hagelsieb, G.; Latimer, K. Choosing BLAST options for better detection of orthologs as reciprocal best hits. Bioinformatics 2008, 24, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.K.; Bencurova, E.; Srivastava, M.; Pahlavan, P.; Balkenhol, J.; Dandekar, T. Improving Re-Annotation of Annotated Eukaryotic Genomes, Big Data Analytics in Genomics; Springer: Cham, Geramny, 2016; pp. 171–195. [Google Scholar]
- Dandekar, T.; Fieselmann, A.; Majeed, S.; Ahmed, Z. Software applications toward quantitative metabolic flux analysis and modeling. Brief. Bioinform. 2014, 15, 91–107. [Google Scholar] [CrossRef] [PubMed]
- von Kamp, A.; Schuster, S. Metatool 5.0: Fast and flexible elementary modes analysis. Bioinformatics 2006, 22, 1930–1931. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets–update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed]
- Kolesnikov, N.; Hastings, E.; Keays, M.; Melnichuk, O.; Tang, Y.A.; Williams, E.; Dylag, M.; Kurbatova, N.; Brandizi, M.; Burdett, T.; et al. ArrayExpress update—Simplifying data submissions. Nucleic Acids Res. 2015, 43, D1113–D1116. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. 2013. Available online: http://www.R-project.org (accessed on 3 July 2018).
- Luo, H.; Lin, Y.; Gao, F.; Zhang, C.T.; Zhang, R. DEG 10, an update of the database of essential genes that includes both protein-coding genes and noncoding genomic elements. Nucleic Acids Res. 2014, 42, D574–D580. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.K.; Gross, R.; Dandekar, T. An antibiotic target ranking and prioritization pipeline combining sequence, structure and network-based approaches exemplified for Serratia marcescens. Gene 2016, 591, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Pop, M. ARDB—Antibiotic Resistance Genes Database. Nucleic Acids Res. 2009, 37, D443–D447. [Google Scholar] [CrossRef] [PubMed]
- McArthur, A.G.; Waglechner, N.; Nizam, F.; Yan, A.; Azad, M.A.; Baylay, A.J.; Bhullar, K.; Canova, M.J.; De Pascale, G.; Ejim, L.; et al. The comprehensive antibiotic resistance database. Antimicrob. Agents Chemother. 2013, 57, 3348–3357. [Google Scholar] [CrossRef] [PubMed]
- Toomey, D.; Hoppe, H.C.; Brennan, M.P.; Nolan, K.B.; Chubb, A.J. Genomes2Drugs: Identifies target proteins and lead drugs from proteome data. PLoS ONE 2009, 4, e6195. [Google Scholar] [CrossRef] [PubMed]
- Bertuzzi, M.; Schrettl, M.; Alcazar-Fuoli, L.; Cairns, T.C.; Munoz, A.; Walker, L.A.; Herbst, S.; Safari, M.; Cheverton, A.M.; Chen, D.; et al. The pH-responsive PacC transcription factor of Aspergillus fumigatus governs epithelial entry and tissue invasion during pulmonary aspergillosis. PLoS Pathog. 2014, 10, e1004413. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Sasse, A.; Hamer, S.N.; Amich, J.; Binder, J.; Krappmann, S. Mutant characterization and in vivo conditional repression identify aromatic amino acid biosynthesis to be essential for Aspergillus fumigatus virulence. Virulence 2016, 7, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Brian, P.; Elson, G.; Hemming, H.; Radley, M. The plant-growth-promoting properties of gibberellic acid, a metabolic product of the fungus Gibberella fujikuroi. J. Sci. Food Agric. 1954, 5, 602–612. [Google Scholar] [CrossRef]
- Contreras-Cornejo, H.A.; Macías-Rodríguez, L.; Cortés-Penagos, C.; López-Bucio, J. Trichoderma virens, a plant beneficial fungus, enhances biomass production and promotes lateral root growth through an auxin-dependent mechanism in Arabidopsis. Plant Physiol. 2009, 149, 1579–1592. [Google Scholar] [CrossRef] [PubMed]
