
Improving the Proteome-Mining of Schizophyllum commune to Enhance Medicinal Mushroom Applications

 ,
,  ,
,  ,
,  ,
,  , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Fungal Strain Isolation, Identification, and Conservation
2.2. Mycelium Growth
2.3. Protein Extraction Procedures
2.4. LC-MS/MS Analysis
2.5. Protein Identification, Functional and Statistical Analysis
3. Results
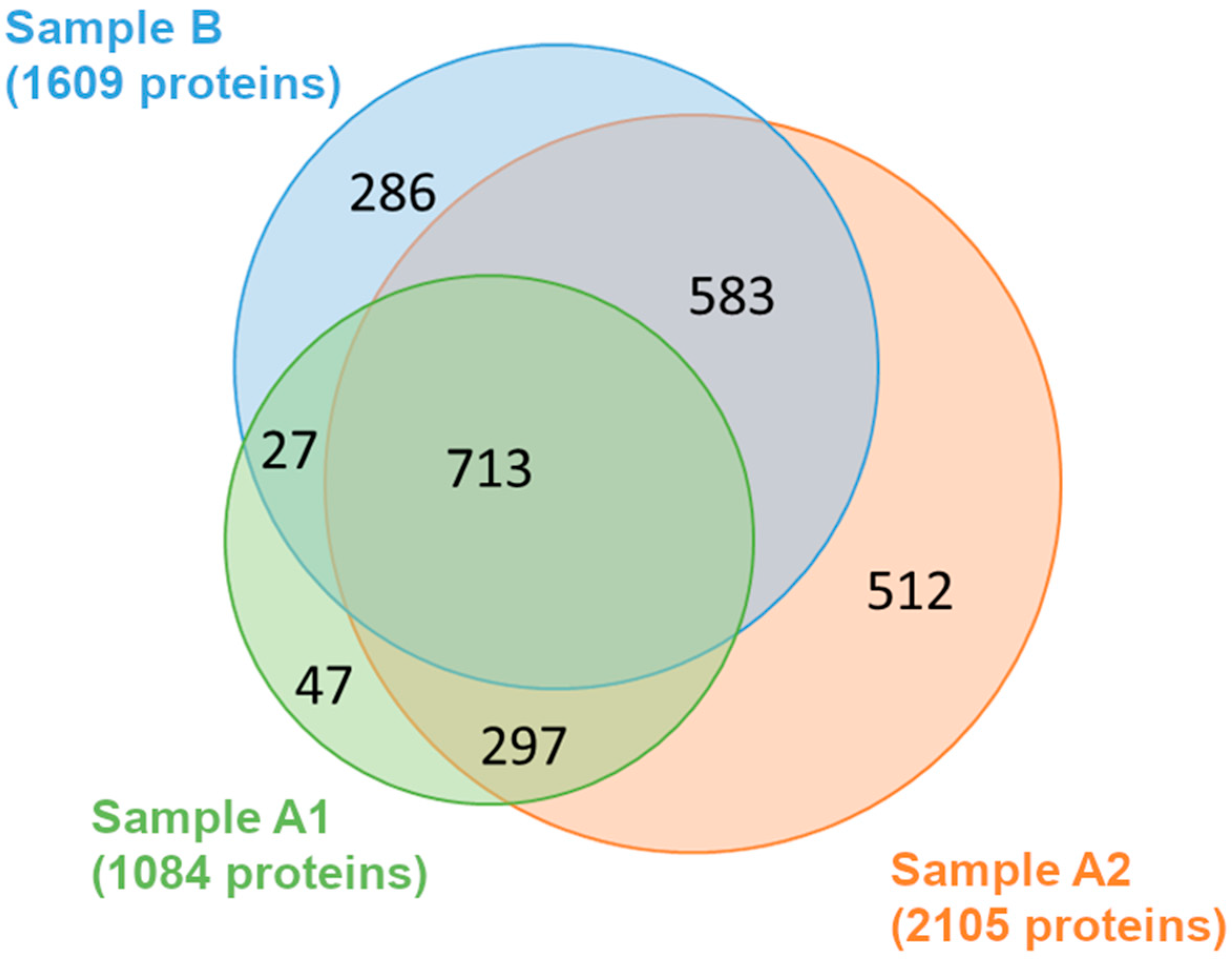
3.1. Different Protein Content and Proteomic Identification in the Three Extracts
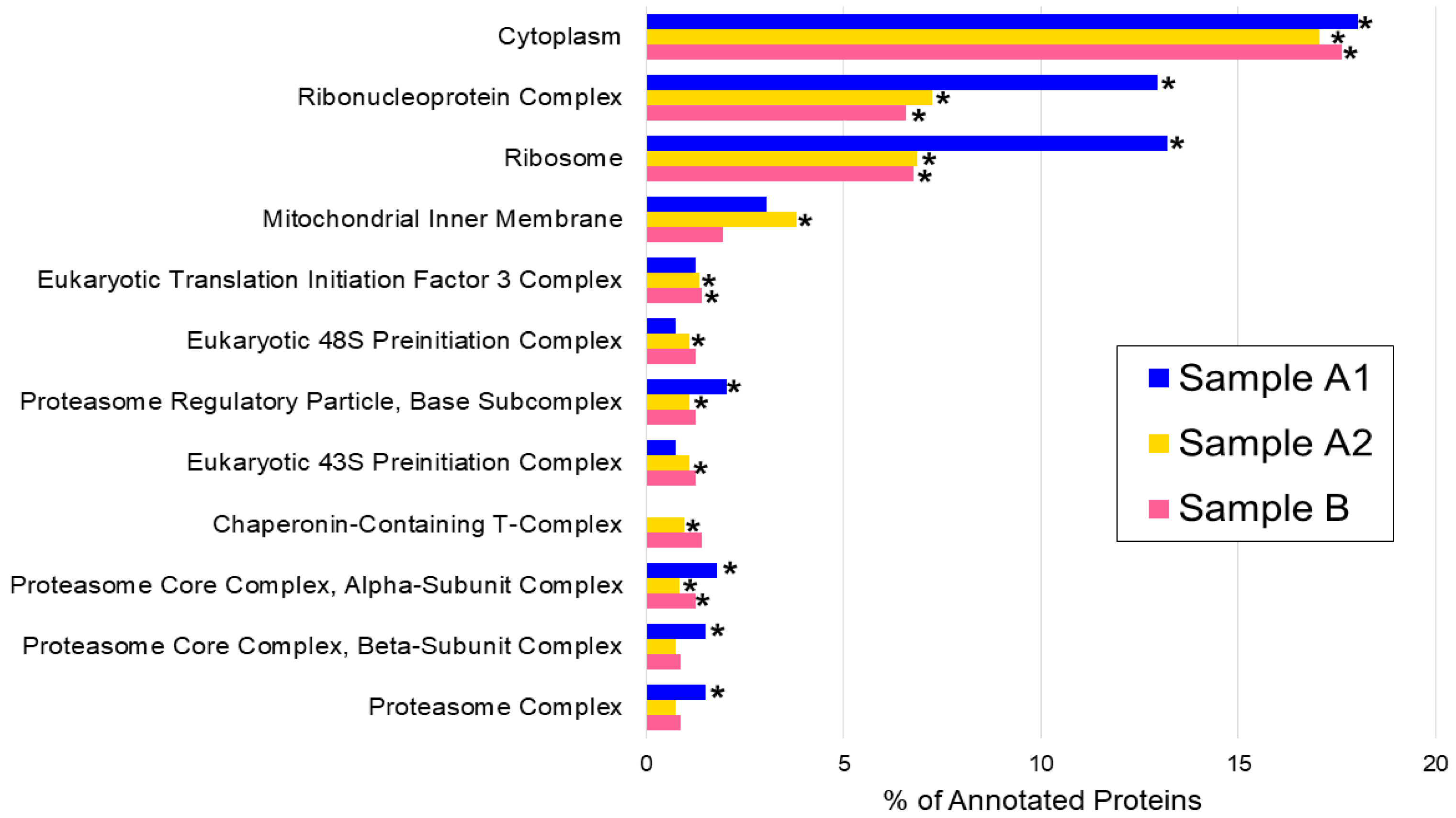
3.2. Cellular Compartmentalization of the Extracted Proteins: The Gene Ontology Cellular Component Analysis
3.3. Functional Analysis and Characterization of the Extracted Proteins
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| BP | Biological process |
| CAZymes | Carbohydrate-active enzymes |
| CC | Cellular Component |
| GO | Gene Ontology |
| LC-MS/MS | Liquid Chromatography with tandem mass spectrometry |
| ME | Malt Extract |
| MEA | Malt Extract Agar |
| PSM | Peptide Spectrum Match |
| TCA | Trichloroacetic acid |
References
- Venturella, G.; Ferraro, V.; Cirlincione, F.; Gargano, M.L. Medicinal mushrooms: Bioactive compounds, use, and clinical trials. Int. J. Mol. Sci. 2021, 22, 634. [Google Scholar] [CrossRef] [PubMed]
- Tee, P.Y.E.; Krishnan, T.; Cheong, X.T.; Maniam, S.A.; Looi, C.Y.; Ooi, Y.Y.; Chua, L.L.C.; Fung, S.Y.; Chia, A.Y.Y. A review on the cultivation, bioactive compounds, health-promoting factors and clinical trials of medicinal mushrooms Taiwanofungus camphoratus, Inonotus obliquus and Tropicoporus linteus. Fungal Biol. Biotechnol. 2024, 11, 7. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Dong, Q.; Chen, M.; Zhao, R.; Zha, L.; Zhao, Y.; Zhang, M.; Zhang, B.; Ma, A. The effect of mushroom dietary fiber on the gut microbiota and related health benefits: A review. J. Fungi 2023, 9, 1028. [Google Scholar] [CrossRef] [PubMed]
- Abd Razak, D.L.; Abd Ghani, A.; Lazim, M.I.M.; Khulidin, K.A.; Shahidi, F.; Ismail, A. Schizophyllum commune (Fries) mushroom: A review on its nutritional components, antioxidative, and anti-inflammatory properties. Curr. Opin. Food Sci. 2024, 56, 101129. [Google Scholar] [CrossRef]
- Zhang, Y.; Kong, H.; Fang, Y.; Nishinari, K.; Phillips, G.O. Schizophyllan: A review on its structure, properties, bioactivities and recent developments. Bioact. Carbohydr. Diet. Fibre. 2013, 1, 53–71. [Google Scholar] [CrossRef]
- Zhong, K.; Tong, L.; Liu, L.; Zhou, X.; Liu, X.; Zhang, Q.; Zhou, S. Immunoregulatory and antitumor activity of schizophyllan under ultrasonic treatment. Int. J. Biol. Macromol. 2015, 80, 302–308. [Google Scholar] [CrossRef]
- Gou, Z.; Peng, Z.; Wang, S.; Chen, L.; Ma, Z.; Kang, Y.; Kang, I.; Sun, L.; Wang, R.; Xu, H.; et al. Efficient production and skincare activity evaluation of schizophyllan, a β-glucan derived from Schizophyllum commune NTU-1. Int. J. Biol. Macromol. 2023, 241, 124504. [Google Scholar] [CrossRef]
- Mišković, J.; Karaman, M.; Rašeta, M.; Krsmanović, N.; Berežni, S.; Jakovljević, D.; Piattoni, F.; Zambonelli, A.; Gargano, M.L.; Venturella, G. Comparison of two Schizophyllum commune strains in production of acetylcholinesterase inhibitors and antioxidants from submerged cultivation. J. Fungi 2021, 7, 115. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, N.; Aslam, S.; Hussain, N.; Bilal, M.; Iqbal, H.M. Transforming lignin biomass to value: Interplay between ligninolytic enzymes and lignocellulose depolymerization. BioEnergy Res. 2023, 16, 1246–1263. [Google Scholar] [CrossRef]
