Fungal Strategies to Evade the Host Immune Recognition

 ,
,  and
and 
Abstract
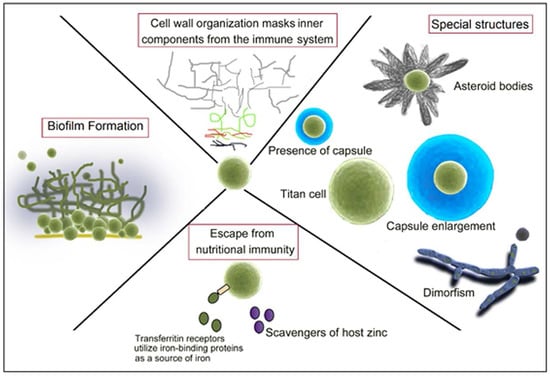
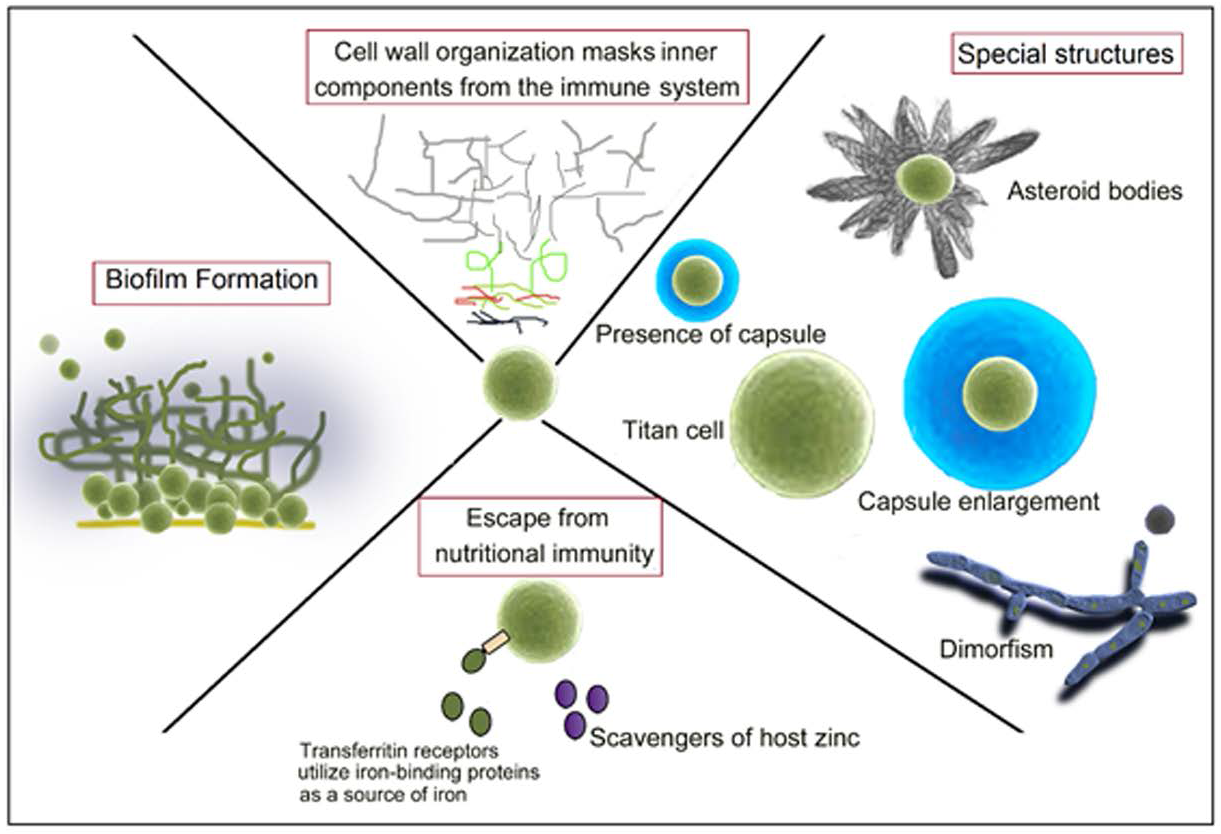
1. Introduction
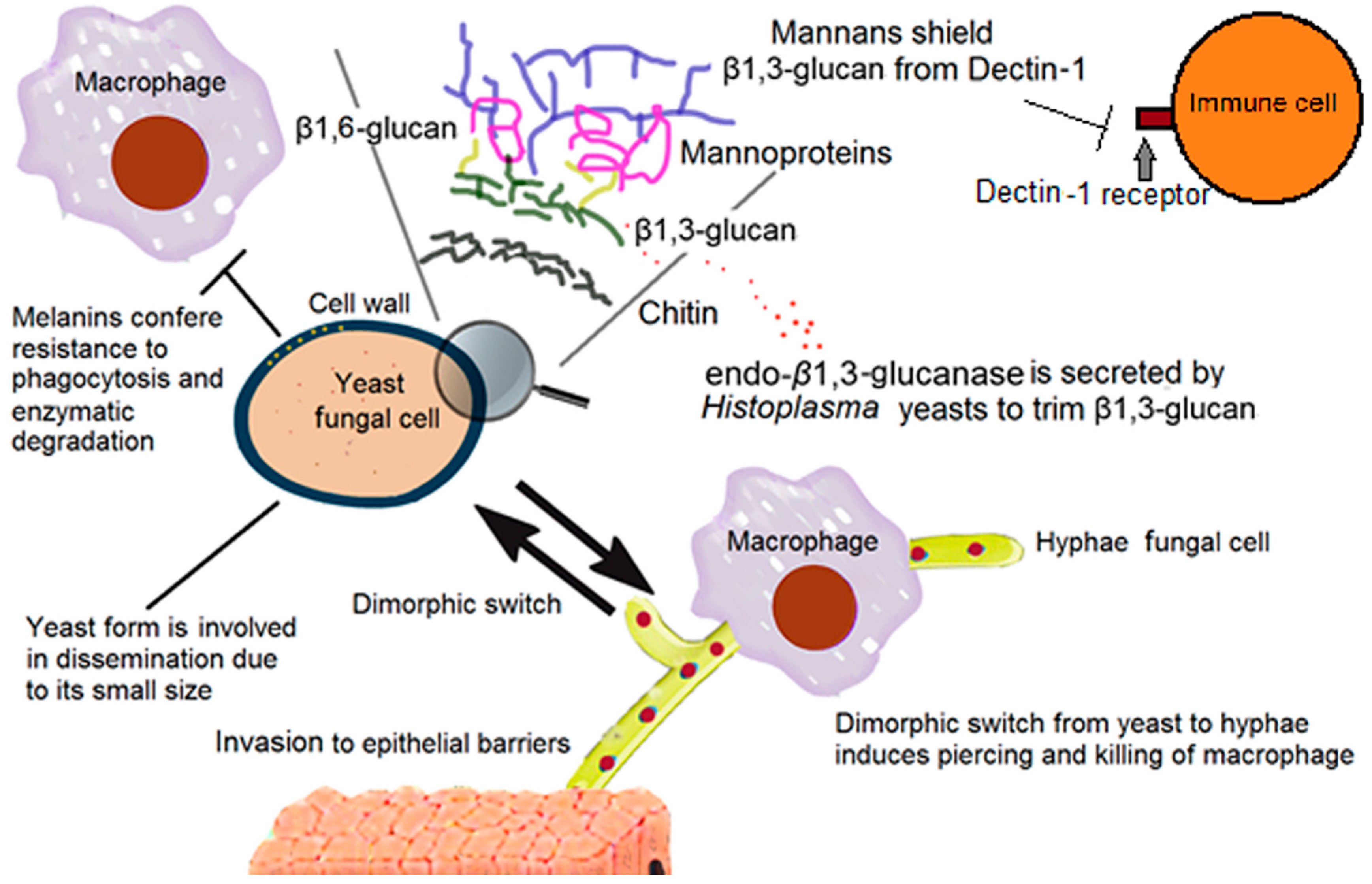
2. The Fungal Cell Wall: Composition and Organization
2.1. The Support: Polysaccharides
2.2. Other Cell Wall Components
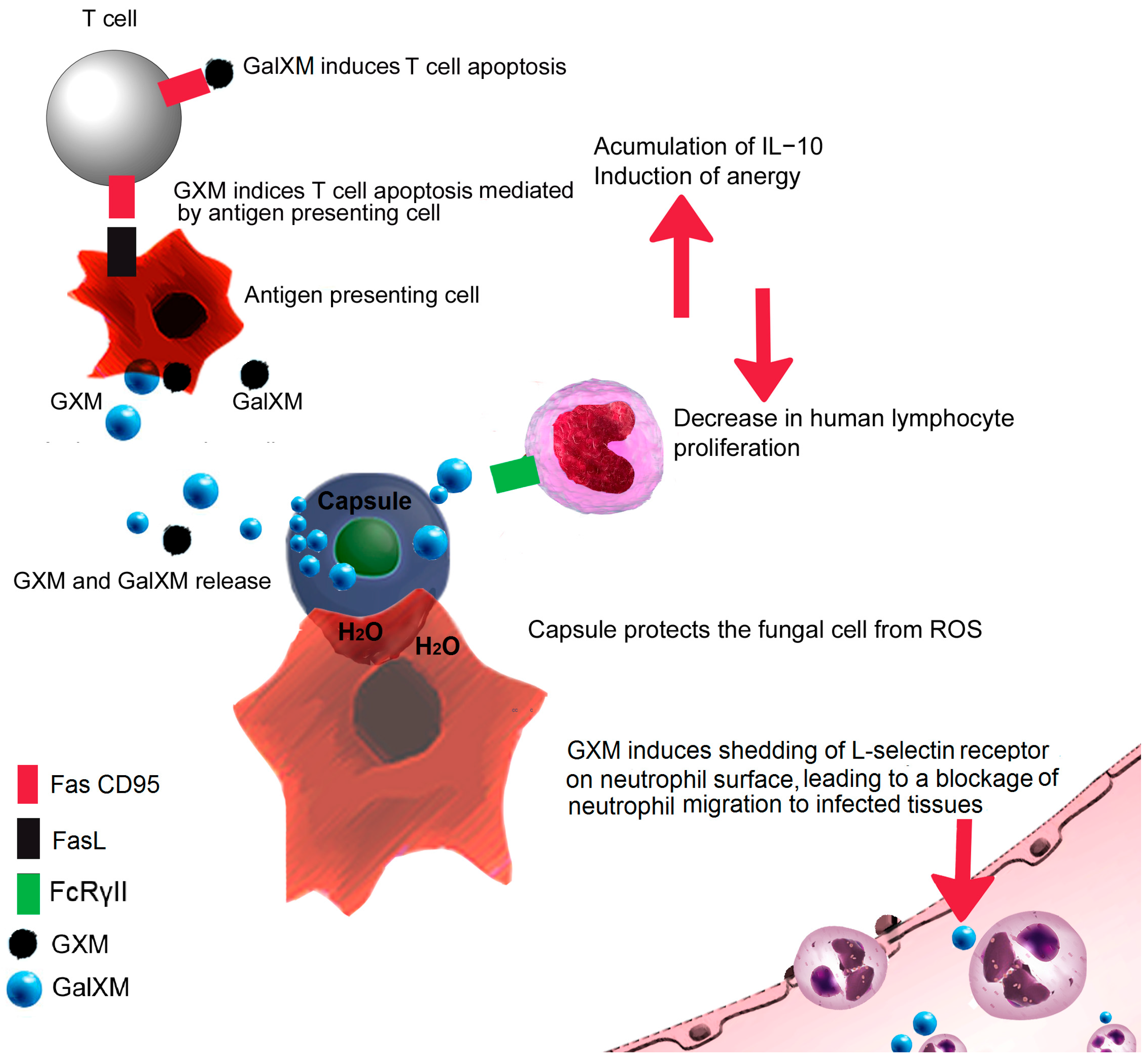
3. Fungal Capsule
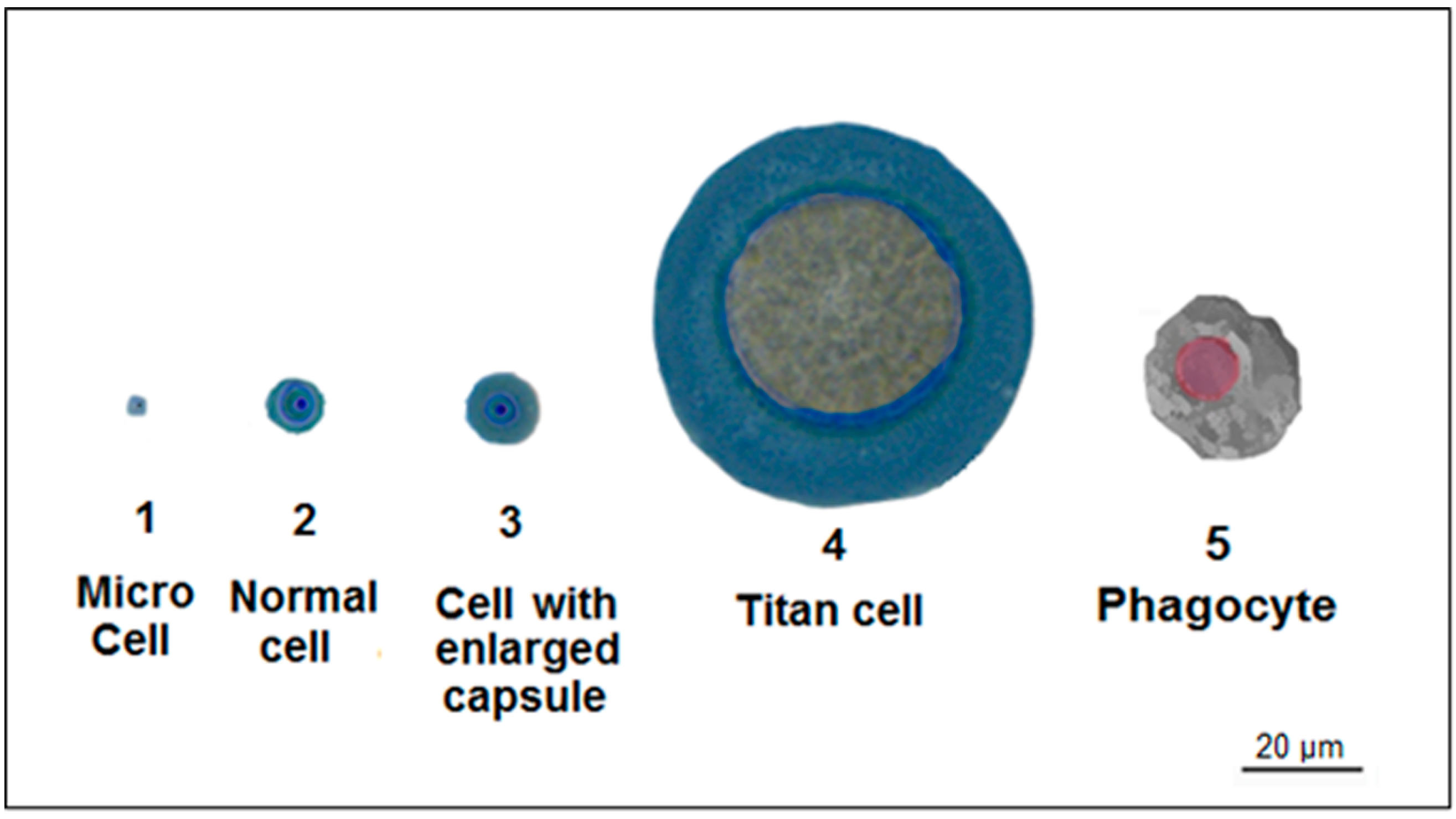
4. Titan Cells
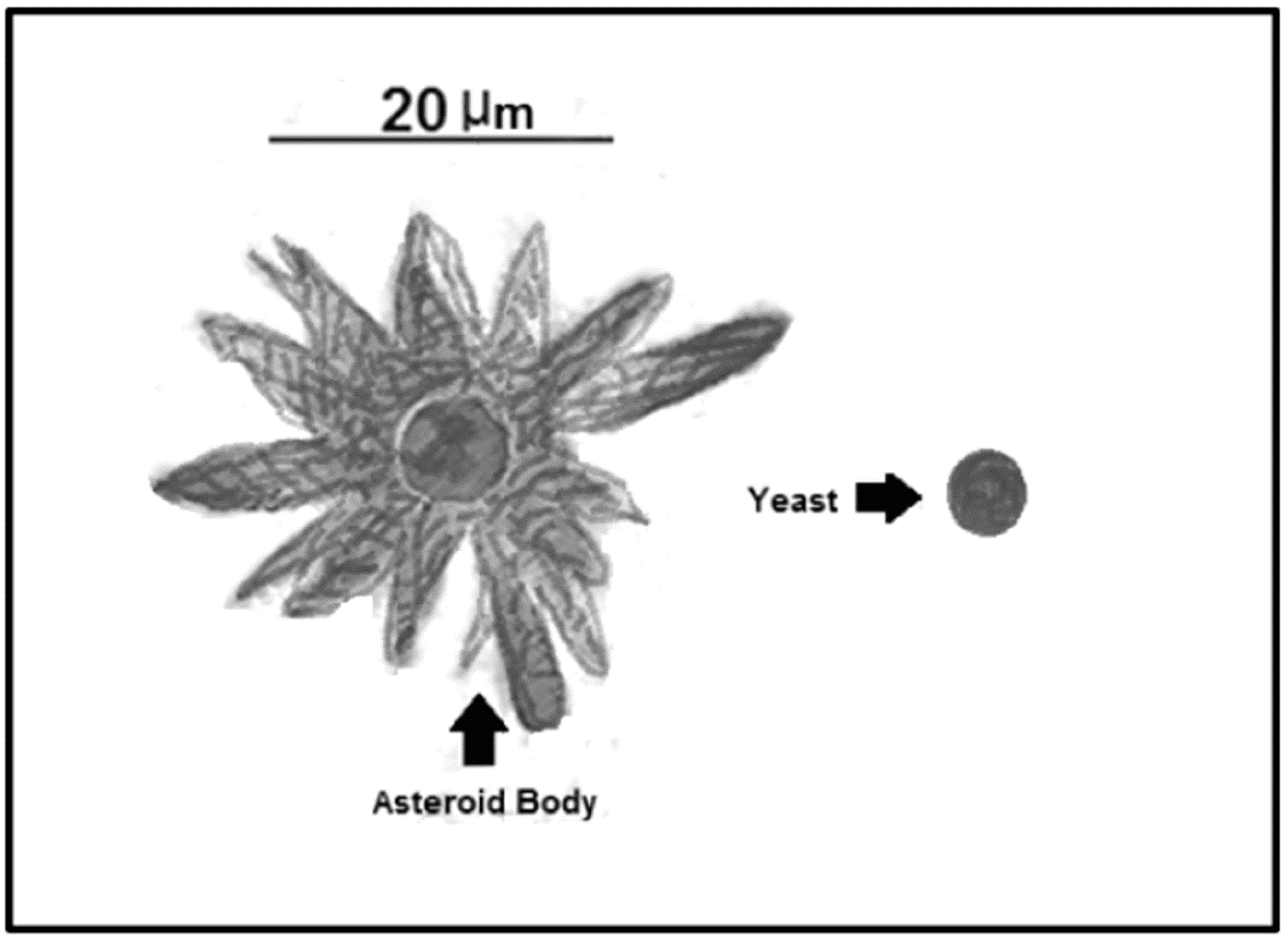
5. Asteroid Bodies
6. Fungal Dimorphism
7. Overcoming Phagocytosis
8. Deceiving the Humoral Immune Response
9. Are You Trapped? Be Like Cryptococcus!
10. Overcoming Ion Starvation or Nutritional Immunity
11. The Role of Fungal Biofilm Formation during Interaction with the Host Immune Response
12. Other Fungal Strategies to Avoid Immune Sensing
13. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Brown, G.D.; Denning, D.W.; Gow, N.A.; Levitz, S.M.; Netea, M.G.; White, T.C. Hidden killers: Human fungal infections. Sci. Transl. Med. 2012, 4, 165rv13. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Jiménez, D.F.; Pérez-García, L.A.; Martínez-Álvarez, J.A.; Mora-Montes, H.M. Role of the fungal cell wall in pathogenesis and antifungal resistance. Curr. Fungal. Infect. Rep. 2012, 6, 275–282. [Google Scholar] [CrossRef]
- Erwig, L.P.; Gow, N.A. Interactions of fungal pathogens with phagocytes. Nat. Rev. Microbiol. 2016, 14, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Coronado, J.E.; Mneimneh, S.; Epstein, S.L.; Qiu, W.G.; Lipke, P.N. Conserved processes and lineage-specific proteins in fungal cell wall evolution. Eukaryot. Cell 2007, 6, 2269–2277. [Google Scholar] [CrossRef] [PubMed]
- Gow, N.A.R.; Latge, J.P.; Munro, C.A. The Fungal Cell Wall: Structure, Biosynthesis, and Function. Microbiol. Spectr. 2017, 5, 5. [Google Scholar] [CrossRef] [PubMed]
- Latge, J.P.; Beauvais, A.; Chamilos, G. The Cell Wall of the Human Fungal Pathogen Aspergillus fumigatus: Biosynthesis, Organization, Immune Response, and Virulence. Annu. Rev. Microbiol. 2017, 71, 99–116. [Google Scholar] [CrossRef] [PubMed]
- Mora-Montes, H.M.; Ponce-Noyola, P.; Villagómez-Castro, J.C.; Gow, N.A.R.; Flores-Carreón, A.; López-Romero, E. Protein glycosylation in Candida. Futur. Microbiol. 2009, 4, 1167–1183. [Google Scholar] [CrossRef] [PubMed]
- Latge, J.P. Tasting the fungal cell wall. Cell Microbiol. 2010, 12, 863–872. [Google Scholar] [CrossRef] [PubMed]
- Camacho, E.; Nino-Vega, G.A. Paracoccidioides spp.: Virulence Factors and Immune-Evasion Strategies. Mediat. Inflamm. 2017, 2017, 5313691. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.D.; Gordon, S. Immune recognition. A new receptor for β-glucans. Nature 2001, 413, 36–37. [Google Scholar] [CrossRef] [PubMed]
- Gantner, B.N.; Simmons, R.M.; Canavera, S.J.; Akira, S.; Underhill, D.M. Collaborative induction of inflammatory responses by dectin-1 and Toll-like receptor 2. J. Exp. Med. 2003, 197, 1107–1117. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.R.; Tsoni, S.V.; Willment, J.A.; Dennehy, K.M.; Rosas, M.; Findon, H.; Haynes, K.; Steele, C.; Botto, M.; Gordon, S.; et al. Dectin-1 is required for β-glucan recognition and control of fungal infection. Nat. Immunol. 2007, 8, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Gow, N.A.R.; Netea, M.G.; Munro, C.A.; Ferwerda, G.; Bates, S.; Mora-Montes, H.M.; Walker, L.; Jansen, T.; Jacobs, L.; Tsoni, V.; et al. Immune recognition of Candida albicans beta-glucan by dectin-1. J. Infect. Dis. 2007, 196, 1565–1571. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, R.T.; Fink, G.R. A drug-sensitive genetic network masks fungi from the immune system. PLoS Pathog. 2006, 2, e35. [Google Scholar] [CrossRef] [PubMed]
- West, L.; Lowman, D.W.; Mora-Montes, H.M.; Grubb, S.; Murdoch, C.; Thornhill, M.H.; Gow, N.A.; Williams, D.; Haynes, K. Differential virulence of Candida glabrata glycosylation mutants. J. Biol. Chem. 2013, 288, 22006–22018. [Google Scholar] [CrossRef] [PubMed]
- Estrada-Mata, E.; Navarro-Arias, M.J.; Perez-Garcia, L.A.; Mellado-Mojica, E.; Lopez, M.G.; Csonka, K.; Gacser, A.; Mora-Montes, H.M. Members of the Candida parapsilosis complex and Candida albicans are differentially recognized by human peripheral blood mononuclear cells. Front. Microbiol. 2015, 6, 1527. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Arias, M.J.; Defosse, T.A.; Dementhon, K.; Csonka, K.; Mellado-Mojica, E.; Dias Valerio, A.; Gónzaez-Hernández, R.J.; Courdavault, V.; Clastre, M.; Hernández, N.V.; et al. Disruption of protein mannosylation affects Candida guilliermondii cell wall, immune sensing, and virulence. Front. Microbiol. 2016, 7, 1951. [Google Scholar] [CrossRef] [PubMed]
- Perez-Garcia, L.A.; Csonka, K.; Flores-Carreon, A.; Estrada-Mata, E.; Mellado-Mojica, E.; Nemeth, T.; López-Ramírez, L.A.; Toth, R.; López, M.G.; Vizler, C.; et al. Role of protein glycosylation in Candida parapsilosis cell wall integrity and host interaction. Front. Microbiol. 2016, 7, 306. [Google Scholar]
- Mora-Montes, H.M.; Bates, S.; Netea, M.G.; Castillo, L.; Brand, A.; Buurman, E.T.; Díaz-Jiménez, D.F.; Jan Kullberg, B.; Brwon, A.J.; Odds, F.C.; et al. A multifunctional mannosyltransferase family in Candida albicans determines cell wall mannan structure and host-fungus interactions. J. Biol. Chem. 2010, 285, 12087–12095. [Google Scholar] [CrossRef] [PubMed]
- Mora-Montes, H.M.; Bates, S.; Netea, M.G.; Diaz-Jimenez, D.F.; Lopez-Romero, E.; Zinker, S.; Ponce-Noyola, P.; Kullberg, B.; Brown, A.J.; Odds, F.C.; et al. Endoplasmic reticulum alpha-glycosidases of Candida albicans are required for N glycosylation, cell wall integrity, and normal host-fungus interaction. Eukaryot. Cell 2007, 6, 2184–2193. [Google Scholar] [CrossRef] [PubMed]
- Rappleye, C.A.; Eissenberg, L.G.; Goldman, W.E. Histoplasma capsulatum alpha-(1,3)-glucan blocks innate immune recognition by the beta-glucan receptor. Proc. Natl. Acad. Sci. USA 2007, 104, 1366–1370. [Google Scholar] [CrossRef] [PubMed]
- Marion, C.L.; Rappleye, C.A.; Engle, J.T.; Goldman, W.E. An alpha-(1,4)-amylase is essential for alpha-(1,3)-glucan production and virulence in Histoplasma capsulatum. Mol. Microbiol. 2006, 62, 970–983. [Google Scholar] [CrossRef] [PubMed]
- Garfoot, A.L.; Shen, Q.; Wuthrich, M.; Klein, B.S.; Rappleye, C.A. The Eng1 β-Glucanase Enhances Histoplasma Virulence by Reducing β-Glucan Exposure. MBio 2016, 7, 7. [Google Scholar] [CrossRef] [PubMed]
- Nosanchuk, J.D.; Casadevall, A. Impact of melanin on microbial virulence and clinical resistance to antimicrobial compounds. Antimicrob. Agents Chemother. 2006, 50, 3519–3528. [Google Scholar] [CrossRef] [PubMed]
- Nosanchuk, J.D.; Stark, R.E.; Casadevall, A. Fungal Melanin: What do We Know About Structure? Front. Microbiol. 2015, 6, 1463. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Hartmann, A.; Dahse, H.-M.; Skerka, C.; Zipfel, P.F. Secreted pH-Regulated Antigen 1 of Candida albicans Blocks Activation and Conversion of Complement C3. J. Immunol. 2010, 185, 2164–2173. [Google Scholar] [CrossRef] [PubMed]
- Zipfel, P.F.; Würzner, R.; Skerka, C. Complement evasion of pathogens: Common strategies are shared by diverse organisms. Mol. Immunol. 2007, 44, 3850–3857. [Google Scholar] [CrossRef] [PubMed]