- Dufosse, L.; Fouillaud, M.; Caro, Y.; Mapari, S.A.; Sutthiwong, N. Filamentous fungi are large-scale producers of pigments and colorants for the food industry. Curr. Opin. Biotechnol. 2014, 26, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Auckloo, B.N.; Pan, C.; Akhter, N.; Wu, B.; Wu, X.; He, S. Stress-Driven Discovery of Novel Cryptic Antibiotics from a Marine Fungus Penicillium sp. BB1122. Front. Microbial. 2017, 8, 1450. [Google Scholar] [CrossRef] [PubMed]
- Munkvold, G.P. Fusarium Species and Their Associated Mycotoxins. Methods Mol. Biol. 2017, 1542, 51–106. [Google Scholar] [PubMed]
- Keller, N.P.; Hohn, T.M. Metabolic pathway gene clusters in filamentous fungi. Fungal Genet. Biol. 1997, 21, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Calvo, A.M.; Wilson, R.A.; Bok, J.W.; Keller, N.P. Relationship between secondary metabolism and fungal development. Microbiol. Mol. Boil. Rev. 2002, 66, 447–459. [Google Scholar] [CrossRef]
- Proschel, M.; Detsch, R.; Boccaccini, A.R.; Sonnewald, U. Engineering of Metabolic Pathways by Artificial Enzyme Channels. Front. Bioeng. Biotechnol. 2015, 3, 168. [Google Scholar] [CrossRef] [PubMed]
- Ratnakar, A.; Shankar, S.; Shikha. An overview of biodegradation of organic pollutants. Int. J. Sci. Innov. Res. 2016, 4, 73–91. [Google Scholar]
- Blumenthal, C.Z. Production of toxic metabolites in Aspergillus niger, Aspergillus oryzae, and Trichoderma reesei: Justification of mycotoxin testing in food grade enzyme preparations derived from the three fungi. Regul. Toxicol. Pharmacol. 2004, 39, 214–228. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, S.; Wei, Q.; Guo, Q.; Bai, Y.; Yang, S.; Song, F.; Zhang, L.; Lei, X. Synthesis and biological evaluation of Aspergillomarasmine A derivatives as novel NDM-1 inhibitor to overcome antibiotics resistance. Bioorg. Med. Chem. 2017, 25, 5133–5141. [Google Scholar] [CrossRef] [PubMed]
- Stanzani, M.; Orciuolo, E.; Lewis, R.; Kontoyiannis, D.P.; Martins, S.L.; St John, L.S.; Komanduri, K.V. Aspergillus fumigatus suppresses the human cellular immune response via gliotoxin-mediated apoptosis of monocytes. Blood 2005, 105, 2258–2265. [Google Scholar] [CrossRef] [PubMed]
- Waring, P.; Eichner, R.D.; Mullbacher, A.; Sjaarda, A. Gliotoxin induces apoptosis in macrophages unrelated to its antiphagocytic properties. J. Biol. Chem. 1988, 263, 18493–18499. [Google Scholar] [PubMed]
- Gardiner, D.M.; Howlett, B.J. Bioinformatic and expression analysis of the putative gliotoxin biosynthetic gene cluster of Aspergillus fumigatus. FEMS Microbiol. Lett. 2005, 248, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Bok, J.W.; Chung, D.; Balajee, S.A.; Marr, K.A.; Andes, D.; Nielsen, K.F.; Frisvad, J.C.; Kirby, K.A.; Keller, N.P. GliZ, a transcriptional regulator of gliotoxin biosynthesis, contributes to Aspergillus fumigatus virulence. Infect. Immun. 2006, 74, 6761–6768. [Google Scholar] [CrossRef] [PubMed]
- Schrettl, M.; Carberry, S.; Kavanagh, K.; Haas, H.; Jones, G.W.; O’Brien, J.; Nolan, A.; Stephens, J.; Fenelon, O.; Doyle, S. Self-protection against gliotoxin—A component of the gliotoxin biosynthetic cluster, GliT, completely protects Aspergillus fumigatus against exogenous gliotoxin. PLoS Pathog. 2010, 6, e1000952. [Google Scholar] [CrossRef] [PubMed]