- Kumar, A.; Bharti, A.K.; Bezie, Y. Schizophyllum commune: A fungal cell-factory for production of valuable metabolites and enzymes. BioResources 2022, 17, 5420–5436. [Google Scholar] [CrossRef]
- Basso, V.; Schiavenin, C.; Mendonça, S.; de Siqueira, F.G.; Salvador, M.; Camassola, M. Chemical features and antioxidant profile by Schizophyllum commune produced on different agroindustrial wastes and byproducts of biodiesel production. Food Chem. 2020, 329, 127089. [Google Scholar] [CrossRef]
- Asgher, M.; Wahab, A.; Bilal, M.; Iqbal, H.M.N. Lignocellulose degradation and production of lignin modifying enzymes by Schizophyllum commune IBL-06 in solid-state fermentation. Biocatal. Agric. Biotechnol. 2016, 6, 195–201. [Google Scholar] [CrossRef]
- Tovar-Herrera, O.E.; Martha-Paz, A.M.; Pérez-Llano, Y.; Aranda, E.; Tacoronte-Morales, J.E.; Pedroso-Cabrera, M.T.; Arévalo-Niño, K.; Folch-Mallol, J.L.; Batista-García, R.A. Schizophyllum commune: An unexploited source for lignocellulose degrading enzymes. MicrobiologyOpen 2018, 7, e00637. [Google Scholar] [CrossRef] [PubMed]
- Vonk, P.J. Functional Genomics in Schizophyllum commune. Ph.D. Thesis, Utrecht University, Utrecht, The Netherlands, 2022. [Google Scholar]
- Appels, F.V.; Dijksterhuis, J.; Lukasiewicz, C.E.; Jansen, K.M.; Wösten, H.A.; Krijgsheld, P. Hydrophobin gene deletion and environmental growth conditions impact mechanical properties of mycelium by affecting the density of the material. Sci. Rep. 2018, 8, 4703. [Google Scholar] [CrossRef] [PubMed]
- Shankar, A.; Sharma, K.K. Fungal Secondary Metabolites in Food and Pharmaceuticals in the Era of Multi-Omics. Appl. Microbiol. Biotechnol. 2022, 106, 3465–3488. [Google Scholar] [CrossRef]
- Giddey, K.; Monod, M.; Barblan, J.; Potts, A.; Waridel, P.; Zaugg, C.; Quadroni, M. Comprehensive analysis of proteins secreted by Trichophyton rubrum and Trichophyton violaceum under in vitro conditions. J. Proteome Res. 2007, 6, 3081–3092. [Google Scholar] [CrossRef] [PubMed]
- Leng, W.; Liu, T.; Li, R.; Yang, J.; Wie, C.; Zhang, W.; Jin, Q. Proteomic profile of dormant Trichophyton rubrum conidia. BMC Genom. 2008, 9, 303. [Google Scholar] [CrossRef] [PubMed]
- Jakopovic, B.; Oršolić, N.; Kraljević Pavelić, S.; Jakopovich, I. Proteomic Research on the Therapeutic Properties of Medicinal Mushrooms. In Mushrooms with Therapeutic Potentials: Recent Advances in Research and Development; Springer Nature: Singapore, 2023; pp. 309–344. [Google Scholar]
- Adav, S.S.; Chao, L.T.; Sze, S.K. Quantitative secretomic analysis of Trichoderma reesei strains reveals enzymatic composition for lignocellulosic biomass degradation. Mol. Cell. Proteom. 2012, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Yin, H.; Peng, H.; Lu, G.; Liu, Z.; Dang, Z. Identification of novel pathways for biotransformation of tetrabromobisphenol A by Phanerochaete chrysosporium, combined with mechanism analysis at proteome level. Sci. Total Environ. 2019, 659, 1352–1361. [Google Scholar] [CrossRef] [PubMed]
- Doria, E.; Altobelli, E.; Girometta, C.; Nielsen, E.; Zhang, T.; Savino, E. Evaluation of lignocellulolytic activities of ten fungal species able to degrade poplar wood. Int. Biodeter. Biodegrad. 2014, 94, 160–166. [Google Scholar] [CrossRef]
- Zhu, N.; Liu, J.; Yang, J.; Lin, Y.; Yang, Y.; Ji, L.; Li, M.; Yuan, H. Comparative analysis of the secretomes of Schizophyllum commune and other wood-decay basidiomycetes during solid-state fermentation reveals its unique lignocellulose-degrading enzyme system. Biotechnol. Biofuels 2016, 9, 42. [Google Scholar] [CrossRef]
- Li, G.; Jian, T.; Liu, X.; Lv, Q.; Zhang, G.; Ling, J. Application of metabolomics in fungal research. Molecules 2022, 27, 7365. [Google Scholar] [CrossRef]
