
Transcriptome Analysis Reveals the Molecular Mechanisms for Mycorrhiza-Enhanced Drought Tolerance in Maize by Regulating the Ca2+ Signaling Pathway

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Design and Statistical Analysis
2.2. Biological Material and Growth Conditions
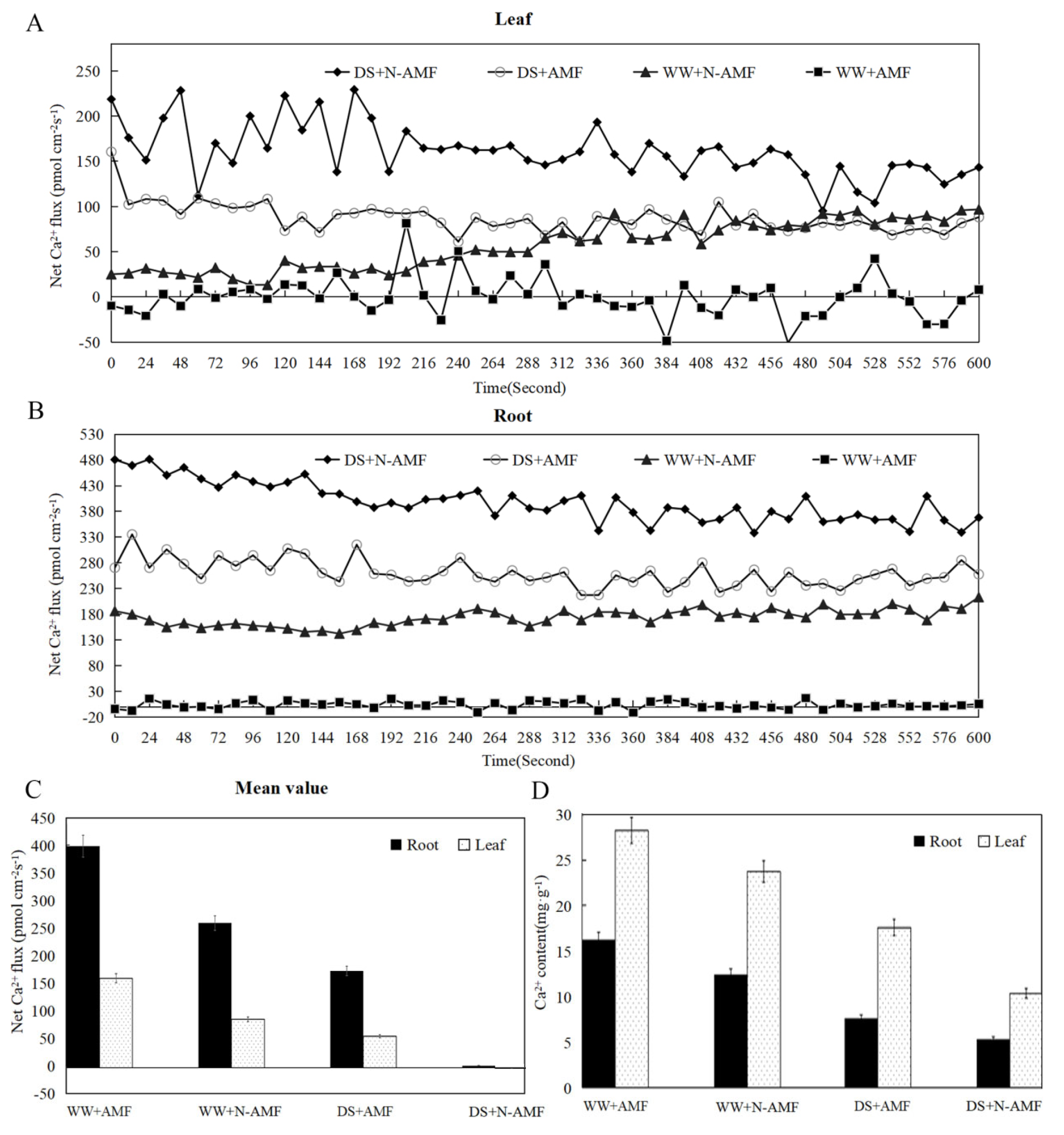
2.3. Detection of Ca2+ Fluxes and Contents in Maize Leaves and Roots
2.4. Total RNA Extraction, Library Preparation, and Illumina Hiseq Sequencing
2.5. De Novo Assembly, Functional Annotation, and Identification of Differential Expression Genes
2.6. Functional Enrichment of DEGS and Identification of Transcription Factor
2.7. Co-Expression Network Analysis
3. Results
3.1. Ca2+ Fluxes and Contents in Maize Roots and Leaves
3.2. Transcriptome Sequencing and Gene Annotation
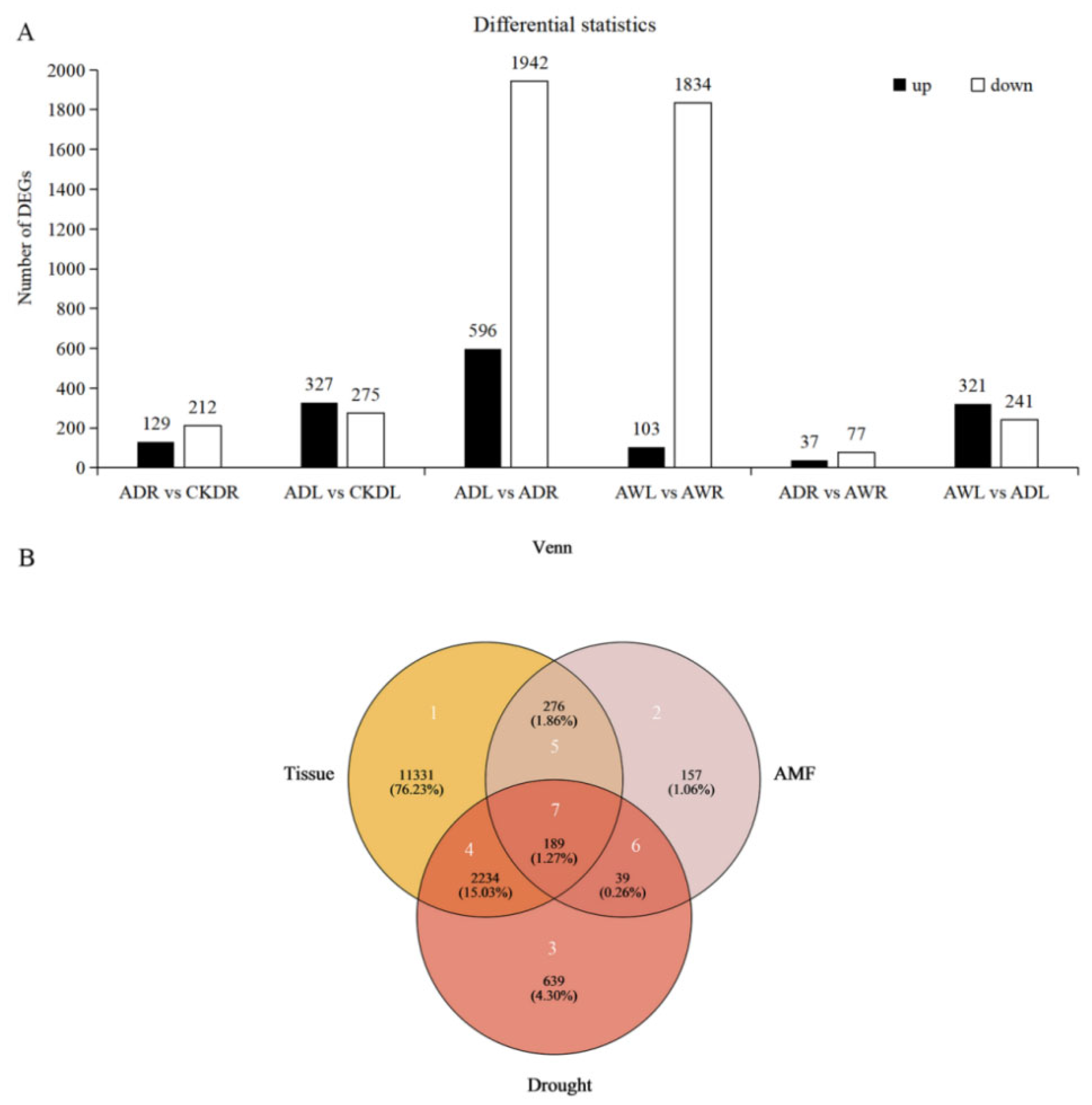
3.3. Identification of Differential Expression Genes (DEGs) Related to F. mosseae Inoculation and Drought Stress in Maize Roots and Leaves
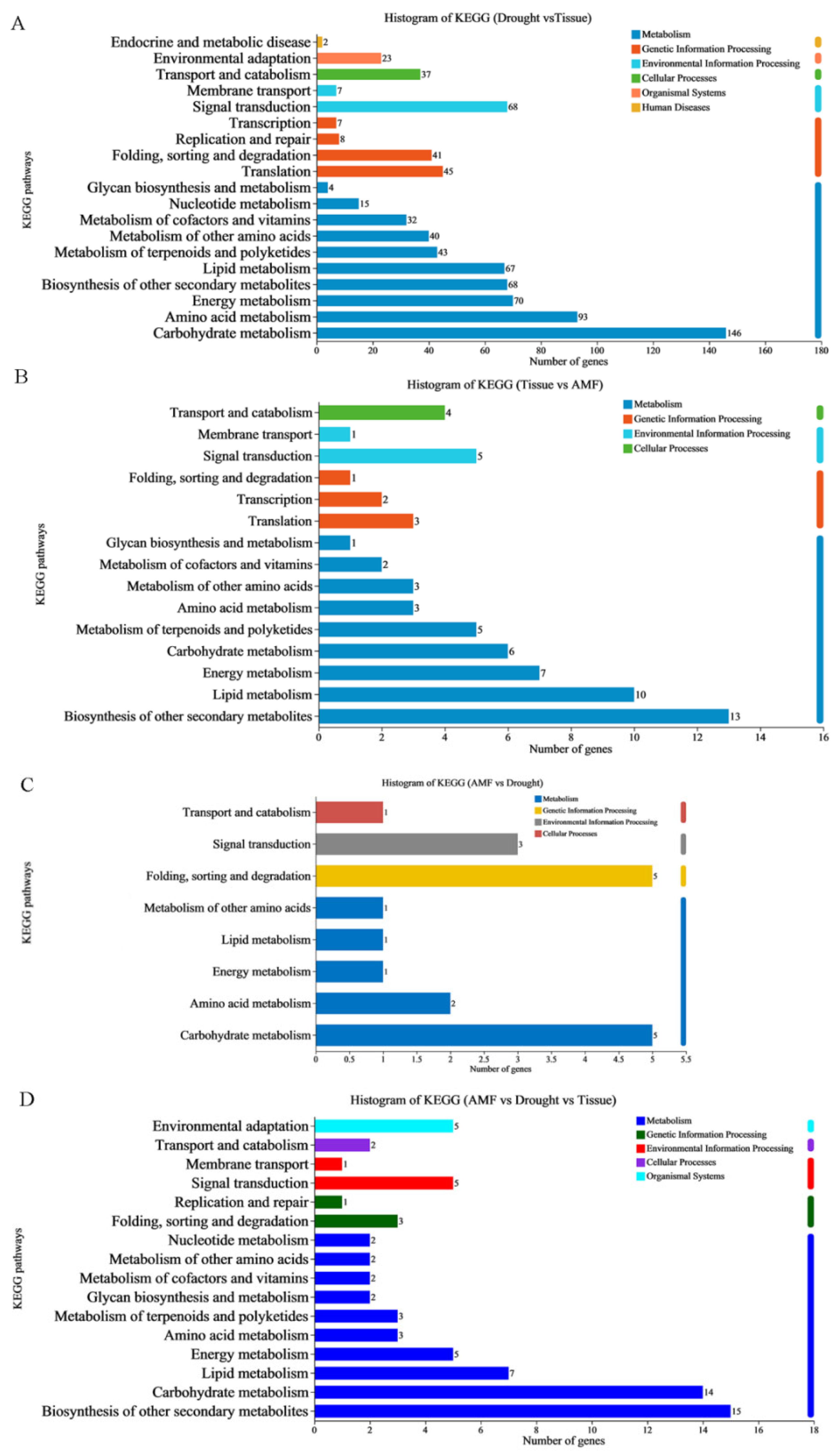
3.4. Functional Annotation of DEGs in Maize Leaves and Roots Induced by F. mosseae Inoculation Under Drought Stress
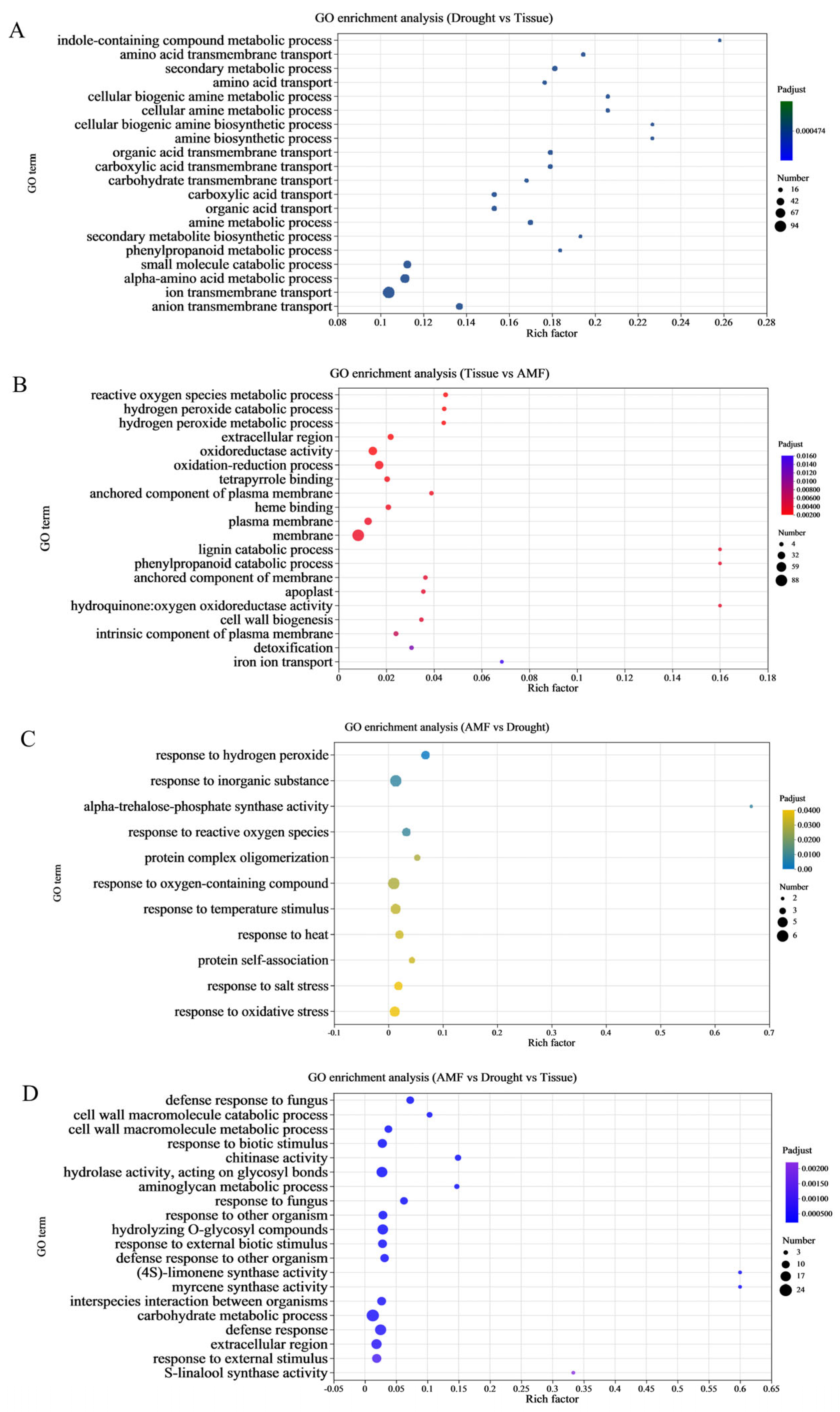
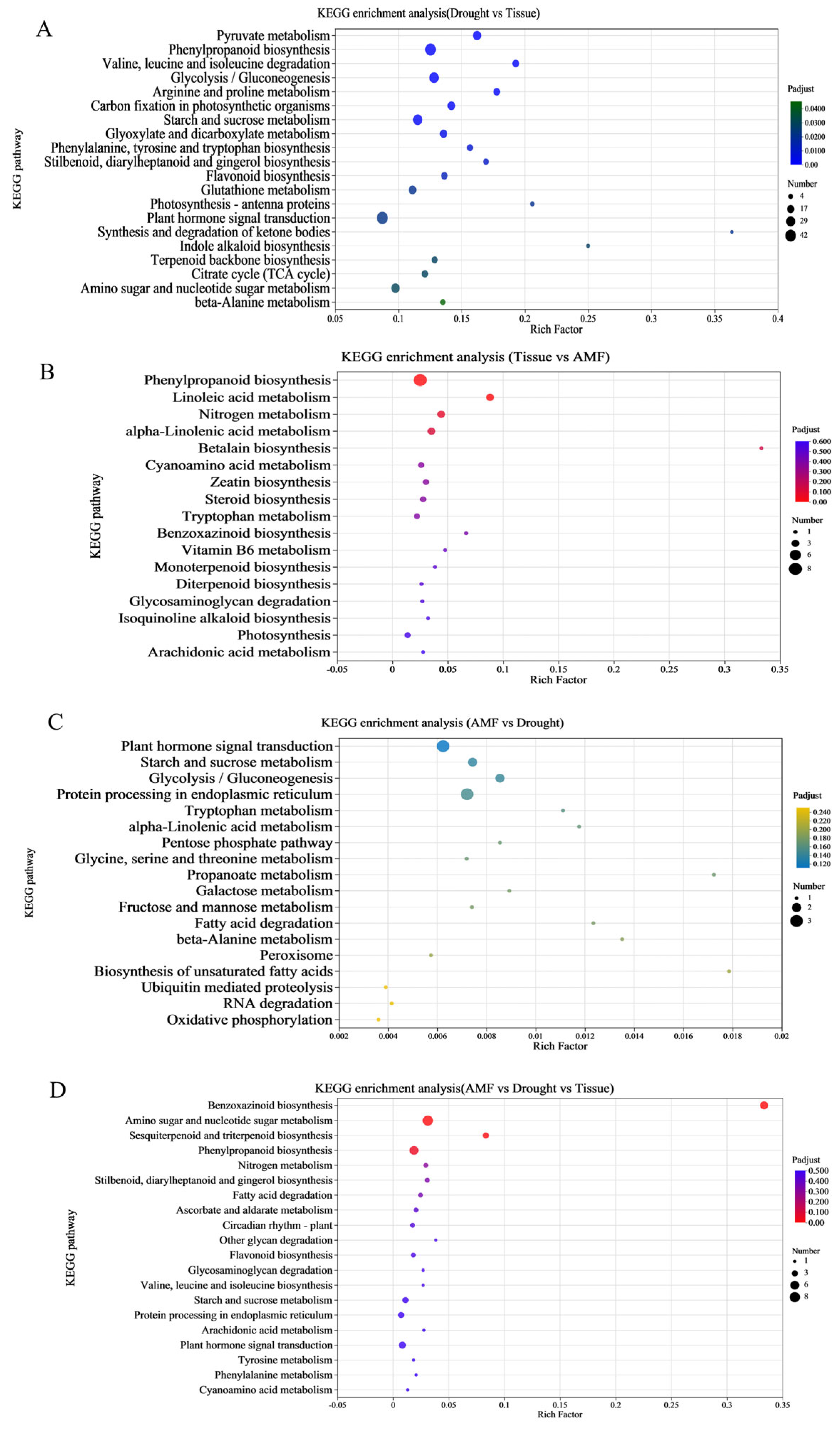
3.5. Functional Enrichment of DEGs in Maize Shoots and Roots Induced by F. mosseae Inoculation Under Drought Stress
3.6. Expression Patterns of DEGs Involved in Calcineurin B-like (CBL) Protein, CBL-Interacting Protein Kinase (CIPK), Mitogen Activated Protein Kinase (MAPK), and Calcium Dependent Protein Kinase (CDPK) Regulated by F. mosseae Inoculation Under Drought Stress
3.7. Identification and Co-Expression Network Analysis of Transcription Factors (Tfs)
4. Discussion
4.1. F. mosseae-Induced Ca2+ Fluxes and Contents in Maize Roots and Leaves Under Drought Stress
4.2. AMF-Induced CBL-CIPK Gene Expression in Maize Roots and Leaves Under Drought Stress
4.3. AMF-Induced MAPK Gene Expression in Maize Roots and Leaves Under Drought Stress
4.4. AMF-Induced CDPK Gene Expression in Maize Roots and Leaves Under Drought Stress
4.5. Regulation of Transcription Factors by F. mosseae Inoculation Under Drought Stress
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Quiroga, G.; Erice, G.; Aroca, R.; Delgado-Huertas, A.; Ruiz-Lozano, J.M. Elucidating the possible involvement of maize aquaporins and arbuscular mycorrhizal symbiosis in the plant ammonium and urea transport under drought stress conditions. Plants 2020, 9, 148. [Google Scholar] [CrossRef] [PubMed]