- Scharf, D.H.; Remme, N.; Heinekamp, T.; Hortschansky, P.; Brakhage, A.A.; Hertweck, C. Transannular disulfide formation in gliotoxin biosynthesis and its role in self-resistance of the human pathogen Aspergillus fumigatus. J. Am. Chem. Soc. 2010, 132, 10136–10141. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Walsh, C.T. Streptomyces clavuligerus HlmI is an intramolecular disulfide-forming dithiol oxidase in holomycin biosynthesis. Biochemistry 2011, 50, 4615–4622. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Baker, A.T.; Raab, A.; Huang, S.; Wang, T.; Yu, Y.; Jaspars, M.; Secombes, C.J.; Deng, H. The fish pathogen Yersinia ruckeri produces holomycin and uses an RNA methyltransferase for self-resistance. J. Biol. Chem. 2013, 288, 14688–14697. [Google Scholar] [CrossRef] [PubMed]
- Chung, E.J.; Lim, H.K.; Kim, J.C.; Choi, G.J.; Park, E.J.; Lee, M.H.; Chung, Y.R.; Lee, S.W. Forest soil metagenome gene cluster involved in antifungal activity expression in Escherichia coli. Appl. Environ. Microbiol. 2008, 74, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Ewald, J.; Bartl, M.; Dandekar, T.; Kaleta, C. Optimality principles reveal a complex interplay of intermediate toxicity and kinetic efficiency in the regulation of prokaryotic metabolism. PLoS Comput. Biol. 2017, 13, e1005371. [Google Scholar] [CrossRef] [PubMed]
- Labena, A.A.; Ye, Y.N.; Dong, C.; Zhang, F.Z.; Guo, F.B. SSER: Species specific essential reactions database. BMC Syst. Boil. 2017, 11, 50. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Sillaots, S.; Lemieux, S.; Davison, J.; Kauffman, S.; Breton, A.; Breton, A.; Linteau, A.; Xin, C.; Bowman, J.; et al. Essential gene identification and drug target prioritization in Aspergillus fumigatus. PLoS Pathog. 2007, 3, e24. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, M.E.; Colombo, A.L.; Paulsen, I.; Ren, Q.; Wortman, J.; Huang, J.; Goldman, M.H.; Goldman, G.H. The ergosterol biosynthesis pathway, transporter genes, and azole resistance in Aspergillus fumigatus. Med. Mycol. 2005, 43 (Suppl. 1), 313–319. [Google Scholar] [CrossRef]
- Firon, A.; Villalba, F.; Beffa, R.; d’Enfert, C. Identification of essential genes in the human fungal pathogen Aspergillus fumigatus by transposon mutagenesis. Eukaryot. Cell 2003, 2, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Thykaer, J.; Andersen, M.R.; Baker, S.E. Essential pathway identification: From in silico analysis to potential antifungal targets in Aspergillus fumigatus. Med. Mycol. 2009, 47 (Suppl. 1), S80–S87. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Sakurada, M.; Okamoto, S.; Ono, M.; Tsukigi, H.; Suzuki, A.; Nagasawa, H.; Sakuda, S. Effects of aflastatin A, an inhibitor of aflatoxin production, on aflatoxin biosynthetic pathway and glucose metabolism in Aspergillus parasiticus. J. Antibiot. 2001, 54, 650–657. [Google Scholar] [CrossRef] [PubMed]
- Yoshinari, T.; Akiyama, T.; Nakamura, K.; Kondo, T.; Takahashi, Y.; Muraoka, Y.; Nonomura, Y.; Nagasawa, H.; Sakuda, S. Dioctatin A is a strong inhibitor of aflatoxin production by Aspergillus parasiticus. Microbiology 2007, 153, 2774–2780. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.; Festa, R.A.; Sun, T.S.; Wang, Z.Y. Iron and copper as virulence modulators in human fungal pathogens. Mol. Microbial. 2014, 93, 10–23. [Google Scholar] [CrossRef] [PubMed]
- Wiemann, P.; Perevitsky, A.; Lim, F.Y.; Shadkchan, Y.; Knox, B.P.; Figueora, J.A.L.; Choera, T.; Niu, M.; Steinberger, A.J.; Wüthrich, M.; et al. Aspergillus fumigatus copper export machinery and reactive oxygen intermediate defense counter host copper-mediated oxidative antimicrobial offense. Cell Rep. 2017, 19, 1008–1021. [Google Scholar] [CrossRef] [PubMed]