- Saw, N.M.M.T.; Suwanchaikasem, P.; Zuniga-Montanez, R.; Qiu, G.; Marzinelli, E.M.; Wuertz, S.; Williams, R.B. Influence of extraction solvent on nontargeted metabolomics analysis of enrichment reactor cultures performing enhanced biological phosphorus removal (EBPR). Metabolites 2021, 11, 269. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.K.; Kumar, M.; Thakur, I.S. Proteomic characterization and schizophyllan production by Schizophyllum commune ISTL04 cultured on Leucaena leucocephala wood under submerged fermentation. Bioresour. Technol. 2017, 236, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Ujor, V.C.; Peiris, D.G.; Monti, M.; Kang, A.S.; Clements, M.O.; Hedger, J.N. Quantitative proteomic analysis of the response of the wood-rot fungus, Schizophyllum commune, to the biocontrol fungus, Trichoderma viride. Lett. Appl. Microbiol. 2012, 54, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Murry, R.; Traxler, L.; Pötschner, J.; Krüger, T.; Kniemeyer, O.; Krause, K.; Kothe, E. Inositol signaling in the basidiomycete fungus Schizophyllum commune. J. Fungi. 2021, 7, 470. [Google Scholar] [CrossRef] [PubMed]
- Girometta, C.E.; Bernicchia, A.; Baiguera, R.M.; Bracco, F.; Buratti, S.; Cartabia, M.; Picco, A.M.; Savino, E. An Italian Research Culture Collection of Wood Decay Fungi. Diversity 2020, 12, 58. [Google Scholar] [CrossRef]
- Cartabia, M.; Girometta, C.E.; Baiguera, R.M.; Buratti, S.; Babbini, S.; Bernicchia, A.; Savino, E. Lignicolous fungi collected in northern Italy: Identification and morphological description of isolates. Diversity 2022, 14, 413. [Google Scholar] [CrossRef]
- Ham, T.H.; Lee, Y.; Kwon, S.W.; Jang, M.J.; Park, Y.J.; Lee, J. Increasing Coverage of Proteome Identification of the Fruiting Body of Agaricus bisporus by Shotgun Proteomics. Foods 2020, 9, 632. [Google Scholar] [CrossRef] [PubMed]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; García-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022, 50, D543–D552. [Google Scholar] [CrossRef]
- Pathan, M.; Keerthikumar, S.; Ang, C.S.; Gangoda, L.; Quek, C.Y.; Williamson, N.A.; Mouradov, D.; Sieber, O.M.; Simpson, R.J.; Salim, A.; et al. FunRich: An open access standalone functional enrichment and interaction network analysis tool. Proteomics 2015, 15, 2597–2601. [Google Scholar] [CrossRef] [PubMed]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. REVIGO Summarizes and Visualizes Long Lists of Gene Ontology Terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef]
- Park, Y.J.; Jeong, Y.U.; Kong, W.S. Genome sequencing and carbohydrate-active enzyme (CAZyme) repertoire of the white rot fungus Flammulina elastica. Int. J. Mol. Sci. 2018, 19, 2379. [Google Scholar] [CrossRef]
- Yoon, J.J.; Hattori, T.; Shimada, M. A metabolic role of the glyoxylate and tricarboxylic acid cycles for development of the copper-tolerant brown-rot fungus Fomitopsis palustris. FEMS Microbiol. Lett. 2002, 217, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.J.; Munir, E.; Miyasou, H.; Hattori, T.; Shimada, M.; Terashita, T. A possible role of the key enzymes of the glyoxylate and gluconeogenesis pathways for fruit-body formation of the wood-rotting basidiomycete Flammulina velutipes. Mycoscience 2002, 43, 327–332. [Google Scholar] [CrossRef]
- Kües, U. From Two to Many: Multiple Mating Types in Basidiomycetes. Fungal Biol. Rev. 2015, 29, 126–166. [Google Scholar] [CrossRef]
- Carreño-Ruiz, S.D.; Lázaro, A.a.Á.; García, S.C.; Hernández, R.G.; Chen, J.; Navarro, G.K.G.; Fajardo, L.V.G.; Pérez, N.D.C.J.; Cruz, M.T.D.L.; Blanco, J.C.; et al. New Record of Schizophyllum (Schizophyllaceae) from Mexico and the Confirmation of Its Edibility in the Humid Tropics. Phytotaxa 2019, 413, 137–148. [Google Scholar] [CrossRef]