- Gong, M.; Bai, N.; Wang, P.; Su, J.; Chang, Q.; Zhang, Q. Co-inoculation with arbuscular mycorrhizal fungi and dark septate endophytes under drought stress: Synergistic or competitive effects on maize growth, photosynthesis, root hydraulic properties and aquaporins? Plants 2023, 12, 2596. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Ma, M.; Wang, Q.; Zhang, M.; Jing, G.; Li, C.; Ma, F. Arbuscular mycorrhizal fungi enhanced drought resistance in apple by regulating genes in the MAPK pathway. Plant Physiol. Biochem. 2020, 149, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Dong, F.; Wang, Y.; Chen, H.; Tang, M. Arbuscular mycorrhizal fungi enhance photosynthesis and drought tolerance by regulating MAPK genes expressions of Populus simonii x P. nigra. Physiol. Plant. 2022, 174, e13829. [Google Scholar] [CrossRef]
- Wang, C.; Yuan, Z.; Li, S.; Wang, W.; Xue, R.; Tai, F. Characterization of eight CBL genes expressions in maize early seeding development. Acta Physiol. Plant. 2014, 36, 3307–3314. [Google Scholar] [CrossRef]
- Zhu, P.; Song, X.; Mao, Y.; Li, Y.; Zhang, C. The flux rate of Ca2+ into embryo can be used to evaluate the vigour level of maize seeds. Qual. Assur. Saf. Crop. 2020, 12, 81–88. [Google Scholar] [CrossRef]
- Sharma, S.S.; Kumar, V.; Dietz, K.J. Emerging trends in metalloid-dependent signaling in plants. Trends Plant Sci. 2020, 26, 452–471. [Google Scholar] [CrossRef]
- Ju, C.; Zhang, Z.; Deng, J.; Miao, C.; Wang, Z.; Wallrad, L.; Javed, L.; Fu, D.; Zhang, T.; Kudla, J. Ca2+ dependent successive phosphorylation of vacuolar transporter MTP8 by CBL2/3-CIPK3/9/26 and CPK5 is critical for manganese homeostasis in Arabidopsis. Mol. Plant 2022, 15, 419–437. [Google Scholar] [CrossRef]
- Shu, B.; Cai, D.; Zhang, F.; Zhang, D.; Liu, C.; Wu, Q.; Luo, C. Identifying citrus CBL and CIPK gene families and their expressions in response to drought and arbuscular mycorrhizal fungi colonization. Biol. Plant 2020, 64, 773–783. [Google Scholar] [CrossRef]
- Shu, B.; Jue, D.; Zhang, F.; Zhang, D.; Liu, C.; Wu, Q.; Luo, C. Genome-wide identification and expression analysis of the citrus calcium-dependent protein kinase (CDPK) genes in response to arbuscular mycorrhizal fungi colonization and drought. Biotechnol. Biotechnol. Equip. 2020, 34, 1304–1314. [Google Scholar] [CrossRef]
- Liu, P.; Duan, Y.; Liu, C.; Xue, Q.; Guo, J.; Qi, T.; Kang, Z.; Guo, J. The calcium sensor TaCBL4 and its interacting protein TaCIPK5 are required for wheat resistance to stripe rust fungus. J. Exp. Bot. 2018, 69, 4443–4457. [Google Scholar] [CrossRef] [PubMed]
- Quiroga, G.; Erice, G.; Ding, L.; Chaumont, F.; Aroca, R.; Ruiz-Lozano, J.M. The arbuscular mycorrhizal symbiosis regulates aquaporins activity and improves root cell water permeability in maize plants subjected to water stress. Plant Cell Environ. 2019, 42, 2274–2290. [Google Scholar] [CrossRef] [PubMed]