- Knight, S.A.B.; Vilaire, G.; Lesuisse, E.; Dancis, A. Iron Acquisition from Transferrin by Candida albicans Depends on the Reductive Pathway. Infect. Immun. 2005, 73, 5482–5492. [Google Scholar] [CrossRef] [PubMed]
- Almeida, R.S.; Brunke, S.; Albrecht, A.; Thewes, S.; Laue, M.; Edwards, J.E., Jr.; Filler, S.G.; Hube, B. The Hyphal-Associated Adhesin and Invasin Als3 of Candida albicans Mediates Iron Acquisition from Host Ferritin. PLoS Pathog. 2008, 4, e1000217. [Google Scholar] [CrossRef] [PubMed]
- Gropp, K.; Schild, L.; Schindler, S.; Hube, B.; Zipfel, P.F.; Skerka, C. The yeast Candida albicans evades human complement attack by secretion of aspartic proteases. Mol. Immunol. 2009, 47, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Behnsen, J.; Lessing, F.; Schindler, S.; Wartenberg, D.; Jacobsen, I.D.; Thoen, M.; Zipfel, P.F.; Brakhage, A.A. Secreted Aspergillus fumigatus Protease Alp1 Degrades Human Complement Proteins C3, C4, and C5. Infect. Immun. 2010, 78, 3585–3594. [Google Scholar] [CrossRef] [PubMed]
- Wintergerst, E.S.; Maggini, S.; Hornig, D.H. Contribution of Selected Vitamins and Trace Elements to Immune Function. Ann. Nutr. Metab. 2007, 51, 301–323. [Google Scholar] [CrossRef] [PubMed]
- Kehl-Fie, T.E.; Skaar, E.P. Nutritional immunity beyond iron: A role for manganese and zinc. Curr. Opin. Chem. Biol. 2010, 14, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Bruns, S.; Kniemeyer, O.; Hasenberg, M.; Aimanianda, V.; Nietzsche, S.; Thywißen, A.; Jeron, A.; Lartgé, J.P.; Brakhage, A.A.; Gunzer, M. Production of Extracellular Traps against Aspergillus fumigatus In Vitro and in Infected Lung Tissue Is Dependent on Invading Neutrophils and Influenced by Hydrophobin RodA. PLoS Pathog. 2010, 6. [Google Scholar] [CrossRef]
- Dague, E.; Alsteens, D.; Latgé, J.P.; Dufrêne, Y.F. High-Resolution Cell Surface Dynamics of Germinating Aspergillus fumigatus Conidia. Biophys. J. 2008, 94, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, C.A.; Chalouni, C.; Williams, A.; Hartl, D.; Lee, C.G.; Elias, J.A. Chitin is a size-dependent regulator of macrophage TNF and IL-10 production. J. Immunol. 2009, 182, 3573–3582. [Google Scholar] [CrossRef] [PubMed]
- Wagener, J.; Malireddi, R.K.; Lenardon, M.D.; Koberle, M.; Vautier, S.; MacCallum, D.M.; Biedermann, T.; Schaller, M.; Netea, M.G.; Kanneganti, T.D.; et al. Fungal chitin dampens inflammation through IL-10 induction mediated by NOD2 and TLR9 activation. PLoS Pathog. 2014, 10, e1004050. [Google Scholar] [CrossRef] [PubMed]
- Mora-Montes, H.M.; Netea, M.G.; Ferwerda, G.; Lenardon, M.D.; Brown, G.D.; Mistry, A.R.; Kullberg, B.J.; O’Callaghan, C.A.; Sheth, C.C.; Odds, F.C.; et al. Recognition and Blocking of Innate Immunity Cells by Candida albicans Chitin. Infect. Immun. 2011, 79, 1961–1970. [Google Scholar] [CrossRef] [PubMed]
- Eisenman, H.C.; Mues, M.; Weber, S.E.; Frases, S.; Chaskes, S.; Gerfen, G.; Casadevall, A. Cryptococcus neoformans laccase catalyses melanin synthesis from both d- and l-DOPA. Microbiology 2007, 153, 3954–3962. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Wakamatsu, K. Quantitative Analysis of Eumelanin and Pheomelanin in Humans, Mice, and Other Animals: A Comparative Review. Pigment. Cell Res. 2003, 16, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, E.S. Pathogenic Roles for Fungal Melanins. Clin. Microbiol. Rev. 2000, 13, 708–717. [Google Scholar] [CrossRef] [PubMed]
- Eisenman, H.C.; Casadevall, A. Synthesis and assembly of fungal melanin. Appl. Microbiol. Biotechnol. 2012, 93, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.J.; Day, A.W. Fungal melanins: A review. Can. J. Microbiol. 1998, 44, 1115–1136. [Google Scholar] [CrossRef]
- Langfelder, K.; Streibel, M.; Jahn, B.; Haase, G.; Brakhage, A.A. Biosynthesis of fungal melanins and their importance for human pathogenic fungi. Fungal. Genet. Biol. 2003, 38, 143–158. [Google Scholar] [CrossRef]
- Almeida-Paes, R.; Frases, S.; Monteiro, P.C.F.; Gutierrez-Galhardo, M.C.; Zancopé-Oliveira, R.M.; Nosanchuk, J.D. Growth conditions influence melanization of brazilian clinical Sporothrix schenckii isolates. Microbes Infect. 2009, 11, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Morris-Jones, R.; Gomez, B.L.; Diez, S.; Uran, M.; Morris-Jones, S.D.; Casadevall, A.; Nosanchuk, J.D.; Hamilton, A.J.; et al. Synthesis of melanin pigment by Candida albicans in vitro and during Infection. Infect. Immun. 2005, 73, 6147–6150. [Google Scholar] [CrossRef] [PubMed]
- Walker, C.A.; Gómez, B.L.; Mora-Montes, H.M.; Mackenzie, K.S.; Munro, C.A.; Brown, A.J.P.; Gow, N.A.; Kibbler, C.C.; Odds, F.C. Melanin externalization in Candida albicans depends on cell wall chitin structures. Eukaryot. Cell 2010, 9, 1329–1342. [Google Scholar] [CrossRef] [PubMed]
- Thywißen, A.; Heinekamp, T.; Dahse, H.-M.; Schmaler-Ripcke, J.; Nietsche, S.; Zipfel, P.; Brakhage, A.A. Conidial dihydroxynaphthalene melanin of the human pathogenic fungus Aspergillus fumigatus interferes with the host endocytosis pathway. Front. Microbiol. 2011, 2, 96. [Google Scholar] [CrossRef] [PubMed]
- Paris, S.; Debeaupuis, J.P.; Crameri, R.; Carey, M.; Charles, F.; Prevost, M.C.; Schmitt, C.; Philippe, B.; Latgé, J.P. Conidial hydrophobins of Aspergillus fumigatus. Appl. Environ. Microbiol. 2003, 69, 1581–1588. [Google Scholar] [CrossRef] [PubMed]
- Aimanianda, V.; Bayry, J.; Bozza, S.; Kniemeyer, O.; Perruccio, K.; Elluru, S.R.; Clavaud, C.; Paris, S.; Brakhage, A.A.; Kaveri, S.V.; et al. Surface hydrophobin prevents immune recognition of airborne fungal spores. Nature 2009, 460, 1117–1121. [Google Scholar] [CrossRef] [PubMed]
- Roberts, I.S.; Saunders, F.K.; Boulnois, G.J. Bacterial capsules and interactions with complement and phagocytes. Biochem. Soc. Trans. 1989, 17, 462–464. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, C.A. Histoplasmosis: A clinical and laboratory update. Clin. Microbiol. Rev. 2007, 20, 115–132. [Google Scholar] [CrossRef] [PubMed]
- Doering, T.L. How sweet it is! Cell wall biogenesis and polysaccharide capsule formation in Cryptococcus neoformans. Annu. Rev. Microbiol. 2009, 63, 223–247. [Google Scholar] [PubMed]
- Vecchiarelli, A. Immunoregulation by capsular components of Cryptococcus neoformans. Med. Mycol. 2000, 38, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Zaragoza, O.; Rodrigues, M.L.; De Jesus, M.; Frases, S.; Dadachova, E.; Casadevall, A. The capsule of the fungal pathogen Cryptococcus neoformans. Adv. Appl. Microbiol. 2009, 68, 133–216. [Google Scholar] [PubMed]
- Syme, R.M.; Bruno, T.F.; Kozel, T.R.; Mody, C.H. The capsule of Cryptococcus neoformans reduces T-lymphocyte proliferation by reducing phagocytosis, which can be restored with anticapsular antibody. Infect. Immun. 1999, 67, 4620–4627. [Google Scholar] [PubMed]
- Monari, C.; Kozel, T.R.; Bistoni, F.; Vecchiarelli, A. Modulation of C5aR expression on human neutrophils by encapsulated and acapsular Cryptococcus neoformans. Infect. Immun. 2002, 70, 3363–3370. [Google Scholar] [CrossRef] [PubMed]