- Ahmed, T.; Suzauddula, M.; Akter, K.; Hossen, M.; Islam, M.N. Green Technology for Fungal Protein Extraction—A Review. Separations 2024, 11, 186. [Google Scholar] [CrossRef]
- Taunk, K.; Paul, D.; Dabhi, R.; Venkatesh, C.; Jajula, S.; Naik, V.; Tamhankar, A.; Naiya, T.; Kumar Santra, M.; Rapole, S.A. Single step and rapid protein extraction protocol developed for cell lines and tissues: Compatible for gel based and gel free proteomic approaches. Methods 2023, 220, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Chi, C.H.; Cho, S.J. Extraction solvent’s effect on protein profiles of processed soybean meal. Food Biosci. 2020, 37, 100734. [Google Scholar] [CrossRef]
- Jaipal Reddy, P.; Anand Rao, A.; Malhotra, D.; Sharma, S.; Kumar, R.; Jain, R.; Gollapalli, K.; Pendharkar, N.; Rapole, S.; Srivastava, S. A simple protein extraction method for proteomic analysis of diverse biological specimens. Curr. Proteom. 2013, 10, 298–311. [Google Scholar] [CrossRef]
- Santa, C.; Anjo, S.I.; Manadas, B. Protein precipitation of diluted samples in SDS-containing buffer with acetone leads to higher protein recovery and reproducibility in comparison with TCA/acetone approach. Proteomics 2016, 16, 1847–1851. [Google Scholar] [CrossRef] [PubMed]
- Ohm, R.A.; de Jong, J.F.; Lugones, L.G.; Aerts, A.; Kothe, E.; Stajich, J.E.; de Vries, R.P.; Record, E.; Levasseur, A.; Baker, S.E.; et al. Genome sequence of the model mushroom Schizophyllum commune. Nat. Biotechnol. 2010, 28, 957–963. [Google Scholar] [CrossRef]
- Ohtake, S.; Wang, Y.J. Trehalose: Current use and future applications. J. Pharm. Sci. 2011, 100, 2020–2053. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wu, X.; Wei, G.; Qu, J.; Qiang, C.; Huang, C.; Zhang, J. Protective roles of trehalose in Pleurotus pulmonarius during heat stress response. J. Integr. Agric. 2019, 18, 428–437. [Google Scholar] [CrossRef]
- Agarwal, N.; Singh, S.P. A novel trehalose synthase for the production of trehalose and trehalulose. Microbiol Spectr. 2021, 9, e0133321. [Google Scholar] [CrossRef]
- Chen, A.; Tapia, H.; Goddard, J.M.; Gibney, P.A. Trehalose and its applications in the food industry. Compr. Rev. Food Sci. Food Saf. 2022, 21, 5004–5037. [Google Scholar] [CrossRef] [PubMed]
- Drula, E.; Garron, M.L.; Dogan, S.; Lombard, V.; Henrissat, B.; Terrapon, N. The carbohydrate-active enzyme database: Functions and literature. Nucleic Acids Res. 2022, 50, D571–D577. [Google Scholar] [CrossRef] [PubMed]
- Chettri, D.; Verma, A.K. Biological significance of carbohydrate active enzymes and searching their inhibitors for therapeutic applications. Carbohydr. Res. 2023, 529, 108853. [Google Scholar] [CrossRef] [PubMed]
- Cantarel, B.L.; Lombard, V.; Henrissat, B. Complex carbohydrate utilization by the healthy human microbiome. PLoS ONE. 2012, 7, e28742. [Google Scholar] [CrossRef]
- Tashkandi, M.; Baz, L. Function of CAZymes encoded by highly abundant genes in rhizosphere microbiome of Moringa oleifera. Saudi J. Biol. Sci. 2023, 30, 103578. [Google Scholar] [CrossRef]
- Ye, S.; Shah, B.R.; Li, J.; Liang, H.; Zhan, F.; Geng, F.; Li, B. A critical review on interplay between dietary fibers and gut microbiota. Trends Food Sci. 2022, 124, 237–249. [Google Scholar] [CrossRef]
- Koenig, J.E.; Spor, A.; Scalfone, N.; Fricker, A.D.; Stombaugh, J.; Knight, R.; Angenent, L.T.; Ley, R.E. Succession of microbial consortia in the developing infant gut microbiome. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. S1), 4578–4585. [Google Scholar] [CrossRef]