- Asadollahi, M.; Iranbakhsh, A.; Ahmadvand, R.; Ebadi, M.; Mehregan, I. Synergetic effect of water deficit and arbuscular mycorrhizal symbiosis on the expression of aquaporins in wheat (Triticum aestivum L.) roots: Insights from NGS RNA-sequencing. Physiol. Mol. Biol. Plants 2023, 29, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.; Chen, Y.H.; Chang, K.C.; Yang, S.Y. Transcriptome analysis reveals the mechanisms for mycorrhiza-enhanced salt tolerance in rice. Front. Plant Sci. 2022, 13, 1072171. [Google Scholar] [CrossRef]
- Li, J.; Meng, B.; Chai, H.; Yang, X.; Song, W.; Li, S.; Lu, A.; Zhang, T.; Sun, W. Arbuscular mycorrhizal fungi alleviate drought stress in C3 (Leymus chinensis) and C4 (Hemarthria altissima) grasses via altering antioxidant enzyme activities and photosynthesis. Front. Plant Sci. 2019, 10, 499. [Google Scholar] [CrossRef]
- Quiroga, G.; Erice, G.; Aroca, R.; Chaumont, F.; Ruiz-Lozano, J.M. Enhanced drought stress tolerance by the arbuscular mycorrhizal symbiosis in a drought-sensitive maize cultivar is related to a broader and differential regulation of host plant aquaporins than in a drought-tolerant cultivar. Front. Plant Sci. 2017, 8, 1056. [Google Scholar] [CrossRef]
- Xie, W.; Hao, Z.; Zhou, J.; Fu, W.; Guo, L.; Zhang, X.; Chen, B. Integrated transcriptomics and metabolomics reveal specific phenolic and flavonoid accumulation in licorice (Glycyrrhiza uralensis Fisch.) induced by arbuscular mycorrhiza symbiosis under drought stress. Plant Physiol. Biochem. 2023, 205, 108173. [Google Scholar] [CrossRef]
- Wang, Y.; Zou, Y.N.; Shu, B.; Wu, Q.S. Deciphering molecular mechanisms regarding enhanced drought tolerance in plants by arbuscular mycorrhizal fungi. Sci. Hortic. 2023, 308, 111591. [Google Scholar] [CrossRef]
- Yang, X.; Lu, M.; Wang, Y.; Wang, Y.; Liu, Z.; Chen, S. Response mechanism of plants to drought stress. Horticulturae 2021, 7, 50. [Google Scholar] [CrossRef]
- Hussain, M.; Farooq, S.; Hasan, W.; Ul-Allah, S.; Tanveer, M.; Farooq, M.; Nawaz, A. Drought stress in sunflower: Physiological effects and its management through breeding and agronomic alternatives. Agric. Water Manag. 2018, 201, 152–166. [Google Scholar] [CrossRef]
- Li, T.; Chen, B.D. Arbuscular mycorrhizal fungi improving drought tolerance of maize plants by up-regulation of aquaporin gene expressions in roots and the fungi themselves. Chin. J. Plant Ecol. 2012, 36, 973. [Google Scholar] [CrossRef]