- Rocha, J.D.B.; Nascimento, M.T.C.; Decote-Ricardo, D.; Côrte-Real, S.; Morrot, A.; Heise, N.; Nunes, M.P.; Previato, J.O.; Mendonca-Previato, L.; DosReis, G.A.; et al. Capsular polysaccharides from Cryptococcus neoformans modulate production of neutrophil extracellular traps (NETs) by human neutrophils. Sci. Rep. 2015, 5, 409. [Google Scholar] [CrossRef]
- Zaragoza, O.; Nielsen, K. Titan cells in Cryptococcus neoformans: Cells with a giant impact. Curr. Opin. Microbiol. 2013, 16, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Okagaki, L.H.; Nielsen, K. Titan cells confer protection from phagocytosis in Cryptococcus neoformans infections. Eukaryot. Cell 2012, 11, 820–826. [Google Scholar] [CrossRef] [PubMed]
- Daniel Da Rosa, W.; Gezuele, E.; Calegari, L.; Goni, F. Asteroid body in sporotrichosis. Yeast viability and biological significance within the host immune response. Med. Mycol. 2008, 46, 443–448. [Google Scholar] [PubMed]
- Costerton, J.W.; Montanaro, L.; Arciola, C.R. Biofilm in implant infections: Its production and regulation. Int. J. Artif. Organs 2005, 28, 1062–1068. [Google Scholar] [PubMed]
- Lynch, A.S.; Robertson, G.T. Bacterial and Fungal Biofilm Infections. Ann. Rev. Med. 2008, 59, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Zelante, T.; Iannitti, R.G.; De Luca, A.; Arroyo, J.; Blanco, N.; Servillo, G.; Sanglard, D.; Reichard, U.; Palmer, G.E.; Latgé, J.P.; et al. Sensing of mammalian IL-17A regulates fungal adaptation and virulence. Nat. Commun. 2012, 3, 683. [Google Scholar] [CrossRef] [PubMed]
- Roilides, E.; Simitsopoulou, M.; Katragkou, A.; Walsh, T.J. How Biofilms Evade Host Defenses. Microbiol. Spectr. 2015, 3, 3. [Google Scholar] [CrossRef]
- Kreindler, J.L.; Steele, C.; Nguyen, N.; Chan, Y.R.; Pilewski, J.M.; Alcorn, J.F.; Vyas, Y.M.; Aujla, SJ.; Finelli, P.; Blanchard, M.; et al. Vitamin D3 attenuates Th2 responses to Aspergillus fumigatus mounted by CD4+ T cells from cystic fibrosis patients with allergic bronchopulmonary aspergillosis. J. Clin. Investig. 2010, 120, 3242–3254. [Google Scholar] [CrossRef] [PubMed]
- Feldmesser, M.; Kress, Y.; Novikoff, P.; Casadevall, A. Cryptococcus neoformans is a facultative intracellular pathogen in murine pulmonary infection. Infect. Immun. 2000, 68, 4225–4237. [Google Scholar] [CrossRef] [PubMed]
- Levitz, S.M.; Specht, C.A. The molecular basis for the immunogenicity of Cryptococcus neoformans mannoproteins. FEMS Yeast Res. 2006, 6, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.M.; Murphy, J.W. Cryptococcal polysaccharides induce L-selectin shedding and tumor necrosis factor receptor loss from the surface of human neutrophils. J. Clin. Investig. 1996, 97, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Bojarczuk, A.; Miller, K.A.; Hotham, R.; Lewis, A.; Ogryzko, N.V.; Kamuyango, A.A.; Frost, H.; Gibson, R.H.; Stillman, E.; May, R.C.; et al. Cryptococcus neoformans intracellular proliferation and capsule size determines early macrophage control of infection. Sci. Rep. 2016, 6, 21489. [Google Scholar] [CrossRef] [PubMed]
- Naslund, P.K.; Miller, W.C.; Granger, D.L. Cryptococcus neoformans fails to induce nitric oxide synthase in primed murine macrophage-like cells. Infect. Immun. 1995, 63, 1298–1304. [Google Scholar] [PubMed]
- Dykstra, M.A.; Friedman, L.; Murphy, J.W. Capsule size of Cryptococcus neoformans: Control and relationship to virulence. Infect. Immun. 1977, 16, 129–135. [Google Scholar] [PubMed]
- Kozel, T.R.; Tabuni, A.; Young, B.J.; Levitz, S.M. Influence of opsonization conditions on C3 deposition and phagocyte binding of large- and small-capsule Cryptococcus neoformans cells. Infect. Immun. 1996, 64, 2336–2338. [Google Scholar] [PubMed]
- Chiapello, L.S.; Aoki, M.P.; Rubinstein, H.R.; Masih, D.T. Apoptosis induction by glucuronoxylomannan of Cryptococcus neoformans. Med. Mycol. 2003, 41, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Monari, C.; Pericolini, E.; Bistoni, G.; Casadevall, A.; Kozel, T.R.; Vecchiarelli, A. Cryptococcus neoformans capsular glucuronoxylomannan induces expression of fas ligand in macrophages. J. Immunol. 2005, 174, 3461–3468. [Google Scholar] [CrossRef] [PubMed]
- Pericolini, E.; Cenci, E.; Monari, C.; De Jesus, M.; Bistoni, F.; Casadevall, A.; Vecchiarelli, A. Cryptococcus neoformans capsular polysaccharide component galactoxylomannan induces apoptosis of human T-cells through activation of caspase-8. Cell Microbiol. 2006, 8, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Monari, C.; Bistoni, F.; Vecchiarelli, A. Glucuronoxylomannan exhibits potent immunosuppressive properties. FEMS Yeast Res. 2006, 6, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Mody, C.H.; Syme, R.M. Effect of polysaccharide capsule and methods of preparation on human lymphocyte proliferation in response to Cryptococcus neoformans. Infect. Immun. 1993, 61, 464–469. [Google Scholar] [PubMed]
- Blackstock, R.; Buchanan, K.L.; Adesina, A.M.; Murphy, J.W. Differential regulation of immune responses by highly and weakly virulent Cryptococcus neoformans isolates. Infect. Immun. 1999, 67, 3601–3609. [Google Scholar] [PubMed]
- Mariano Andrade, R.; Monteiro Almeida, G.; Alexandre DosReis, G.; Alves Melo Bento, C. Glucuronoxylomannan of Cryptococcus neoformans exacerbates in vitro yeast cell growth by interleukin 10-dependent inhibition of CD4+ T lymphocyte responses. Cell Immunol. 2003, 222, 116–125. [Google Scholar] [CrossRef]
- Okagaki, L.H.; Wang, Y.; Ballou, E.R.; O’Meara, T.R.; Bahn, Y.S.; Alspaugh, J.A.; Xue, C.; Nielsen, K. Cryptococcal titan cell formation is regulated by G-protein signaling in response to multiple stimuli. Eukaryot. Cell 2011, 10, 1306–1316. [Google Scholar] [CrossRef] [PubMed]
- Chrisman, C.J.; Albuquerque, P.; Guimaraes, A.J.; Nieves, E.; Casadevall, A. Phospholipids trigger Cryptococcus neoformans capsular enlargement during interactions with amoebae and macrophages. PLoS Pathog. 2011, 7, e1002047. [Google Scholar] [CrossRef] [PubMed]
- Okagaki, L.H.; Strain, A.K.; Nielsen, J.N.; Charlier, C.; Baltes, N.J.; Chretien, F.; Heitman, J.; Dromer, F.; Nielsen, K. Cryptococcal cell morphology affects host cell interactions and pathogenicity. PLoS Pathog. 2010, 6, e1000953. [Google Scholar] [CrossRef]
- Crabtree, J.N.; Okagaki, L.H.; Wiesner, D.L.; Strain, A.K.; Nielsen, J.N.; Nielsen, K. Titan cell production enhances the virulence of Cryptococcus neoformans. Infect. Immun. 2012, 80, 3776–3785. [Google Scholar] [CrossRef] [PubMed]
- Baker, R.D. The primary pulmonary lymph node complex of crytptococcosis. Am. J. Clin. Pathol. 1976, 65, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Hermoso, D.; Janbon, G.; Dromer, F. Epidemiological evidence for dormant Cryptococcus neoformans infection. J. Clin. Microbiol. 1999, 37, 3204–3209. [Google Scholar] [PubMed]
- Hiruma, M.; Kawada, A.; Ishibashi, A. Ultrastructure of asteroid bodies in sporotrichosis. Mycoses 1991, 34, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Hiruma, M.; Kawada, A.; Noda, T.; Yamazaki, M.; Ishibashi, A. Tissue response in sporotrichosis: Light and electron microscopy studies. Mycoses 1992, 35, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Lurie, H.I.; Still, W.J. The “capsule” of Sporotrichum schenckii and the evolution of the asteroid body. A light and electron microscopic study. Sabouraudia 1969, 7, 64–70. [Google Scholar] [PubMed]