- Onyango, S.O.; Juma, J.; De Paepe, K.; Van de Wiele, T. Oral and gut microbial carbohydrate-active enzymes landscape in health and disease. Front. Microbiol. 2021, 12, 653448. [Google Scholar] [CrossRef] [PubMed]
- Amin, K.; Tranchimand, S.; Benvegnu, T.; Abdel-Razzak, Z.; Chamieh, H. Glycoside hydrolases and glycosyltransferases from hyperthermophilic archaea: Insights on their characteristics and applications in biotechnology. Biomolecules 2021, 11, 1557. [Google Scholar] [CrossRef]
- Morosanova, M.A.; Fedorova, T.V.; Polyakova, A.S.; Morosanova, E.I. Agaricus bisporus crude extract: Characterization and analytical application. Molecules 2020, 25, 5996. [Google Scholar] [CrossRef] [PubMed]
- Ban, X.; Dhoble, A.S.; Li, C.; Gu, Z.; Hong, Y.; Cheng, L.; Holler, T.P.; Kaustubh, B.; Li, Z. Bacterial 1, 4-α-glucan branching enzymes: Characteristics, preparation and commercial applications. Crit. Rev. Biotechnol. 2020, 40, 380–396. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Lin, R.; Xu, K.; Guo, L.; Yu, H. Comparative proteomic analysis within the developmental stages of the mushroom white Hypsizygus marmoreus. J. Fungi 2021, 7, 1064. [Google Scholar] [CrossRef] [PubMed]
- Shankar, A.; Jain, K.K.; Kuhad, R.C.; Sharma, K.K. Comparison of lignocellulosic enzymes and CAZymes between ascomycetes (Trichoderma) and basidiomycetes (Ganoderma) species: A proteomic approach. Z. Naturforsch. C 2023, 80, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Roukas, T.; Kotzekidou, P. From food industry wastes to second generation bioethanol: A review. Rev. Environ. Sci. Bio/Technol. 2022, 21, 299–329. [Google Scholar] [CrossRef]
- Geng, X.; Tian, G.; Zhao, Y.; Zhao, L.; Wang, H.; Ng, T.B. A Fungal α-Galactosidase from Tricholoma matsutake with broad substrate specificity and good hydrolytic activity on raffinose family oligosaccharides. Molecules 2015, 20, 13550–13562. [Google Scholar] [CrossRef]
- Bhatia, S.; Singh, A.; Batra, N.; Singh, J. Microbial production and biotechnological applications of α-galactosidase. Int. J. Biol. Macromol. 2020, 150, 1294–1313. [Google Scholar] [CrossRef]
- Bangoria, P.; Patel, A.; Shah, A.R. Characterization of a fungal α-galactosidase and its synergistic effect with β-mannanase for hydrolysis of galactomannan. Carbohydr. Res. 2023, 531, 108893. [Google Scholar] [CrossRef]
- Saqib, S.; Akram, A.; Halim, S.A.; Tassaduq, R. Sources of β-galactosidase and its applications in food industry. Biotech 2017, 7, 79. [Google Scholar] [CrossRef]
- Volford, B.; Varga, M.; Szekeres, A.; Kotogán, A.; Nagy, G.; Vágvölgyi, C.; Papp, T.; Takó, M. β-galactosidase-producing isolates in mucoromycota: Screening, enzyme production, and applications for functional oligosaccharide synthesis. J. Fungi 2021, 7, 229. [Google Scholar] [CrossRef]
- Singh, R.V.; Sambyal, K. β-Galactosidase as an industrial enzyme: Production and potential. Chem. Pap. 2023, 77, 11–31. [Google Scholar] [CrossRef]
- Lu, L.; Guo, L.; Wang, K.; Liu, Y.; Xiao, M. β-Galactosidases: A great tool for synthesizing galactose-containing carbohydrates. Biotechnol. Adv. 2020, 39, 107465. [Google Scholar] [CrossRef]
- Ibrahim, O. Erythritol chemical structure, biosynthesis pathways, properties, applications, and production. Int. J. Microbiol. Biotechnol. 2021, 6, 59. [Google Scholar] [CrossRef]
- Kuroda, K.; Hirakawa, S.; Suzuki, M.; Shinji, K.; Ogasa, K.; Uraji, T.; Amachi, T.; Ueda, M. Growth acceleration of plants and mushroom by erythritol. Plant Biotechnol. 2008, 25, 489–492. [Google Scholar] [CrossRef]