- Moradi Tarnabi, Z.; Iranbakhsh, A.; Mehregan, I.; Ahmadvand, R. Impact of arbuscular mycorrhizal fungi (AMF) on gene expression of some cell wall and membrane elements of wheat (Triticum aestivum L.) under water deficit using transcriptome analysis. Physiol. Mol. Biol. Plants 2020, 26, 143–162. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Gong, M.G.; Liu, K.Y.; Wei, Y.N.; Bai, N.; Qiu, Z.J.; Zhang, Q.M. Effects of arbuscular mycorrhizal fungi on photosynthetic characteristics and mesophyll cell ultrastructure of cotton under arsenic stress. Cotton Sci. 2022, 34, 256–266. [Google Scholar]
- Sanders, D.; Pelloux, J.; Brownlee, C.; Harper, J.F. Calcium at the crossroads of signaling. Plant Cell 2002, 14, S401–S417. [Google Scholar] [CrossRef]
- Lecourieux, D.; Ranjeva, R.; Pugin, A. Calcium in plant defence-signalling pathways. New Phytol. 2006, 171, 249–269. [Google Scholar] [CrossRef]
- Kurusu, T.; Hamada, J.; Nokajima, H.; Kitagawa, Y.; Kiyoduka, M.; Takahashi, A.; Hanamata, S.; Ohno, R.; Hayashi, T.; Okada, K.; et al. Regulation of microbe-associated molecular pattern-induced hypersensitive cell death, phytoalexin production, and defense gene expression by calcineurin B-Like protein-interacting protein kinases, OsCIPK14/15, in rice cultured cells. Plant Physiol. 2010, 153, 678–692. [Google Scholar] [CrossRef]
- Shao, H.B.; Song, W.Y.; Chu, L.Y. Advances of calcium signals involved in plant anti-drought. Comptes Rendus Biol. 2008, 331, 587–596. [Google Scholar] [CrossRef]
- Gong, M.; Bai, N.; Su, J.; Wang, Y.; Wei, Y.; Zhang, Q. Transcriptome analysis of Gossypium reveals the molecular mechanisms of Ca2+ signaling pathway on arsenic tolerance induced by arbuscular mycorrhizal fungi. Front. Microbiol. 2024, 15, 1362296. [Google Scholar] [CrossRef]
- Wang, G.; Jin, Z.; George, T.S.; Feng, G.; Zhang, L. Arbuscular mycorrhizal fungi enhance plant phosphorus uptake through stimulating hyphosphere soil microbiome functional profiles for phosphorus turnover. New Phytol. 2023, 238, 2578–2593. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Li, Q.H.; Yu, Y.N.; Qiao, Y.M.; Haq, S.U.; Gong, Z.H. The CBL-CIPK pathway in plant response to stress signals. Int. J. Mol. Sci. 2020, 21, 5668. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Li, H.; Liu, Z.N.; Wang, X.H.; Xu, P.; Dai, S.J.; Cao, X.; Cui, X.Y. The soybean CBL-interacting protein kinase, GmCIPK2, positively regulates drought tolerance and ABA signaling. Plant Physiol. Biochem. 2021, 167, 980–989. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Huang, Y.; Xiong, L. Characterization of stress-responsive CIPK genes in rice for stress tolerance improvement. Plant Physiol. 2007, 144, 1416–1428. [Google Scholar] [CrossRef]