- Linde, J.; Hortschansky, P.; Fazius, E.; Brakhage, A.A.; Guthke, R.; Haas, H. Regulatory interactions for iron homeostasis in aspergillus fumigatus inferred by a systems biology approach. BMC Syst. Biol. 2012, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Brandon, M.; Howard, B.; Lawrence, C.; Laubenbacher, R. Iron acquisition and oxidative stress response in Aspergillus fumigatus. BMC Syst. Boil. 2015, 9, 19. [Google Scholar] [CrossRef] [PubMed]
- Kurucz, V.; Krüger, T.; Antal, K.; Dietl, A.M.; Haas, H.; Pócsi, I.; Kniemeyer, O.; Emri, T. Additional oxidative stress reroutes the global response of Aspergillus fumigatus to iron depletion. BMC Genom. 2018, 19, 357. [Google Scholar] [CrossRef] [PubMed]
- Braymer, J.J.; Lill, R. Iron-sulfur cluster biogenesis and trafficking in mitochondria. J Biol. Chem. 2017, 292, 12754–12763. [Google Scholar] [CrossRef] [PubMed]
- Kroll, K.; Shekhova, E.; Mattern, D.J.; Thywissen, A.; Jacobsen, I.D.; Strassburger, M.; Heinekamp, T.; Shelest, E.; Brakhage, A.A.; Kniemeyer, O. The hypoxia-induced dehydrogenase HorA is required for coenzyme Q10 biosynthesis, azole sensitivity and virulence of Aspergillus fumigatus. Mol. Microbiol. 2016, 101, 92–108. [Google Scholar] [CrossRef] [PubMed]
- Ismail, T.; Fatima, N.; Muhammad, S.A.; Zaidi, S.S.; Rehman, N.; Hussain, I.; Tariq, N.U.S.; Amirzada, I.; Mannan, A. Prioritizing and modelling of putative drug target proteins of Candida albicans by systems biology approach. Acta Biochim. Pol. 2018, 65, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Müller, C.; Binder, U.; Maurer, E.; Grimm, C.; Giera, M.; Bracher, F. Fungal sterol C22-desaturase is not an antimycotic target as shown by selective inhibitors and testing on clinical isolates. Steroids 2015, 101, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Santos, A.; von Mering, C.; Jensen, L.J.; Bork, P.; Kuhn, M. STITCH 5: Augmenting protein-chemical interaction networks with tissue and affinity data. Nucleic Acids Res. 2016, 44, D380–D384. [Google Scholar] [CrossRef] [PubMed]
- Khedr, M.A.; Massarotti, A.; Mohamed, M.E. Rational Discovery of (+) (S) Abscisic Acid as a Potential Antifungal Agent: A Repurposing Approach. Sci. Rep. 2018, 8, 8565. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.P.; Rees, S.; Kalindjian, S.B.; Philpott, K.L. Principles of Early Drug Discovery. Br. J. Pharmacol. 2011, 162, 1239–1249. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Adelstein, S.J.; Kassis, A.I. Target discovery from data mining approaches. Drug Discov. Today 2009, 14, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Marton, M.J.; DeRisi, J.L.; Bennett, H.A.; Iyer, V.R.; Meyer, M.R.; Roberts, C.J.; Stoughton, R.; Burchard, J.; Slade, D.; Dai, H.; et al. Drug target validation and identification of secondary drug target effects using DNA microarrays. Nat. Med. 1998, 4, 1293–1301. [Google Scholar] [CrossRef] [PubMed]
- Joachimiak, M.P.; Chang, C.; Rosenthal, P.J.; Cohen, F.E. The impact of whole genome sequence data on drug discovery-a malaria case study. Mol. Med. 2001, 7, 698–710. [Google Scholar] [PubMed]
- Walker, T.M.; Kohl, T.A.; Omar, S.V.; Hedge, J.; Elias, C.D.O.; Bradley, P.; Iqbal, Z.; Feuerriegel, S.; Niehaus, K.E.; Wilson, D.J.; et al. Whole-genome sequencing for prediction of Mycobacterium tuberculosis drug susceptibility and resistance: A retrospective cohort study. Lancet Infect. Dis. 2015, 15, 1193–1202. [Google Scholar] [CrossRef]