- Berman, J.; Sudbery, P.E. Candida albicans: A molecular revolution built on lessons from budding yeast. Nat. Rev. Genet. 2002, 3, 918–930. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.J.; Kohler, J.R.; DiDomenico, B.; Loebenberg, D.; Cacciapuoti, A.; Fink, G.R. Nonfilamentous C. albicans mutants are avirulent. Cell 1997, 90, 939–949. [Google Scholar] [CrossRef]
- Braun, B.R.; Johnson, A.D. Control of filament formation in Candida albicans by the transcriptional repressor TUP1. Science 1997, 277, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, G.M. Dimorphism in fungal pathogens of mammals, plants, and insects. PLoS Pathog. 2015, 11, e1004608. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.; Li, A.; Calo, S.; Heitman, J. Calcineurin plays key roles in the dimorphic transition and virulence of the human pathogenic zygomycete Mucor circinelloides. PLoS Pathog. 2013, 9, e1003625. [Google Scholar] [CrossRef] [PubMed]
- Bonifaz, A.; Gomez-Daza, F.; Paredes, V.; Ponce, R.M. Tinea versicolor, tinea nigra, white piedra, and black piedra. Clin. Dermatol. 2010, 28, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Inglis, D.O.; Voorhies, M.; Hocking Murray, D.R.; Sil, A. Comparative transcriptomics of infectious spores from the fungal pathogen Histoplasma capsulatum reveals a core set of transcripts that specify infectious and pathogenic states. Eukaryot. Cell 2013, 12, 828–852. [Google Scholar] [CrossRef] [PubMed]
- Newman, S.L.; Gootee, L.; Kidd, C.; Ciraolo, G.M.; Morris, R. Activation of human macrophage fungistatic activity against Histoplasma capsulatum upon adherence to type 1 collagen matrices. J. Immunol. 1997, 158, 1779–1786. [Google Scholar] [PubMed]
- Hung, C.Y.; Yu, J.J.; Seshan, K.R.; Reichard, U.; Cole, G.T. A parasitic phase-specific adhesin of Coccidioides immitis contributes to the virulence of this respiratory Fungal pathogen. Infect. Immun. 2002, 70, 3443–3456. [Google Scholar] [CrossRef] [PubMed]
- Soll, D.R. Why does Candida albicans switch? FEMS Yeast Res. 2009, 9, 973–989. [Google Scholar] [CrossRef] [PubMed]
- Marcil, A.; Harcus, D.; Thomas, D.Y.; Whiteway, M. Candida albicans killing by RAW 264.7 mouse macrophage cells: Effects of Candida genotype, infection ratios, and gamma interferon treatment. Infect. Immun. 2002, 70, 6319–6329. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, C.G.; Koser, U.; Lewis, L.E.; Bain, J.M.; Mora-Montes, H.M.; Barker, R.N.; Gow, N.A.; Erwig, L.P. Contribution of Candida albicans cell wall components to recognition by and escape from murine macrophages. Infect. Immun. 2010, 78, 1650–1658. [Google Scholar] [CrossRef] [PubMed]
- Mukaremera, L.; Lee, K.K.; Mora-Montes, H.M.; Gow, N.A.R. Candida albicans yeast, pseudohyphal, and hyphal morphogenesis differentially affects immune recognition. Front. Immunol. 2017, 8, 629. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, A.; Hung, C.Y.; Cole, G.T. Coccidioides releases a soluble factor that suppresses nitric oxide production by murine primary macrophages. Microb. Pathog. 2011, 50, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Almeida, A.J.; Cunha, C.; Carmona, J.A.; Sampaio-Marques, B.; Carvalho, A.; Malavazi, I.; Steensma, H.Y.; Johnson, D.I.; Leao, C.; Logarinho, E.; et al. Cdc42p controls yeast-cell shape and virulence of Paracoccidioides brasiliensis. Fungal. Genet. Biol. 2009, 46, 919–926. [Google Scholar] [CrossRef] [PubMed]
- Marcil, A.; Gadoury, C.; Ash, J.; Zhang, J.; Nantel, A.; Whiteway, M. Analysis of PRA1 and its relationship to Candida albicans- macrophage interactions. Infect. Immun. 2008, 76, 4345–4358. [Google Scholar] [CrossRef] [PubMed]
- Cockayne, A.; Odds, F.C. Interactions of Candida albicans yeast cells, germ tubes and hyphae with human polymorphonuclear leucocytes in vitro. J. Gen. Microbiol. 1984, 130, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Keppler-Ross, S.; Douglas, L.; Konopka, J.B.; Dean, N. Recognition of yeast by murine macrophages requires mannan but not glucan. Eukaryot. Cell 2010, 9, 1776–1787. [Google Scholar] [CrossRef] [PubMed]
- Lewis, L.E.; Bain, J.M.; Lowes, C.; Gillespie, C.; Rudkin, F.M.; Gow, N.A.; Erwig, L.P. Stage specific assessment of Candida albicans phagocytosis by macrophages identifies cell wall composition and morphogenesis as key determinants. PLoS Pathog. 2012, 8, e1002578. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.D. Innate antifungal immunity: The key role of phagocytes. Annu. Rev. Immunol. 2011, 29, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Sorrell, T.C.; Juillard, P.G.; Djordjevic, J.T.; Kaufman-Francis, K.; Dietmann, A.; Milonig, A.; Combes, V.; Grau, G.E. Cryptococcal transmigration across a model brain blood-barrier: Evidence of the Trojan horse mechanism and differences between Cryptococcus neoformans var. grubii strain H99 and Cryptococcus gattii strain R265. Microbes Infect. 2016, 18, 57–67. [Google Scholar] [PubMed]
- Zaragoza, O.; Chrisman, C.J.; Castelli, M.V.; Frases, S.; Cuenca-Estrella, M.; Rodriguez-Tudela, J.L.; Casadevall, A. Capsule enlargement in Cryptococcus neoformans confers resistance to oxidative stress suggesting a mechanism for intracellular survival. Cell Microbiol. 2008, 10, 2043–2057. [Google Scholar] [PubMed]
- Komalapriya, C.; Kaloriti, D.; Tillmann, A.T.; Yin, Z.; Herrero-de-Dios, C.; Jacobsen, M.D.; Belmonte, R.C.; Cameron, G.; Haynes, K.; Grebogi, C.; et al. Integrative Model of Oxidative Stress Adaptation in the Fungal Pathogen Candida albicans. PLoS ONE 2015, 10, e0137750. [Google Scholar] [CrossRef] [PubMed]
- Tavares, A.H.; Silva, S.S.; Dantas, A.; Campos, E.G.; Andrade, R.V.; Maranhao, A.Q.; Brígido, M.M.; Passos-Silva, D.G.; Fachin, A.L.; Teixeira, S.M.; et al. Early transcriptional response of Paracoccidioides brasiliensis upon internalization by murine macrophages. Microbes Infect. 2007, 9, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Parente-Rocha, J.A.; Parente, A.F.; Baeza, L.C.; Bonfim, S.M.; Hernandez, O.; McEwen, J.G.; Bailao, A.M.; Taborda, C.P.; Borges, C.L.; Soares, C.M. Macrophage Interaction with Paracoccidioides brasiliensis Yeast Cells Modulates Fungal Metabolism and Generates a Response to Oxidative Stress. PLoS ONE 2015, 10, e0137619. [Google Scholar] [CrossRef] [PubMed]
- de Arruda Grossklaus, D.; Bailao, A.M.; Vieira Rezende, T.C.; Borges, C.L.; de Oliveira, M.A.; Parente, J.A.; de Almeida Soares, C.M. Response to oxidative stress in Paracoccidioides yeast cells as determined by proteomic analysis. Microbes Infect. 2013, 15, 347–364. [Google Scholar] [CrossRef] [PubMed]
- Parente, A.F.; Naves, P.E.; Pigosso, L.L.; Casaletti, L.; McEwen, J.G.; Parente-Rocha, J.A.; Soares, C.M. The response of Paracoccidioides spp. to nitrosative stress. Microbes Infect. 2015, 17, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Fleischmann, J.; Wu-Hsieh, B.; Howard, D.H. The intracellular fate of Histoplasma capsulatum in human macrophages is unaffected by recombinant human interferon-gamma. J. Infect. Dis. 1990, 161, 143–145. [Google Scholar] [CrossRef] [PubMed]