- Diamantopoulou, P.; Papanikolaou, S. Biotechnological production of sugar-alcohols: Focus on Yarrowia lipolytica and edible/medicinal mushrooms. Process Biochem. 2023, 124, 113–131. [Google Scholar] [CrossRef]
- Munir, E.; Yoon, J.J.; Tokimatsu, T.; Hattori, T.; Shimada, M. New role for glyoxylate cycle enzymes in wood-rotting basidiomycetes in relation to biosynthesis of oxalic acid. J. Wood Sci. 2001, 47, 368–373. [Google Scholar] [CrossRef]
- Murry, R.; Kniemeyer, O.; Krause, K.; Saiardi, A.; Kothe, E. Crosstalk between Ras and inositol phosphate signaling revealed by lithium action on inositol monophosphatase in Schizophyllum commune. Adv. Biol. Regul. 2019, 72, 78–88. [Google Scholar] [CrossRef]
- Xu, L.; Guo, L.; Yu, H. Label-free comparative proteomics analysis revealed heat stress responsive mechanism in Hypsizygus marmoreus. Front. Microbiol. 2021, 11, 541967. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Sample A1 | Sample A2 | Sample B | ||||
|---|---|---|---|---|---|---|
| Cellular Component | # Proteins | p-Value | # Proteins | p-Value | # Proteins | p-Value |
| Cytoplasm | 71 | 8.39 × 10−12 * | 139 | 4.03 × 10−26 * | 99 | 2.53 × 10−17 * |
| Ribonucleoprotein complex | 51 | 1.15 × 10−18 * | 59 | 8.21 × 10−10 * | 37 | 8.88 × 10−4 * |
| Ribosome | 52 | 5.46 × 10−24 * | 56 | 4.38 × 10−12 * | 38 | 1.71 × 10−6 * |
| Mitochondrial inner membrane | 12 | 1.00 | 31 | 6.23 × 10−4 * | 11 | 1.00 |
| Eukaryotic translation initiation factor 3 complex | 5 | 1.00 | 11 | 1.74 × 10−5 * | 8 | 9.40 × 10−3 * |
| Eukaryotic 48S preinitiation complex | 3 | 1.00 | 9 | 3.73 × 10−4 * | 7 | 0.02 |
| Proteasome regulatory particle, base subcomplex | 8 | 4.06 × 10−5 * | 9 | 3.73 × 10−4 * | 7 | 0.02 |
| Eukaryotic 43S preinitiation complex | 3 | 1.00 | 9 | 3.73 × 10−4 * | 7 | 0.02 |
| Chaperonin-containing T-complex | 0 | 1.00 | 8 | 1.73 × 10−3 * | 8 | 8.69 × 10−5 * |
| Proteasome core complex, alpha-subunit complex | 7 | 4.80 × 10−5 * | 7 | 7.99 × 10−3 * | 7 | 5.85 × 10−4 * |
| Proteasome core complex, beta-subunit complex | 6 | 4.62 × 10−4 * | 6 | 0.04 | 5 | 0.14 |
| Proteasome complex | 6 | 4.62 × 10−4 * | 6 | 0.04 | 5 | 0.14 |
| Cellular Component | # Identified Proteins | Quantification (PSMs) | Fold Change Value | ||
|---|---|---|---|---|---|
| Sample A2 | Sample A1 | Sample A2 | Sample A1 | ||
| Chaperonin-Containing T-Complex | 8 | 0 | 147.5 | 0 | 3280.7 |
| Microtubule | 9 | 3 | 75.5 | 2.4 | 7.0 |
| Arp2/3 Protein Complex | 6 | 3 | 21.9 | 1.3 | 3.7 |
| Eukaryotic 43S Preinitiation Complex | 9 | 3 | 72 | 4.3 | 3.7 |
| Eukaryotic 48S Preinitiation Complex | 9 | 3 | 72 | 4.3 | 3.7 |
| Eukaryotic Translation Initiation Factor 3 Complex | 11 | 5 | 84.1 | 5.3 | 3.5 |
| Golgi Membrane | 14 | 5 | 85.6 | 5.6 | 3.4 |
| Mitochondrial Inner Membrane | 31 | 12 | 129.2 | 14.1 | 2.0 |
| Plasma Membrane | 17 | 6 | 106.5 | 12.3 | 1.9 |
| Proteasome Regulatory Particle, Base Subcomplex | 9 | 8 | 120.7 | 15.7 | 1.7 |
| Cytosol | 10 | 3 | 42.1 | 7 | 1.3 |
| Endoplasmic Reticulum Membrane | 24 | 8 | 75.1 | 12.5 | 1.3 |
| Proteasome Complex | 6 | 6 | 42 | 7.3 | 1.2 |
| Nucleus | 81 | 38 | 392.6 | 70 | 1.2 |
| Spliceosomal Complex | 16 | 7 | 32.7 | 6 | 1.2 |
| Mitochondrion | 29 | 17 | 207.9 | 41.3 | 1.1 |
| Respirasome | 9 | 5 | 28.7 | 5.7 | 1.1 |
| Cytoplasm | 139 | 71 | 1051.5 | 210 | 1.1 |
| Membrane | 183 | 76 | 601.2 | 124 | 1.1 |
| Cellular Component | # Identified Proteins | Quantification (PSMs) | Fold Change Value | ||
|---|---|---|---|---|---|
| Sample A2 | Sample B | Sample A2 | Sample B | ||
| Respirasome | 9 | 3 | 28.7 | 5 | 3.0 |
| Endoplasmic Reticulum Membrane | 24 | 9 | 75.1 | 17.2 | 2.3 |
| Golgi Membrane | 14 | 9 | 85.6 | 20.7 | 2.2 |
| Ribosome | 56 | 38 | 449.7 | 108.9 | 2.2 |