- Aslam, M.; Fakher, B.; Jakada, B.H.; Zhao, L.; Cao, S.; Cheng, Y.; Qin, Y. Genome-wide identification and expression profiling of CBL-CIPK gene family in pineapple (Ananas comosus) and the role of AcCBL1 in abiotic and biotic stress response. Biomolecules 2019, 9, 293. [Google Scholar] [CrossRef]
- Tai, F.; Wang, Q.; Yuan, Z.; Yuan, Z.; Li, H.; Wang, W. Characterization of five CIPK gene expressions in maize under water stress. Acta Physiol. Plant. 2013, 35, 1555–1564. [Google Scholar] [CrossRef]
- Tai, F.; Yuan, Z.; Li, S.; Wang, Q.; Liu, F.; Wang, W. ZmCIPK8, a CBL-interacting protein kinase, regulates maize response to drought stress. Plant Cell Tissue Organ Cult. 2016, 124, 459–469. [Google Scholar] [CrossRef]
- Yao, Y.; Zhao, H.; Sun, L.; Wu, W.; Li, C.; Wu, Q. Genome-wide identification of MAPK gene family members in Fagopyrum tataricum and their expression during development and stress responses. BMC Genom. 2022, 23, 96. [Google Scholar] [CrossRef]
- Moustafa, K.; AbuQamar, S.; Jarrar, M.; Al-Rajab, A.J.; Trémouillaux-Guiller, J. MAPK cascades and major abiotic stresses. Plant Cell Rep. 2014, 33, 1217–1225. [Google Scholar] [CrossRef]
- Xiong, L.Z.; Yang, Y.N. Disease resistance and abiotic stress tolerance in rice are inversely modulated by an abscisic acid-inducible mitogen-activated protein kinase. Plant Cell 2003, 15, 745–759. [Google Scholar] [CrossRef]
- Liu, Z.; Li, Y.; Ma, L.; Wei, H.; Zhang, J.; He, X.; Tian, C. Coordinated regulation of arbuscular mycorrhizal fungi and soybean MAPK pathway genes improved mycorrhizal soybean drought tolerance. Mol. Plant-Microbe Interact. 2015, 28, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Hu, W.; Deng, X.; Zhang, Y.; Liu, X.; Zhao, X.; Luo, Q.; Jin, Z.; Li, Y.; Zhou, S. A rice calcium-dependent protein kinase OsCPK9 positively regulates drought stress tolerance and spikelet fertility. BMC Plant Biol. 2014, 14, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Yip Delormel, T.; Boudsocq, M. Properties and functions of calcium-dependent protein kinases and their relatives in Arabidopsis thaliana. New Phytol. 2019, 224, 585–604. [Google Scholar] [CrossRef]
- Coca, M.; San Segundo, B. AtCPK1 calcium-dependent protein kinase mediates pathogen resistance in Arabidopsis. Plant J. 2010, 63, 526–540. [Google Scholar] [CrossRef]
- Zhu, S.Y.; Yu, X.C.; Wang, X.J.; Zhao, R.; Li, Y.; Fan, R.C.; Shang, Y.; Du, S.Y.; Wang, X.F.; Wu, F.Q. Two calcium-dependent protein kinases, CPK4 and CPK11, regulate abscisic acid signal transduction in Arabidopsis. Plant Cell 2007, 19, 3019–3036. [Google Scholar] [CrossRef]
- Saijo, Y.; Hata, S.; Kyozuka, J.; Shimamoto, K.; Izui, K. Over-expression of a single Ca2+ dependent protein kinase confers both cold and salt/drought tolerance on rice plants. Plant J. 2000, 23, 319–327. [Google Scholar] [CrossRef]