- Brummer, E.; Stevens, D.A. Antifungal mechanisms of activated murine bronchoalveolar or peritoneal macrophages for Histoplasma capsulatum. Clin. Exp. Immunol. 1995, 102, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Newman, S.L.; Bucher, C.; Rhodes, J.; Bullock, W.E. Phagocytosis of Histoplasma capsulatum yeasts and microconidia by human cultured macrophages and alveolar macrophages. Cellular cytoskeleton requirement for attachment and ingestion. J. Clin. Investig. 1990, 85, 223–230. [Google Scholar] [PubMed]
- Kurita, N.; Terao, K.; Brummer, E.; Ito, E.; Nishimura, K.; Miyaji, M. Resistance of Histoplasma capsulatum to killing by human neutrophils. Evasion of oxidative burst and lysosomal-fusion products. Mycopathologia 1991, 115, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Schaffner, A.; Davis, C.E.; Schaffner, T.; Markert, M.; Douglas, H.; Braude, A.I. In vitro susceptibility of fungi to killing by neutrophil granulocytes discriminates between primary pathogenicity and opportunism. J. Clin. Investig. 1986, 78, 511–524. [Google Scholar] [CrossRef] [PubMed]
- Brummer, E.; Kurita, N.; Yosihida, S.; Nishimura, K.; Miyaji, M. Fungistatic activity of human neutrophils against Histoplasma capsulatum: Correlation with phagocytosis. J. Infect. Dis. 1991, 164, 158–162. [Google Scholar] [CrossRef] [PubMed]
- Youseff, B.H.; Holbrook, E.D.; Smolnycki, K.A.; Rappleye, C.A. Extracellular superoxide dismutase protects Histoplasma yeast cells from host-derived oxidative stress. PLoS Pathog. 2012, 8, e1002713. [Google Scholar] [CrossRef] [PubMed]
- Giles, S.S.; Stajich, J.E.; Nichols, C.; Gerrald, Q.D.; Alspaugh, J.A.; Dietrich, F.; Perfect, J.R. The Cryptococcus neoformans Catalase Gene Family and Its Role in Antioxidant Defense. Eukaryot. Cell 2006, 5, 1447–1459. [Google Scholar] [CrossRef] [PubMed]
- Cox, G.M.; Harrison, T.S.; McDade, H.C.; Taborda, C.P.; Heinrich, G.; Casadevall, A.; Perfect, J.R. Superoxide Dismutase Influences the Virulence of Cryptococcus neoformans by Affecting Growth within Macrophages. Infect. Immun. 2003, 71, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Missall, T.A.; Cherry-Harris, J.F.; Lodge, J.K. Two glutathione peroxidases in the fungal pathogen Cryptococcus neoformans are expressed in the presence of specific substrates. Microbiology 2005, 151, 2573–2581. [Google Scholar] [CrossRef] [PubMed]
- Missall, T.A.; Lodge, J.K. Function of the thioredoxin proteins in Cryptococcus neoformans during stress or virulence and regulation by putative transcriptional modulators. Mol. Microbiol. 2005, 57, 847–858. [Google Scholar] [CrossRef] [PubMed]
- Gerik, K.J.; Bhimireddy, S.R.; Ryerse, J.S.; Specht, C.A.; Lodge, J.K. PKC1 Is Essential for Protection against both Oxidative and Nitrosative Stresses, Cell Integrity, and Normal Manifestation of Virulence Factors in the Pathogenic Fungus Cryptococcus neoformans. Eukaryot. Cell 2008, 7, 1685–1698. [Google Scholar] [CrossRef] [PubMed]
- Philippe, B.; Ibrahim-Granet, O.; Prévost, M.C.; Gougerot-Pocidalo, M.A.; Sanchez Perez, M.; Van der Meeren, A.; Latgé, J.P. Killing of Aspergillus fumigatus by Alveolar Macrophages Is Mediated by Reactive Oxidant Intermediates. Infect. Immun. 2003, 71, 3034–3042. [Google Scholar] [CrossRef] [PubMed]
- Lambou, K.; Lamarre, C.; Beau, R.; Dufour, N.; Latge, J.-P. Functional analysis of the superoxide dismutase family in Aspergillus fumigatus. Mol. Microbiol. 2010, 75, 910–923. [Google Scholar] [CrossRef] [PubMed]
- Miyasaka, N.R.S.; Unterkircher, C.S.; Shimizu, M.T. Catalase activity of different Candida species after exposition to specific antiserum. Br. J. Microbiol. 2008, 39, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Frohner, I.E.; Bourgeois, C.; Yatsyk, K.; Majer, O.; Kuchler, K. Candida albicans cell surface superoxide dismutases degrade host-derived reactive oxygen species to escape innate immune surveillance. Mol. Microbiol. 2009, 71, 240–252. [Google Scholar] [CrossRef] [PubMed]
- Briones-Martin-Del-Campo, M.; Orta-Zavalza, E.; Juarez-Cepeda, J.; Gutierrez-Escobedo, G.; Cañas-Villamar, I.; Castaño, I.; De Las Peñas, A. The oxidative stress response of the opportunistic fungal pathogen Candida glabrata. Rev. Iberoam. Micol. 2014, 31, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Chagas, R.F.; Bailão, A.M.; Pereira, M.; Winters, M.S.; Smullian, A.G.; Deepe, G.S., Jr.; de Almeida Soares, C.M. The catalases of Paracoccidioides brasiliensis are differentially regulated: protein activity and transcript analysis. Fungal Genet. Biol. 2008, 45, 1470–1478. [Google Scholar]
- Moreira, S.F.I.; Bailão, A.M.; Barbosa, M.S.; Jesuino, R.S.A.; Felipe, M.S.S.; Pereira, M.; de Almeida Soares, C.M. Monofunctional catalase P of Paracoccidioides brasiliensis: Identification, characterization, molecular cloning and expression analysis. Yeast 2004, 21, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Levitz, S.M.; Harrison, T.S.; Tabuni, A.; Liu, X. Chloroquine induces human mononuclear phagocytes to inhibit and kill Cryptococcus neoformans by a mechanism independent of iron deprivation. J. Clin. Investig. 1997, 100, 1640–1646. [Google Scholar] [CrossRef] [PubMed]
- DeLeon-Rodriguez, C.M.; Casadevall, A. Cryptococcus neoformans: Tripping on Acid in the Phagolysosome. Front. Microbiol. 2016, 7, 164. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.J.; Eastman, A.J.; Qiu, Y.; Gregorka, B.; Kozel, T.R.; Osterholzer, J.J.; Curtis, J.L.; Swanson, J.A.; Olszewski, M.A. Cryptococcus neoformans-induced macrophage lysosome damage crucially contributes to fungal virulence. J. Immunol. 2015, 194, 2219–2231. [Google Scholar] [CrossRef] [PubMed]
- Eissenberg, L.G.; Goldman, W.E.; Schlesinger, P.H. Histoplasma capsulatum modulates the acidification of phagolysosomes. J. Exp. Med. 1993, 177, 1605–1611. [Google Scholar] [CrossRef] [PubMed]
- Beck, M.R.; Dekoster, G.T.; Cistola, D.P.; Goldman, W.E. NMR structure of a fungal virulence factor reveals structural homology with mammalian saposin B. Mol. Microbiol. 2009, 72, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Vylkova, S.; Lorenz, M.C. Modulation of phagosomal pH by Candida albicans promotes hyphal morphogenesis and requires STP2p, a regulator of amino acid transport. PLoS Pathog. 2014, 10, e1003995. [Google Scholar] [CrossRef] [PubMed]
- Okai, B.; Lyall, N.; Gow, N.A.; Bain, J.M.; Erwig, L.P. Rab14 regulates maturation of macrophage phagosomes containing the fungal pathogen Candida albicans and outcome of the host-pathogen interaction. Infect. Immun. 2015, 83, 1523–1535. [Google Scholar] [CrossRef] [PubMed]
- Rivas-Santiago, B.; Sada, E.; Hernández-Pando, R.; Tsutsumi, V. Péptidos antimicrobianos en la inmunidad innata de enfermedades infecciosas. Salud Pública México 2006, 48, 62–71. [Google Scholar] [CrossRef]
- Peschel, A.; Sahl, H.-G. The co-evolution of host cationic antimicrobial peptides and microbial resistance. Nat. Rev. Microbiol. 2006, 4, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Lehrer, R.I.; Ganz, T. Antimicrobial peptides in mammalian and insect host defence. Curr. Opin. Immunol. 1999, 11, 23–27. [Google Scholar] [CrossRef]
- Steinstraesser, L.; Kraneburg, U.; Jacobsen, F.; Al-Benna, S. Host defense peptides and their antimicrobial-immunomodulatory duality. Immunobiology 2011, 216, 322–333. [Google Scholar] [CrossRef] [PubMed]