- Katsila, T.; Spyroulias, G.A.; Patrinos, G.P.; Matsoukas, M.T. Computational Approaches in Target Identification and Drug Discovery. Comput. Struct. Biotechnol. J. 2016, 14, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Imoto, S.; Tamada, Y.; Savoie, C.J.; Miyanoaa, S. Analysis of gene networks for drug target discovery and validation. Methods Mol. Biol. 2007, 360, 33–56. [Google Scholar] [PubMed]
- Jamshidi, N.; Palsson, B.Ø. Investigating the metabolic capabilities of Mycobacterium tuberculosis H37Rv using the in silico strain iNJ 661 and proposing alternative drug targets. BMC Syst. Boil. 2017, 1, 26. [Google Scholar]
- Denayer, T.; Stöhr, T.; Van Roy, M. Animal models in translational medicine: Validation and prediction. New Horiz. Transl. Med. 2014, 2, 5–11. [Google Scholar] [CrossRef]
- Andricopulo, A.D.; Salum, L.B.; Abraham, D.J. Structure-based drug design strategies in medicinal chemistry. Curr. Top. Med. Chem. 2009, 9, 771–790. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Lai, L. Towards structure-based protein drug design. Biochem. Soc. Trans. 2011, 39, 1382–1386. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.G.; dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular docking and structure-based drug design strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef] [PubMed]
- Mandal, S.; Moudgil, M.N.; Mandal, S.K. Rational drug design. Eur. J. Pharmacol. 2009, 625, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Biasini, M.; Bienert, S.; Waterhouse, A.; Arnold, K.; Studer, G.; Schmidt, T.; Kiefer, F.; Gallo Cassarino, T.; Bertoni, M.; Bordoli, L.; et al. SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014, 42, W252–W258. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Molecular Operating Environment (MOE). 2013.08. Chemical Computing Group ULC, 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7. 2018. Available online: https://www.chemcomp.com/announcements/2013-10-23-MOE2013.08.pdf (accessed on 3 July 2018).
- Šali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraint. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. Cambridge Structural Database. Acta Cryst. 2016, B72, 171–179. [Google Scholar]
- Chandrika, B.R.; Subramanian, J.; Sharma, S.D. Managing protein flexibility in docking and its applications. Drug Discov. Today 2009, 14, 394–400. [Google Scholar]
- Durrant, J.D.; McCammon, J.A. Computer-aided drug-discovery techinques that account for receptor flexibility. Curr. Opin. Pharmacol. 2010, 10, 770–774. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.Y.; Zou, X. Advances and challenges in protein–ligand docking. Int. J. Mol. Sci. 2010, 11, 3016–3034. [Google Scholar] [CrossRef] [PubMed]
- Harvey, M.J.; De Fabritiis, G. High-throughput molecular dynamics: The powerful new tool for drug discovery. Drug Discov. Today 2012, 17, 1059–1062. [Google Scholar] [CrossRef] [PubMed]
- Alonso, H.; Bliznyuk, A.A.; Gready, J.E. Combining docking and molecular dynamic simulations in drug design. Med. Res. Rev. 2006, 26, 531–568. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C. Beware of docking! Trends Pharmacol. Sci. 2015, 36, 78–95. [Google Scholar] [CrossRef] [PubMed]
- Watson, K.A.; Chrysina, E.D.; Tsitsanou, K.E.; Zographos, S.E.; Archontis, G.; Fleet, G.W.; Oikonomakos, N.G. Kinetic and crystallographic studies of glucopyranose spirohydantoin and glucopyranosylamine analogs inhibitors of glycogen phosphorylase. Proteins 2005, 61, 966–983. [Google Scholar] [CrossRef] [PubMed]