- Asano, T.; Hayashi, N.; Kobayashi, M.; Aoki, N.; Miyao, A.; Mitsuhara, I.; Ichikawa, H.; Komatsu, S.; Hirochika, H.; Kikuchi, S.; et al. A rice calcium-dependent protein kinase OsCPK12 oppositely modulates salt-stress tolerance and blast disease resistance. Plant J. 2012, 69, 26–36. [Google Scholar] [CrossRef]
- Wang, Y.; Dong, F.; Tang, M. Transcriptome analysis of arbuscular mycorrhizal Casuarina glauca in damage mitigation of roots on NaCl stress. Microorganisms 2021, 10, 15. [Google Scholar] [CrossRef]
- Cervantes-Gámez, R.G.; Bueno-Ibarra, M.A.; Cruz-Mendívil, A.; Calderón-Vázquez, C.L.; Ramírez-Douriet, C.M.; Maldonado-Mendoza, I.E.; Villalobos-López, M.Á.; Valdez-Ortiz, Á.; López-Meyer, M. Arbuscular mycorrhizal symbiosis-induced expression changes in Solanum lycopersicum leaves revealed by RNA-seq analysis. Plant Mol. Biol. Rep. 2016, 34, 89–102. [Google Scholar] [CrossRef]
- Corso, M.; Doccula, F.G.; de Melo, J.R.F.; Costa, A.; Verbruggen, N. Endoplasmic reticulum-localized CCX2 is required for osmotolerance by regulating ER and cytosolic Ca²⁺ dynamics in Arabidopsis. Proc. Natl. Acad. Sci. USA 2018, 115, 3966–3971. [Google Scholar] [CrossRef]
- Ouziad, F.; Wilde, P.; Schmelzer, E.; Hildebrandt, U.; Bothe, H. Analysis of expression of aquaporins and Na⁺/H⁺ transporters in tomato colonized by arbuscular mycorrhizal fungi and affected by salt stress. Environ. Exp. Bot. 2006, 57, 177–186. [Google Scholar] [CrossRef]
- Aroca, R.; Porcel, R.; Ruiz-Lozano, J.M. How does arbuscular mycorrhizal symbiosis regulate root hydraulic properties and plasma membrane aquaporins in Phaseolus vulgaris under drought, cold or salinity stresses? New Phytol. 2007, 173, 808–816. [Google Scholar] [CrossRef] [PubMed]
- Puccio, G.; Ingraffia, R.; Mercati, F.; Amato, G.; Giambalvo, D.; Martinelli, F.; Sunseri, F.; Frenda, A.S. Transcriptome changes induced by arbuscular mycorrhizal symbiosis in leaves of durum wheat (Triticum durum Desf.) promote higher salt tolerance. Sci. Rep. 2023, 13, 116. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.E.; Basnayake, B.V.S.; Zhang, H.; Li, G.; Li, W.; Virk, N.; Mengiste, T.; Song, F. The Arabidopsis ATAF1, a NAC transcription factor, is a negative regulator of defense responses against necrotrophic fungal and bacterial pathogens. Mol. Plant-Microbe Interact. 2009, 22, 1227–1238. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (A) Statistics of Transcriptome Sequencing | ||||||||||
| Sample | Raw Reads | Clean Reads | Clean Reads Ratio (%) | Q20% | Q30% | GC% | Total Reads | Total Mapped | Multiple Mapped | Uniquely Mapped |