| Ribonucleoprotein Complex | 59 | 37 | 438.8 | 109.6 | 2.1 |
| Mitochondrial Inner Membrane | 31 | 11 | 129.2 | 36.4 | 1.9 |
| Cellular Anatomical Entity | 7 | 5 | 53.4 | 16.2 | 1.7 |
| Mitochondrion | 29 | 20 | 207.9 | 63.2 | 1.7 |
| Spliceosomal Complex | 16 | 7 | 32.7 | 10 | 1.7 |
| Chaperonin-Containing T-Complex | 8 | 8 | 147.5 | 47.7 | 1.6 |
| Small Ribosomal Subunit | 7 | 6 | 78.8 | 25.7 | 1.6 |
| Cytoplasm | 139 | 99 | 1051.5 | 438.6 | 1.3 |
| Eukaryotic Translation Initiation Factor 3 Complex | 11 | 8 | 84.1 | 36.8 | 1.2 |
| Eukaryotic 43S Preinitiation Complex | 9 | 7 | 72 | 31.7 | 1.2 |
| Eukaryotic 48S Preinitiation Complex | 9 | 7 | 72 | 31.7 | 1.2 |
| Proteasome Regulatory Particle, Base Subcomplex | 9 | 7 | 120.7 | 53.7 | 1.2 |
| Membrane | 183 | 109 | 601.2 | 289.8 | 1.1 |
| Microtubule | 9 | 7 | 75.5 | 37 | 1.1 |
| Plasma Membrane | 17 | 15 | 106.5 | 53.4 | 1.1 |
| Endoplasmic Reticulum | 13 | 9 | 92 | 47 | 1.0 |
| Key Enzyme | Trehalose Biosynthesis | PSMs Sample A1 | PSMs Sample A2 | PSMs Sample B |
| D8QC44 | Trehalose-6-phosphate synthase (EC 2.4.1.15) (UDP-glucose-glucosephosphate glucosyltransferase) | 0.0 * | 10.7 | 2.0 * |
| D8QIA1 | Trehalase (EC 3.2.1.28) (Alpha-trehalose glucohydrolase) | 0.0 | 2.3 | 0.0 |
| Q2HZZ3 | Trehalose phosphorylase (EC 2.4.1.231) | 13.7 * | 42.0 | 23.0 * |
| Response to oxidative stress | PSMs Sample A1 | PSMs Sample A2 | PSMs Sample B | |
| D8PT51 | Superoxide dismutase (EC 1.15.1.1) | 8.3 * | 20.0 | 3.0 * |
| D8Q7V6 | Superoxide dismutase (EC 1.15.1.1) | 1.7 * | 8.0 | 0.0 |
| S5VRV3 | Superoxide dismutase (EC 1.15.1.1) | 0.7 | 2.3 | 0.0 * |
| D8PMW1 | Catalase (EC 1.11.1.6) | 0.0 | 2.7 | 4.3 |
| D8PTL6 | Catalase (EC 1.11.1.6) | 1.3 * | 23.0 | 4.0 * |
| D8QLS5 | Heme peroxidase | 0.0 | 1.3 | 0.0 |
| D8QBR0 | Peroxidase (EC 1.11.1.-) | 1.0 * | 12.7 | 5.7 * |
| Pentose-phosphate Shunt | PSMs Sample A1 | PSMs Sample A2 | PSMs Sample B | |
| D8PMS0 | 6-phosphogluconate dehydrogenase, decarboxylating (EC 1.1.1.44) | 15.7 * | 35.7 | 14.3 * |
| D8PP98 | Ribulose-phosphate 3-epimerase (EC 5.1.3.1) | 1.7 | 2.0 | 2.7 |
| D8PNV4 | Glucose-6-phosphate 1-dehydrogenase (EC 1.1.1.49) | 0.7 * | 15.7 | 1.7 * |
| D8PNV6 | Glucose-6-phosphate 1-dehydrogenase (EC 1.1.1.49) | 1.0 | 3.7 | 0.0 * |
| D8PN33 | 6-phosphogluconolactonase (6PGL) (EC 3.1.1.31) | 1.7 * | 5.0 | 0.0 * |
| D8PXY7 | Transaldolase (EC 2.2.1.2) | 14.3 | 22.7 | 21.7 |
| D8PWF8 | Transketolase (EC 2.2.1.1) | 5.7 * | 24.0 | 4.3 * |
| Tricarboxylic acid cycle | PSMs Sample A1 | PSMs Sample A2 | PSMs Sample B | |
| D8QDQ7 | Isocitrate dehydrogenase [NADP] (EC 1.1.1.42) | 1.7* | 5.3 | 0.0 * |
| D8Q9D5 | Malate synthase (EC 2.3.3.9) | 9.7 * | 33.7 | 14.7 * |
| D8Q486 | Oxalate oxidase | 3.3 * | 13.3 | 10.3 * |
| D8QFS7 | Malate dehydrogenase (EC 1.1.1.37) | 3.0 * | 15.3 | 21.7 * |
| D8QLH9 | Malate dehydrogenase (EC 1.1.1.37) | 3.0 * | 15.7 | 19.3 * |
| D8Q525 | Citrate synthase | 6.7 * | 22.7 | 2.0 * |
| D8PX28 | Citrate synthase | 0.0 * | 8.7 | 0.7 * |
| D8QEN1 | ATP citrate synthase (EC 2.3.3.8) (ATP-citrate (pro-S-)-lyase) (Citrate cleavage enzyme) | 0.7 * | 54.7 | 31.0 * |
| D8PMP1 | Isocitrate lyase | 1.7 | 1.7 | 0.0 * |
| D8Q077 | Isocitrate lyase | 8.7 * | 26.7 | 13.7 * |
| D8PZN4 | Trehalase (EC 3.2.1.28) (Alpha-trehalose glucohydrolase) | 6.7 * | 22.7 | 0.0 * |
| Carbohydrate-Active Enzymes (CAZymes) | ||||
| Carbohydrate metabolic processes (GHs class) | PSMs Sample A1 | PSMs Sample A2 | PSMs Sample B | |
| D8PUK9 | Glycoside hydrolase family 31 protein/ beta-glucosidase | 2.7 * | 12.0 | 15.7 |
| D8PJY8 | Glycoside hydrolase family 125 protein | 0.3 | 4.7 | 6.7 |