- Meiller, T.F.; Hube, B.; Schild, L.; Shirtliff, M.E.; Scheper, M.A.; Winkler, R.; Ton, A.; Jabra-Rizk, M.A. A Novel Immune Evasion Strategy of Candida albicans: Proteolytic Cleavage of a Salivary Antimicrobial Peptide. PLoS ONE 2009, 4, 4. [Google Scholar] [CrossRef] [PubMed]
- Szafranski-Schneider, E.; Swidergall, M.; Cottier, F.; Tielker, D.; Román, E.; Pla, J.; Ernst, J.F. Msb2 Shedding Protects Candida albicans against Antimicrobial Peptides. PLoS Pathog. 2012, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Swidergall, M.; Ernst, A.M.; Ernst, J.F. Candida albicans Mucin Msb2 Is a Broad-Range Protectant against Antimicrobial Peptides. Antimicrob. Agents Chemother. 2013, 57, 3917–3922. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Kumar, R.; Tati, S.; Puri, S.; Edgerton, M. Candida albicans Flu1-Mediated Efflux of Salivary Histatin 5 Reduces Its Cytosolic Concentration and Fungicidal Activity. Antimicrob. Agents Chemother. 2013, 57, 1832–1839. [Google Scholar] [CrossRef] [PubMed]
- Vylkova, S.; Jang, W.S.; Li, W.; Nayyar, N.; Edgerton, M. Histatin 5 Initiates Osmotic Stress Response in Candida albicans via Activation of the Hog1 Mitogen-Activated Protein Kinase Pathway. Eukaryot. Cell 2007, 6, 1876–1888. [Google Scholar] [CrossRef] [PubMed]
- Argimón, S.; Fanning, S.; Blankenship, J.R.; Mitchell, A.P. Interaction between the Candida albicans High-Osmolarity Glycerol (HOG) Pathway and the Response to Human β-Defensins 2 and 3. Eukaryot. Cell 2011, 10, 272–275. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Monge, R.; Carvaihlo, S.; Nombela, C.; Rial, E.; Pla, J. The Hog1 MAP kinase controls respiratory metabolism in the fungal pathogen Candida albicans. Microbiology 2009, 155, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.I.; Finkel, J.S.; Solis, N.V.; Chaili, S.; Mitchell, A.P.; Yeaman, M.R.; Filler, S.G. Bcr1 Functions Downstream of Ssd1 To Mediate Antimicrobial Peptide Resistance in Candida albicans. Eukaryot. Cell 2013, 12, 411–419. [Google Scholar] [PubMed]
- Gank, K.D.; Yeaman, M.R.; Kojima, S.; Yount, N.Y.; Park, H.; Edwards, J.E.; Filler, S.G.; Fu, Y. SSD1 Is Integral to Host Defense Peptide Resistance in Candida albicans. Eukaryot. Cell 2008, 7, 1318–1327. [Google Scholar] [PubMed]
- Rambach, G.; Speth, C. Complement in Candida albicans infections. Front. BioSci. 2009, 1, 1–12. [Google Scholar]
- Peltz, G.; Zaas, A.K.; Zheng, M.; Solis, N.V.; Zhang, M.X.; Liu, H.H.; Hu, Y.; Boxx, G.M.; Phan, Q.T.; Dill, D. Next-Generation Computational Genetic Analysis: Multiple Complement Alleles Control Survival after Candida albicans Infection. Infect. Immun. 2011, 79, 4472–4479. [Google Scholar] [PubMed]
- Ashman, R.B.; Papadimitriou, J.M.; Fulurija, A.; Drysdale, K.E.; Farah, C.S.; Naidoo, O.; Gotjamanos, T. Role of complement C5 and T lymphocytes in pathogenesis of disseminated and mucosal candidiasis in susceptible DBA/2 mice. Microb. Pathog. 2003, 34, 103–113. [Google Scholar] [CrossRef]
- Tsoni, S.V.; Kerrigan, A.M.; Marakalala, M.J.; Srinivasan, N.; Duffield, M.; Taylor, P.R.; Botto, M.; Steele, C.; Brown, G.D. Complement C3 Plays an Essential Role in the Control of Opportunistic Fungal Infections. Infect. Immun. 2009, 77, 3679–3685. [Google Scholar] [PubMed]
- Luo, S.; Hoffmann, R.; Skerka, C.; Zipfel, P.F. Glycerol-3-Phosphate Dehydrogenase 2 Is a Novel Factor H–, Factor H–like Protein 1–, and Plasminogen-Binding Surface Protein of Candida albicans. J. Infect. Dis. 2013, 207, 594–603. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Poltermann, S.; Kunert, A.; Rupp, S.; Zipfel, P.F. Immune evasion of the human pathogenic yeast Candida albicans: Pra1 is a Factor H, FHL-1 and plasminogen binding surface protein. Mol. Immunol. 2009, 47, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Zipfel, P.F.; Jokiranta, T.S.; Hellwage, J.; Koistinen, V.; Meri, S. The factor H protein family. Immunopharmacology 1999, 42, 53–60. [Google Scholar] [CrossRef]
- Luo, S.; Blom, A.M.; Rupp, S.; Hipler, U.C.; Hube, B.; Skerka, C.; Zipfel, P.F. The pH-regulated Antigen 1 of Candida albicans Binds the Human Complement Inhibitor C4b-binding Protein and Mediates Fungal Complement Evasion. J. Biol. Chem. 2011, 286, 8021–8029. [Google Scholar] [PubMed]
- Poltermann, S.; Kunert, A.; von der Heide, M.; Eck, R.; Hartmann, A.; Zipfel, P.F. Gpm1p Is a Factor H-, FHL-1-, and Plasminogen-binding Surface Protein of Candida albicans. J. Biol. Chem. 2007, 282, 37537–37544. [Google Scholar] [CrossRef] [PubMed]
- Lesiak-Markowicz, I.; Vogl, G.; Schwarzmüller, T.; Speth, C.; Lass-Flörl, C.; Dierich, M.P.; Kuchler, K.; Würzner, R. Candida albicans Hgt1p, a Multifunctional Evasion Molecule: Complement Inhibitor, CR3 Analogue, and Human Immunodeficiency Virus–Binding Molecule. J. Infect. Dis. 2011, 204, 802–809. [Google Scholar] [CrossRef] [PubMed]
- Vogl, G.; Lesiak, I.; Jensen, D.B.; Perkhofer, S.; Eck, R.; Speth, C.; Lass-Flörl, C.; Zipfel, P.F.; Blom, A.M.; Dierich, M.P. Immune evasion by acquisition of complement inhibitors: The mould Aspergillus binds both factor H and C4b binding protein. Mol. Immunol. 2008, 45, 1485–1493. [Google Scholar] [CrossRef] [PubMed]
- Behnsen, J.; Hartmann, A.; Schmaler, J.; Gehrke, A.; Brakhage, A.A.; Zipfel, P.F. The Opportunistic Human Pathogenic Fungus Aspergillus fumigatus Evades the Host Complement System. Infect. Immun. 2008, 76, 820–827. [Google Scholar] [CrossRef] [PubMed]
- Henwick, S.; Hetherington, S.V.; Patrick, C.C. Complement binding to Aspergillus conidia correlates with pathogenicity. J. Lab. Clin. Med. 1993, 122, 27–35. [Google Scholar] [PubMed]
- Tsai, H.-F.; Washburn, R.G.; Chang, Y.C.; Kwon-Chung, K.J. Aspergillus fumigatus arp1 modulates conidial pigmentation and complement deposition. Mol. Microbiol. 1997, 26, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.-F.; Chang, Y.C.; Washburn, R.G.; Wheeler, M.H.; Kwon-Chung, K.J. The Developmentally Regulated alb1 Gene of Aspergillus fumigatus: Its Role in Modulation of Conidial Morphology and Virulence. J. Bacteriol. 1998, 180, 3031–3038. [Google Scholar] [PubMed]
- Langfelder, K.; Jahn, B.; Gehringer, H.; Schmidt, A.; Wanner, G.; Brakhage, A.A. Identification of a polyketide synthase gene (pksP) of Aspergillus fumigatus involved in conidial pigment biosynthesis and virulence. Med. Microbiol. Immunol. 1998, 187, 79–89. [Google Scholar] [CrossRef] [PubMed]
- da Silva, M.B.; Marques, A.F.; Nosanchuk, J.D.; Casadevall, A.; Travassos, L.R.; Taborda, C.P. Melanin in the dimorphic fungal pathogen Paracoccidioides brasiliensis: Effects on phagocytosis, intracellular resistance and drug susceptibility. Microbes Infect. 2006, 8, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Rosbjerg, A.; Genster, N.; Pilely, K.; Skjoedt, M.O.; Stahl, G.L.; Garred, P. Complementary roles of the classical and lectin complement pathways in the defense against Aspergillus fumigatus. Front. Immunol. 2016, 7, 473. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.X.; Brandhorst, T.T.; Kozel, T.R.; Klein, B.S. Role of Glucan and Surface Protein BAD1 in Complement Activation by Blastomyces dermatitidisYeast. Infect. Immun. 2001, 69, 7559–7564. [Google Scholar] [CrossRef] [PubMed]