- Wasko, M.J.; Pellegrene, K.A.; Madura, J.D.; Surratt, C.K. A role of fragment-based drug design in developing novel lead compounds for central nervous system targets. Front. Neurol. 2015, 6, 197. [Google Scholar] [CrossRef] [PubMed]
- Goodford, P.J. A Computational Procedure for Determining Energetically Favorable Binding Sites on Biologically Important Macromolecules. J. Med. Chem. 1985, 28, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Miranker, A.; Karplus, M. Functionality maps of binding sites: A multiple copy simultaneous search method. Proteins Struct. Funct. Bioinform. 1991, 11, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Böhm, H.J. The computer program LUDI: A new method for the de novo design of enzyme inhibitors. J. Comput. Aided. Mol. Des. 1992, 6, 61–78. [Google Scholar] [CrossRef] [PubMed]
- Gillet, V.; Johnson, A.P.; Mata, P.; Sike, S.; Williams, P. SPROUT: A program for structure generation. J. Comput. Aided. Mol. Des. 1993, 7, 127–153. [Google Scholar] [CrossRef] [PubMed]
- Verdonk, M.L.; Cole, J.C.; Taylor, R. SuperStar: A knowledge-based approach for identifying interaction sites in proteins. J. Mol. Biol. 1999, 289, 1093–1108. [Google Scholar] [CrossRef] [PubMed]
- Carlson, H.A.; Masukawa, K.M.; Rubins, K.; Bushman, F.D.; Jorgensen, W.L.; Lins, R.D.; Briggs, J.M.; McCammon, J.A. Developing a dynamic pharmacophore model for HIV-1 integrase. J. Med. Chem. 2000, 43, 2100–2114. [Google Scholar] [CrossRef] [PubMed]
- Shang, E.; Yuan, Y.; Chen, X.; Liu, Y.; Pei, J.; Lai, L. De novo design of multitarget ligands with an iterative fragment-growing strategy. J. Chem. Inf. Model. 2014, 54, 1235–1241. [Google Scholar] [CrossRef] [PubMed]
- Durrant, J.D.; Amaro, R.E.; McCammon, J.A. AutoGrow: A novel algorithm for protein inhibitor design. Chem. Biol. Drug Des. 2009, 73, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Lionta, E.; Spyrou, G.; K Vassilatis, D.; Cournia, Z. Structure-based virtual screening for drug discovery: Principles, applications and recent advances. Curr. Top. Med. Chem. 2014, 14, 1923–1938. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. ZINC: A free tool to discover chemistry for biology. J. Chem. Inf. Model. 2012, 52, 1757–1768. [Google Scholar] [CrossRef] [PubMed]
- Pence, H.E.; Williams, A. ChemSpider: An online chemical information resource. J. Chem. Educ. 2010, 87, 1123–1124. [Google Scholar] [CrossRef]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. DrugBank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34, D668–DD672. [Google Scholar] [CrossRef] [PubMed]
- Kunz, M.; Liang, C.; Nilla, S.; Cecil, A.; Dandekar, T. The drug-minded protein interaction database (DrumPID) for efficient target analysis and drug development. Database 2016, 2016, Baw041. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, P.; Bhalla, S.; Chaudhary, K.; Kumar, R.; Sharma, M.; Raghava, G.P.S. In Silico Approach for Prediction of Antifungal Peptides. Front. Microbiol. 2018, 9, 323. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Ni, T.; Chai, X.; Wang, T.; Wang, H.; Chen, J.; Jin, Y.; Zhang, D.; Yu, S.; Jiang, Y. Molecular docking, design, synthesis and antifungal activity study of novel triazole derivatives. Eur. J. Med. Chem. 2018, 143, 1840–1846. [Google Scholar] [CrossRef] [PubMed]
- Dühring, S.; Ewald, J.; Germerodt, S.; Kaleta, C.; Dandekar, T.; Schuster, S. Modelling the host–pathogen interactions of macrophages and Candida albicans using Game Theory and dynamic optimization. J. R. Soc. Interface 2017, 14, 20170095. [Google Scholar] [CrossRef] [PubMed]