| D8PK12 | Glycoside hydrolase family 43 protein | 0.0 | 0.0 | 1.0 |
| D8PL55 | Glycoside hydrolase family 43 and carbohydrate-binding module family 35 protein | 1.3 | 1.3 | 2.7 |
| D8PLY0 | Glycoside hydrolase family 76 protein | 0.0 | 0.3 | 0.7 |
| D8PMG3 | Glycoside hydrolase family 92 protein | 1.7 | 6.0 | 8.0 |
| D8PMG6 | Glycoside hydrolase family 92 protein | 0.3 | 3.0 | 4.7 |
| D8PMH4 | Glycoside hydrolase family 2 protein | 1.7 | 1.7 | 0.0 * |
| D8PPV4 | Glycoside hydrolase family 92 protein | 3.3 | 0.7 | 5.7 * |
| D8PS75 | Glycoside hydrolase family 18 protein | 1.0 * | 5.0 | 3.3 |
| D8PSJ0 | Glycoside hydrolase family 2 protein | 0.0 | 0.0 | 0.7 |
| D8PVQ7 | Glycoside hydrolase family 5 protein/ glucan 1,3- beta-glucosidase | 0.7 | 0.7 | 1.0 |
| D8PXL2 | Glycoside hydrolase family 5 protein/ glucan 1,3- beta-glucosidase | 0.3 | 0.0 | 0.0 |
| D8PXU7 | Glycoside hydrolase family 15 protein | 1.7 | 4.0 | 0.7 * |
| D8QJY6 | Glycoside hydrolase family 95 protein | 0.0 | 1.0 | 1.3 |
| D8PJV0 | Beta-glucosidase (EC 3.2.1.21) | 0.0 | 1.3 | 0.0 |
| D8PS34 | 1,4-alpha-glucan-branching enzyme (EC 2.4.1.18) (Glycogen-branching enzyme) | 1.3 | 20.3 | 4.0 |
| D8PNW5 | Beta-glucosidase | 0.0 | 1.3 | 2.3 |
| D8PQP1 | glucan 1,3-beta-glucosidase | 0.7 * | 3.7 | 0.0 * |
| D8PU51 | Beta-glucosidase | 5.3 | 10.0 | 22.0 * |
| D8PUK9 | Beta-glucosidase | 2.7 * | 12.0 | 15.7 |
| D8Q8Q6 | Beta-glucosidase | 0.0 | 2.0 | 0.0 |
| D8Q3Q9 | Beta-galactosidase (EC 3.2.1.23) | 1.0 | 7.3 | 8.0 |
| D8PK76 | Beta-galactosidase (EC 3.2.1.23) | 2.0 | 5.7 | 7.7 |
| D8PM67 | Beta-galactosidase (EC 3.2.1.23) | 0.0 | 0.3 | 0.3 |
| D8Q3Q9 | Beta-galactosidase (EC 3.2.1.23) | 1.0 | 7.3 | 8.0 |
| D8PQ92 | Alpha-galactosidase (EC 3.2.1.22) (Melibiase) | 3.3 * | 12.3 | 15.7 |
| D8QGJ4 | Alpha-mannosidase (EC 3.2.1.24) | 16.7 | 32.0 | 7.3 * |
| Carbohydrate metabolic processes (GTs class) | PSMs Sample A1 | PSMs Sample A2 | PSMs Sample B | |
| D8Q7P4 | Glycosyltransferase family 32 protein | 0.0 | 1.0 | 0.0 |
| D8QHQ3 | Glycosyltransferase family 20 protein | 0.3 * | 10.7 | 0.0 * |
| D8PXY7 | Transaldolase (EC 2.2.1.2) | 14.3 | 22.7 | 21.7 |
| Inositol phosphatase dephosphorylation | PSMs Sample A1 | PSMs Sample A2 | PSMs Sample B | |
| D8PM90 | Inositol-1-monophosphatase (EC 3.1.3.25) | 2.0 | 1.7 | 3.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Desiderio, A.; Goppa, L.; Santambrogio, C.; Brocca, S.; Buratti, S.; Girometta, C.E.; Sarkar, M.; Venuti, M.T.; Savino, E.; Rossi, P.; et al. Improving the Proteome-Mining of Schizophyllum commune to Enhance Medicinal Mushroom Applications. J. Fungi 2025, 11, 120. https://doi.org/10.3390/jof11020120
Desiderio A, Goppa L, Santambrogio C, Brocca S, Buratti S, Girometta CE, Sarkar M, Venuti MT, Savino E, Rossi P, et al. Improving the Proteome-Mining of Schizophyllum commune to Enhance Medicinal Mushroom Applications. Journal of Fungi. 2025; 11(2):120. https://doi.org/10.3390/jof11020120
Chicago/Turabian StyleDesiderio, Anthea, Lorenzo Goppa, Carlo Santambrogio, Stefania Brocca, Simone Buratti, Carolina Elena Girometta, Meghma Sarkar, Maria Teresa Venuti, Elena Savino, Paola Rossi, and et al. 2025. "Improving the Proteome-Mining of Schizophyllum commune to Enhance Medicinal Mushroom Applications" Journal of Fungi 11, no. 2: 120. https://doi.org/10.3390/jof11020120
APA StyleDesiderio, A., Goppa, L., Santambrogio, C., Brocca, S., Buratti, S., Girometta, C. E., Sarkar, M., Venuti, M. T., Savino, E., Rossi, P., & Ferrari, E. (2025). Improving the Proteome-Mining of Schizophyllum commune to Enhance Medicinal Mushroom Applications. Journal of Fungi, 11(2), 120. https://doi.org/10.3390/jof11020120