- Li, X.S.; Reddy, M.S.; Baev, D.; Edgerton, M. Candida albicans Ssa1/2p Is the Cell Envelope Binding Protein for Human Salivary Histatin 5. J. Biol. Chem. 2003, 278, 28553–28561. [Google Scholar] [CrossRef] [PubMed]
- Vylkova, S.; Li, X.S.; Berner, J.C.; Edgerton, M. Distinct Antifungal Mechanisms: β-Defensins Require Candida albicans Ssa1 Protein, while Trk1p Mediates Activity of Cysteine-Free Cationic Peptides. Antimicrob. Agents Chemother. 2006, 50, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Aerts, A.M.; François, I.E.J.A.; Cammue, B.P.A.; Thevissen, K. The mode of antifungal action of plant, insect and human defensins. Cell. Mol. Life Sci. 2008, 65, 2069–2079. [Google Scholar] [CrossRef] [PubMed]
- Doering, T.L.; Nosanchuk, J.D.; Roberts, W.K.; Casadevall, A. Melanin as a potential cryptococcal defence against microbicidal proteins. Med. Mycol. 1999, 37, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Zawrotniak, M.; Rapala-Kozik, M. Neutrophil extracellular traps (NETs)—Formation and implications. Acta Biochim. Pol. 2013, 60, 277–284. [Google Scholar] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil Extracellular Traps Kill Bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Urban, C.F.; Reichard, U.; Brinkmann, V.; Zychlinsky, A. Neutrophil extracellular traps capture and kill Candida albicans yeast and hyphal forms. Cell. Microbiol. 2006, 8, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, M.; Hakkim, A.; Brinkmann, V.; Siler, U.; Seger, R.A.; Zychlinsky, A.; Reichenbach, J. Restoration of NET formation by gene therapy in CGD controls aspergillosis. Blood 2009, 114, 2619–2622. [Google Scholar] [CrossRef] [PubMed]
- Springer, D.J.; Ren, P.; Raina, R.; Dong, Y.; Behr, M.J.; McEwen, B.F.; Bowser, S.S.; Samsonoff, W.A.; Chaturvedi, S.; Chaturvedi, V. Extracellular Fibrils of Pathogenic Yeast Cryptococcus gattii Are Important for Ecological Niche, Murine Virulence and Human Neutrophil Interactions. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Andrews, N.C. Disorders of Iron Metabolism. N. Engl. J. Med. 1999, 341, 1986–1995. [Google Scholar] [CrossRef] [PubMed]
- Noble, S. Candida albicans specializations for iron homeostasis: From commensalism to virulence. Curr. Opin. Microbiol. 2013, 16, 708–715. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Liu, X.-B.; Quinones, M.; Melby, P.C.; Ghio, A.; Haile, D.J. Regulation of Reticuloendothelial Iron Transporter MTP1 (Slc11a3) by Inflammation. J. Biol. Chem. 2002, 277, 39786–39791. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Takano, M.; Murakami, M.; Tanaka, H.; Matsuhisa, A.; Nakao, N.; Mikami, T.; Suzuki, M.; Matsumoto, T. Characterization of a haemolytic factor from Candida albicans. Microbiology 1999, 145, 689–694. [Google Scholar] [PubMed]
- Weissman, Z.; Kornitzer, D. A family of Candida cell surface haem-binding proteins involved in haemin and haemoglobin-iron utilization. Mol. Microbiol. 2004, 53, 1209–1220. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.; Buisson, N.; Knight, S.; Dancis, A.; Camadro, J.-M.; Lesuisse, E. Haemin uptake and use as an iron source by Candida albicans: role of CaHMX1-encoded haem oxygenase. Microbiology 2003, 149, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Knight, S.A.B.; Lesuisse, E.; Stearman, R.; Klausner, R.D.; Dancis, A. Reductive iron uptake by Candida albicans: role of copper, iron and the TUP1 regulator. Microbiology 2002, 148, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, L.; Terzulli, A.; Gaur, R.; McCarthy, R.; Kosman, D.J. Functional Characterization of the Ferroxidase, Permease High Affinity Iron Transport Complex from Candida albicans. Mol. Microbiol. 2011, 81, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Ramanan, N.; Wang, Y. A High-Affinity Iron Permease Essential for Candida albicans Virulence. Science 2000, 288, 1062–1064. [Google Scholar] [CrossRef] [PubMed]
- Heymann, P.; Gerads, M.; Schaller, M.; Dromer, F.; Winkelmann, G.; Ernst, J.F. The Siderophore Iron Transporter of Candida albicans (Sit1p/Arn1p) Mediates Uptake of Ferrichrome-Type Siderophores and Is Required for Epithelial Invasion. Infect. Immun. 2002, 70, 5246–5255. [Google Scholar] [CrossRef] [PubMed]
- Hissen, A.H.T.; Wan, A.N.C.; Warwas, M.L.; Pinto, L.J.; Moore, M.M. The Aspergillus fumigatus Siderophore Biosynthetic Gene sidA, Encoding l-Ornithine N5-Oxygenase, Is Required for Virulence. Infect. Immun. 2005, 73, 5493–5503. [Google Scholar] [CrossRef] [PubMed]
- Hilty, J.; George Smulian, A.; Newman, S.L. Histoplasma capsulatum utilizes siderophores for intracellular iron acquisition in macrophages. Med. Mycol. 2011, 49, 633–642. [Google Scholar] [PubMed]
- Silva, M.; Schrank, A.; Bailão, E.F.; Bailão, A.; Borges, C.; Staats, C.; Parente, J.A.; Pereira, M.; Salem-Izacc, S.M.; Mendes-Giannini, M.J. The Homeostasis of Iron, Copper, and Zinc in Paracoccidioides brasiliensis, Cryptococcus neoformans Var. Grubii, and Cryptococcus Gattii: A Comparative Analysis. Front. Microbiol. 2011, 2. [Google Scholar] [CrossRef] [PubMed]
- Jung, W.H.; Kronstad, J.W. Iron and fungal pathogenesis: A case study with Cryptococcus neoformans. Cell. Microbiol. 2008, 10, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Cadieux, B.; Lian, T.; Hu, G.; Wang, J.; Biondo, C.; Teti, G.; Liu, V.; Murphy, M.E.; Creagh, A.L.; Kronstad, J.W. The Mannoprotein Cig1 Supports Iron Acquisition From Heme and Virulence in the Pathogenic Fungus Cryptococcus neoformans. J. Infect. Dis. 2013, 207, 1339–1347. [Google Scholar] [PubMed]
- Citiulo, F.; Jacobsen, I.D.; Miramón, P.; Schild, L.; Brunke, S.; Zipfel, P.; Brack, M.; Hube, B.; Wilson, D. Candida albicans Scavenges Host Zinc via Pra1 during Endothelial Invasion. PLoS Pathog. 2012, 8, e1002777. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.-E.; Nevitt, T.; Thiele, D.J. Mechanisms for copper acquisition, distribution and regulation. Nat. Chem. Biol. 2008, 4, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Georgatsou, E.; Mavrogiannis, L.A.; Fragiadakis, G.S.; Alexandraki, D. The Yeast Fre1p/Fre2p Cupric Reductases Facilitate Copper Uptake and Are Regulated by the Copper-modulated Mac1p Activator. J. Biol. Chem. 1997, 272, 13786–13792. [Google Scholar] [CrossRef] [PubMed]
- Dancis, A.; Yuan, D.S.; Haile, D.; Askwith, C.; Eide, D.; Moehle, C.; Kaplan, J.; Klausner, R.D. Molecular characterization of a copper transport protein in S. cerevisiae: An unexpected role for copper in iron transport. Cell 1994, 76, 393–402. [Google Scholar] [CrossRef]
- Peña, M.M.O.; Puig, S.; Thiele, D.J. Characterization of the Saccharomyces cerevisiae High Affinity Copper Transporter Ctr3. J. Biol. Chem. 2000, 275, 33244–33251. [Google Scholar] [CrossRef] [PubMed]
- Costerton, J.W.; Stewart, P.S.; Greenberg, E.P. Bacterial Biofilms: A Common Cause of Persistent Infections. Science 1999, 284, 1318–1322. [Google Scholar] [CrossRef] [PubMed]
- Donlan, R.M.; Costerton, J.W. Biofilms: Survival Mechanisms of Clinically Relevant Microorganisms. Clin. Microbiol. Rev. 2002, 15, 167–193. [Google Scholar] [CrossRef] [PubMed]