| AWR_3 | 49004258 | 48267150 | 0.98 | 97.74 | 93.78 | 52.69 | 48267150 | 39998058(82.87%) | 2246287(4.65%) | 37751771(78.21%) |
| AWR_2 | 43278510 | 42415960 | 0.98 | 97.88 | 94.07 | 52.59 | 42415960 | 28480510(67.15%) | 1675997(3.95%) | 26804513(63.19%) |
| AWR_1 | 44510978 | 43872336 | 0.99 | 97.83 | 93.98 | 52.26 | 43872336 | 34821451(79.37%) | 3450861(7.87%) | 31370590(71.5%) |
| AWL_3 | 49623792 | 48947880 | 0.99 | 97.74 | 93.8 | 53.91 | 48947880 | 42329513(86.48%) | 2918391(5.96%) | 39411122(80.52%) |
| AWL_2 | 41304944 | 40581566 | 0.98 | 97.69 | 93.84 | 53.36 | 40581566 | 32763271(80.73%) | 2481943(6.12%) | 30281328(74.62%) |
| AWL_1 | 55064656 | 54262806 | 0.99 | 98.16 | 94.67 | 51.53 | 54262806 | 42882808(79.03%) | 1924899(3.55%) | 40957909(75.48%) |
| ADR_3 | 46985536 | 46484262 | 0.99 | 97.98 | 94.3 | 53.55 | 46484262 | 37870479(81.47%) | 1800566(3.87%) | 36069913(77.6%) |
| ADR_2 | 43801530 | 43222778 | 0.99 | 98 | 94.3 | 52.01 | 43222778 | 35468866(82.06%) | 1715287(3.97%) | 33753579(78.09%) |
| ADR_1 | 46029238 | 45542410 | 0.99 | 97.93 | 94.13 | 53.15 | 45542410 | 39682609(87.13%) | 1576485(3.46%) | 38106124(83.67%) |
| ADL_3 | 46078662 | 45479426 | 0.99 | 98.03 | 94.43 | 53.75 | 45479426 | 38905176(85.54%) | 3218143(7.08%) | 35687033(78.47%) |
| ADL_2 | 41448764 | 40812670 | 0.98 | 97.97 | 94.3 | 52.4 | 40812670 | 32832809(80.45%) | 2032086(4.98%) | 30800723(75.47%) |
| ADL_1 | 44821098 | 44248464 | 0.99 | 97.82 | 93.91 | 53.71 | 44248464 | 37521200(84.8%) | 2519531(5.69%) | 35001669(79.1%) |
| CKWR_3 | 41299570 | 40841682 | 0.99 | 97.97 | 94.23 | 51.98 | 40841682 | 33667906(82.44%) | 1656725(4.06%) | 32011181(78.38%) |
| CKWR_2 | 44071930 | 43511138 | 0.99 | 97.8 | 93.87 | 51.96 | 43511138 | 34440119(79.15%) | 1427552(3.28%) | 33012567(75.87%) |
| CKWR_1 | 44310396 | 43754414 | 0.99 | 97.89 | 94.01 | 51.74 | 43754414 | 34045241(77.81%) | 1375476(3.14%) | 32669765(74.67%) |
| CKWL_3 | 45821846 | 45246014 | 0.99 | 97.92 | 94.16 | 53.51 | 45246014 | 36643359(80.99%) | 3637806(8.04%) | 33005553(72.95%) |
| CKWL_2 | 47213008 | 46625466 | 0.99 | 98.08 | 94.53 | 53.53 | 46625466 | 39198700(84.07%) | 2417415(5.18%) | 36781285(78.89%) |
| CKWL_1 | 43116158 | 42534804 | 0.99 | 98.02 | 94.41 | 53.24 | 42534804 | 34386366(80.84%) | 3106046(7.3%) | 31280320(73.54%) |
| CKDR_3 | 46665196 | 46112606 | 0.99 | 97.94 | 94.17 | 53.12 | 46112606 | 33263877(72.14%) | 1249731(2.71%) | 32014146(69.43%) |
| CKDR_2 | 45748734 | 45274614 | 0.99 | 98.07 | 94.52 | 53.74 | 45274614 | 38031581(84.0%) | 2062495(4.56%) | 35969086(79.45%) |