- Hummert, S.; Glock, C.; Lang, S.N.; Hummert, C.; Skerka, C.; Zipfel, P.F.; Germerodt, S.; Schuster, S. Playing ‘hide-and-seek’ with factor H: Game-theoretical analysis of a single nucleotide polymorphism. J. R. Soc. Interface 2018, 15, 20170963. [Google Scholar] [CrossRef] [PubMed]
- Schelenz, S.; Hagen, F.; Rhodes, J.L.; Abdolrasouli, A.; Chowdhary, A.; Hall, A.; Ryan, L.; Shackleton, J.; Trimlett, R.; Meis, J.F.; et al. First hospital outbreak of the globally emerging Candida auris in a European hospital. Antimicrob. Resist. Infect. Control. 2016, 5, 35. [Google Scholar] [CrossRef] [PubMed]
- Cavalcante, C.S.P.; de Aguiar, F.L.L.; Fontenelle, R.O.; de Menezes, R.R.D.P.; Martins, A.M.C.; Falcão, C.B.; Andreu, D.; Rádis-Baptista, G. Insights into the candidacidal mechanism of Ctn[15-34]—A carboxyl-terminal, crotalicidin-derived peptide related to cathelicidins. J. Med. Microbiol. 2018, 67, 129–138. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Host Species | Agent | Diseases | Description | Treatment |
|---|---|---|---|---|
| Honeybee (Apis mellifera) | Ascosphaera apis | Chalkbrood | Fungus affects the gut of larvae. Outcompetes larvae for nutrition and turns the larvae into “chalk-like” mummies. Very infectious. | Apiguard, thymol-based treatment |
| Honeybee (Apis mellifera) | A. fumigatus, A. flavus, A. niger | Stonebrood | The fungus causing the green or yellow mummification of larvae. Very infectious but not common. | No treatment |
| All mammals, birds | A. fumigatus, A. flavus, A. nidulans, A. niger | Aspergillosis, Keratomycosis (horses) | Fungal infection attack mostly disease-weakened pets, disease or drug therapy is possible. The nasal and pulmonary form is most typical. | Ketoconazole, Itraconazole, Fluconazole |
| Dogs, horses (rarely cats) | Blastomyces dermatitis | Blastomycosis | Systematic mycosis described in humans, dogs, and horses, endemic in North America. | Amphotericin B and ketoconazole (dogs), no treatment for horses |
| Horses, mules, donkey, cattle | Histoplasma farciminosum | Epizootic Lymphangitis | The chronic fungal disease causes inflammation and suppuration of the cutaneous and subcutaneous lymphatic vessels and glands. | No treatment (only surgical excision of all affected nodes) |
| All mammals, birds | C. albicans, C. fumata | Candidiasis | Mucocutaneous disease infecting mostly the nasopharynx, gastrointestinal tract and external genitalia. Often occurs in birds, in cats candidiasis is rare. Cause of arthritis in horses and mastitis in cattle. | Amphotericin B, Fluconazole, Chlorhexidine (birds) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bencurova, E.; Gupta, S.K.; Sarukhanyan, E.; Dandekar, T. Identification of Antifungal Targets Based on Computer Modeling. J. Fungi 2018, 4, 81. https://doi.org/10.3390/jof4030081
Bencurova E, Gupta SK, Sarukhanyan E, Dandekar T. Identification of Antifungal Targets Based on Computer Modeling. Journal of Fungi. 2018; 4(3):81. https://doi.org/10.3390/jof4030081
Chicago/Turabian StyleBencurova, Elena, Shishir K. Gupta, Edita Sarukhanyan, and Thomas Dandekar. 2018. "Identification of Antifungal Targets Based on Computer Modeling" Journal of Fungi 4, no. 3: 81. https://doi.org/10.3390/jof4030081
APA StyleBencurova, E., Gupta, S. K., Sarukhanyan, E., & Dandekar, T. (2018). Identification of Antifungal Targets Based on Computer Modeling. Journal of Fungi, 4(3), 81. https://doi.org/10.3390/jof4030081