- Ning, Y.; Hu, X.; Ling, J.; Du, Y.; Liu, J.; Liu, H.; Peng, Z. Candida albicans survival and biofilm formation under starvation conditions. Int. Endod. J. 2013, 46, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Chandra, J.; McCormick, T.S.; Imamura, Y.; Mukherjee, P.K.; Ghannoum, M.A. Interaction of Candida albicans with Adherent Human Peripheral Blood Mononuclear Cells Increases C. albicans Biofilm Formation and Results in Differential Expression of Pro- and Anti-Inflammatory Cytokines. Infect. Immun. 2007, 75, 2612–2620. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Thompson, A.; Sobue, T.; Kashleva, H.; Xu, H.; Vasilakos, J.; Dongari-Bagtzoglou, A. Candida albicans Biofilms Do Not Trigger Reactive Oxygen Species and Evade Neutrophil Killing. J. Infect. Dis. 2012, 206, 1936–1945. [Google Scholar]
- Martinez, L.R.; Casadevall, A. Cryptococcus neoformans Cells in Biofilms Are Less Susceptible than Planktonic Cells to Antimicrobial Molecules Produced by the Innate Immune System. Infect. Immun. 2006, 74, 6118–6123. [Google Scholar] [CrossRef] [PubMed]
- Limper, A.H.; Standing, J.E.; Hoffman, O.A.; Castro, M.; Neese, L.W. Vitronectin binds to Pneumocystis carinii and mediates organism attachment to cultured lung epithelial cells. Infect. Immun. 1993, 61, 4302–4309. [Google Scholar] [PubMed]
- O'Riordan, D.M.; Standing, J.E.; Limper, A.H. Pneumocystis carinii glycoprotein A binds macrophage mannose receptors. Infect. Immun. 1995, 63, 779–784. [Google Scholar] [PubMed]
- Lasbury, M.E.; Lin, P.; Tschang, D.; Durant, P.J.; Lee, C.-H. Effect of Bronchoalveolar Lavage Fluid from Pneumocystis carinii- Infected Hosts on Phagocytic Activity of Alveolar Macrophages. Infect. Immun. 2004, 72, 2140–2147. [Google Scholar] [CrossRef] [PubMed]
- Swain, S.D.; Lee, S.J.; Nussenzweig, M.C.; Harmsen, A.G. Absence of the Macrophage Mannose Receptor in Mice Does Not Increase Susceptibility to Pneumocystis carinii Infection In Vivo. Infect. Immun. 2003, 71, 6213–6221. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.-Y.; Seshan, K.R.; Yu, J.-J.; Schaller, R.; Xue, J.; Basrur, V.; Gardner, M.J.; Cole, GT. A Metalloproteinase of Coccidioides posadasii Contributes to Evasion of Host Detection. Infect. Immun. 2005, 73, 6689–6703. [Google Scholar] [CrossRef] [PubMed]
- Stanley, V.C.; Hurley, R. The growth of Candida species in cultures of mouse peritoneal macrophages. J. Pathol. 1969, 97, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Ermert, D.; Niemiec, M.J.; Röhm, M.; Glenthøj, A.; Borregaard, N.; Urban, C.F. Candida albicans escapes from mouse neutrophils. J. Leukoc. Biol. 2013, 94, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Dementhon, K.; El-Kirat-Chatel, S.; Noël, T. Development of an In Vitro Model for the Multi-Parametric Quantification of the Cellular Interactions between Candida Yeasts and Phagocytes. PLoS ONE 2012, 7, e32621. [Google Scholar] [CrossRef] [PubMed]
- Kaur, R.; Ma, B.; Cormack, B.P. A family of glycosylphosphatidylinositol-linked aspartyl proteases is required for virulence of Candida glabrata. Proc. Natl. Acad. Sci. USA 2007, 104, 7628–7633. [Google Scholar] [CrossRef] [PubMed]
- Seider, K.; Brunke, S.; Schild, L.; Jablonowski, N.; Wilson, D.; Majer, O.; Brz, D.; Hass, A.; Kuchler, K.; Schaller, M. The Facultative Intracellular Pathogen Candida glabrata Subverts Macrophage Cytokine Production and Phagolysosome Maturation. J. Immunol. 2011, 187, 3072–3086. [Google Scholar] [PubMed]
- Rai, M.N.; Sharma, V.; Balusu, S.; Kaur, R. An essential role for phosphatidylinositol 3-kinase in the inhibition of phagosomal maturation, intracellular survival and virulence in Candida glabrata. Cell. Microbiol. 2015, 17, 269–287. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecules Involved in Host Immune Evasion | Function | Effect on the Human Immune System | Fungi Where They Have Been Described |
|---|---|---|---|
| Mannans | Proper cell wall architecture | Shielding of β-1,3-glucans from Dectin-1 receptor [13,14,15,16,17,18,19,20] | Candida spp, H. capsulatum yeast [13,14,15,16,17,18,19,20] |
| Melanin | Mechanical strength of the cell wall, enzymatic degradation resistance and UV protection [24] | Resistance to phagocytosis [9,24,25] | C. neoformans, Paracoccidioides brasiliensis, and Sporothrix schenckii [24] |
| α-1,3-Glucan | Proper cell wall structure and architecture [21] | Shielding of β-1,3-glucans from Dectin-1 receptor [21] | H. capsulatum yeast [21] |
| Endo β-1,3-glucanase | Trimming of β-1,3-glucan segments exposed on the fungal cell surface [23] | Reduced recognition of the yeast via Dectin-1 and as a consequence, a reduction in stimulation of proinflammatory cytokines [23] | H. capsulatum yeast [23] |
| Transferrin Receptors | Utilizing host iron-binding proteins as a source of iron [26,27] | Overcoming the host nutritional immunity of iron by the host [28,29] | C. albicans [28,29] |
| Pra 1 | Scavenger of host zinc [30,31] | Overcoming the nutritional immunity of zinc by the host [32,33] | C. albicans [32,33] |
| Rod A | Cell wall hydrophobin | Diminished host NETs formation [34,35] | A. fumigatus [34,35] |
| Structures and Architectural Changes Associated in Host Immune Evasion | Function | Effect on the Human Immune System | Fungi Where They Have Been Described |
|---|---|---|---|
| Fungal Capsule | Protection form environment and source of virulence factors [51] | Anti-phagocytic properties [56], suppression of T lymphocyte proliferation [56,57], induction of the anti-inflammatory cytokine IL-10, reduction in pro-inflammatory cytokines [56] and survival inside macrophage environment [55]. NETs formation is impeded by the capsular component GXM [58] | C. neoformans [53,58] |
| Titan-cell Formation | Protection from hostile environments [59,60] | Evasion of phagocytosis and resistance to oxidative and nitrosative stresses [59,60] | C. neoformans [59,60] |
| Asteroid Bodies | Resistance structure that protects the central yeast from the environment | Trapping of IgGs and IgMs, interfering with the proper immune system action [61] | Sporothrix spp.; Lacazia spp.; Candida spp.; Histoplasma spp.; Paracoccidioides spp.; Aspergillus spp. [61] |
| Biofilm Formation | Adhesion on biotic or abiotic surfaces, survival on hostile environments [62,63] | Resistance to neutrophil attack, avoids ROS triggering, and increases resistance to the antifungal activity of PBMNCs | Candida spp.; Aspergillus spp.; C. neoformans [64,65,66] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hernández-Chávez, M.J.; Pérez-García, L.A.; Niño-Vega, G.A.; Mora-Montes, H.M. Fungal Strategies to Evade the Host Immune Recognition. J. Fungi 2017, 3, 51. https://doi.org/10.3390/jof3040051
Hernández-Chávez MJ, Pérez-García LA, Niño-Vega GA, Mora-Montes HM. Fungal Strategies to Evade the Host Immune Recognition. Journal of Fungi. 2017; 3(4):51. https://doi.org/10.3390/jof3040051
Chicago/Turabian StyleHernández-Chávez, Marco J., Luis A. Pérez-García, Gustavo A. Niño-Vega, and Héctor M. Mora-Montes. 2017. "Fungal Strategies to Evade the Host Immune Recognition" Journal of Fungi 3, no. 4: 51. https://doi.org/10.3390/jof3040051
APA StyleHernández-Chávez, M. J., Pérez-García, L. A., Niño-Vega, G. A., & Mora-Montes, H. M. (2017). Fungal Strategies to Evade the Host Immune Recognition. Journal of Fungi, 3(4), 51. https://doi.org/10.3390/jof3040051