| CKDR_1 | 41104330 | 40618086 | 0.99 | 97.99 | 94.29 | 52.39 | 40618086 | 34492466(84.92%) | 1618976(3.99%) | 32873490(80.93%) |
| CKDL_3 | 40917860 | 40519570 | 0.99 | 98.13 | 94.63 | 53.74 | 40519570 | 34016680(83.95%) | 2245287(5.54%) | 31771393(78.41%) |
| CKDL_2 | 43367096 | 42950066 | 0.99 | 98.11 | 94.61 | 53.62 | 42950066 | 34668701(80.72%) | 2233725(5.2%) | 32434976(75.52%) |
| CKDL_1 | 44047556 | 43590294 | 0.99 | 98.05 | 94.49 | 54.14 | 43590294 | 34692789(79.59%) | 2555542(5.86%) | 32137247(73.73%) |
| (B) Annotation Analysis of Expressed Genes and Transcripts | ||||||||||
| DB Name | Expressed Gene Number (Percent) | Expressed Transcript Number (Percent) | All Gene Number (Percent) | All Transcript Number (Percent) | ||||||
| GO | 35283(0.7551) | 138412(0.8598) | 40189(0.7059) | 154937(0.8424) | ||||||
| KEGG | 16514(0.3534) | 80595(0.5006) | 18538(0.3256) | 89473(0.4865) | ||||||
| COG | 37828(0.8096) | 149246(0.9271) | 42635(0.7489) | 166372(0.9046) | ||||||
| NR | 42327(0.9059) | 155685(0.9671) | 48510(0.8521) | 174495(0.9488) | ||||||
| Swiss-Prot | 28570(0.6114) | 125447(0.7792) | 31367(0.5509) | 138779(0.7546) | ||||||
| Pfam | 29227(0.6255) | 116275(0.7223) | 31933(0.5609) | 127784(0.6948) | ||||||
| Total annotation | 42446(0.9084) | 155829(0.968) | 48662(0.8547) | 174675(0.9497) | ||||||
| Total | 46725(1.0) | 160987(1.0) | 56933(1) | 183919(1) | ||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Q.; Yang, W.; Wang, M.; Chen, J.; Zhang, Z.; Wei, Y.; Chang, Q.; Gong, M. Transcriptome Analysis Reveals the Molecular Mechanisms for Mycorrhiza-Enhanced Drought Tolerance in Maize by Regulating the Ca2+ Signaling Pathway. J. Fungi 2025, 11, 375. https://doi.org/10.3390/jof11050375
Zhang Q, Yang W, Wang M, Chen J, Zhang Z, Wei Y, Chang Q, Gong M. Transcriptome Analysis Reveals the Molecular Mechanisms for Mycorrhiza-Enhanced Drought Tolerance in Maize by Regulating the Ca2+ Signaling Pathway. Journal of Fungi. 2025; 11(5):375. https://doi.org/10.3390/jof11050375
Chicago/Turabian StyleZhang, Qiaoming, Wenjing Yang, Miaomiao Wang, Junwei Chen, Zhaoran Zhang, Yanan Wei, Qingshan Chang, and Minggui Gong. 2025. "Transcriptome Analysis Reveals the Molecular Mechanisms for Mycorrhiza-Enhanced Drought Tolerance in Maize by Regulating the Ca2+ Signaling Pathway" Journal of Fungi 11, no. 5: 375. https://doi.org/10.3390/jof11050375
APA StyleZhang, Q., Yang, W., Wang, M., Chen, J., Zhang, Z., Wei, Y., Chang, Q., & Gong, M. (2025). Transcriptome Analysis Reveals the Molecular Mechanisms for Mycorrhiza-Enhanced Drought Tolerance in Maize by Regulating the Ca2+ Signaling Pathway. Journal of Fungi, 11(5), 375. https://doi.org/10.3390/jof11050375